
Multi-Omics and Experimental Validation Identify GPX7 and Glutathione-Associated Oxidative Stress as Potential Biomarkers in Ischemic Stroke

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
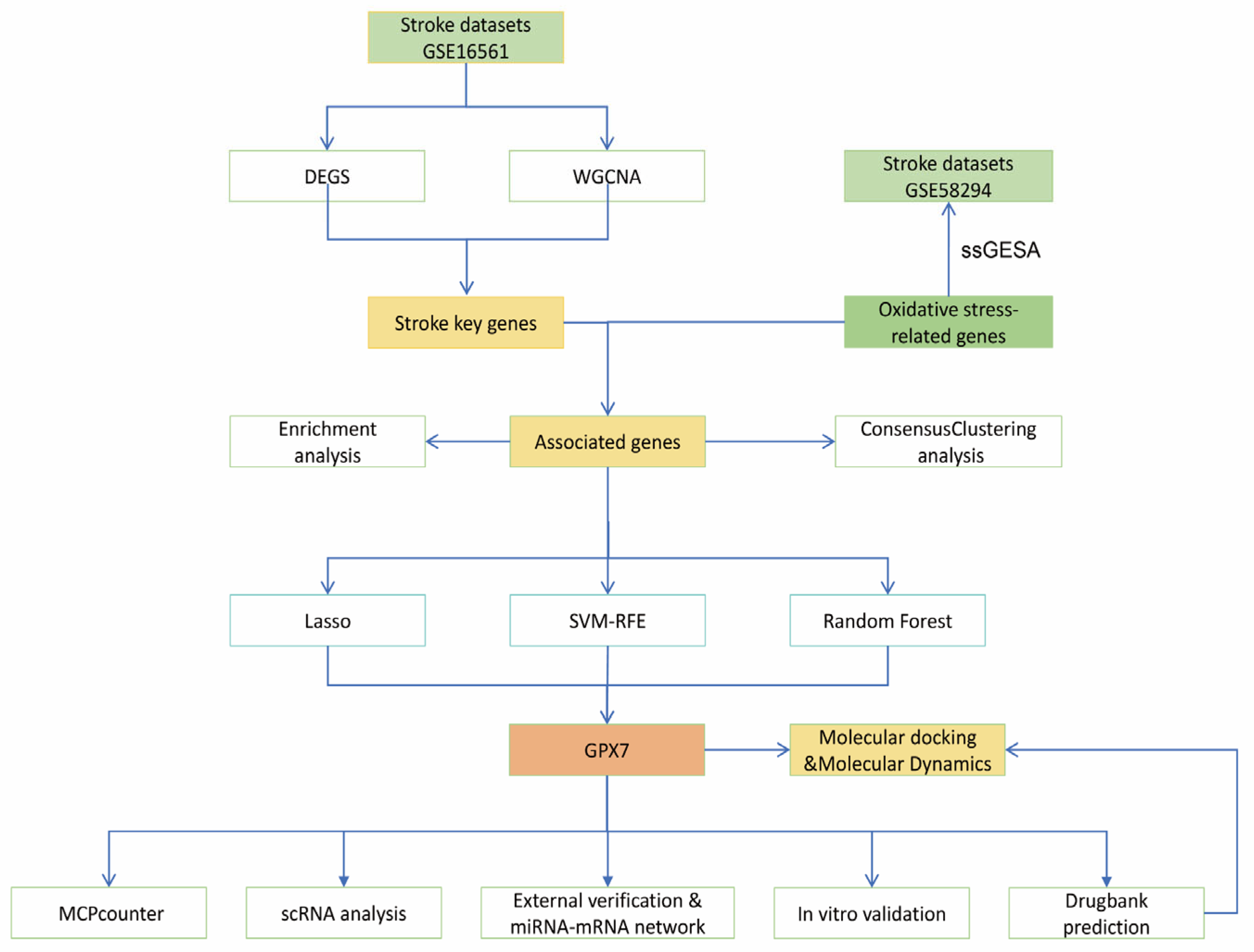
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Identification and Functional Annotation of Differentially Expressed Genes (DEGs)
2.3. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.4. Functional Enrichment Analysis
2.5. Consensus Clustering Analysis and Subtype Differences
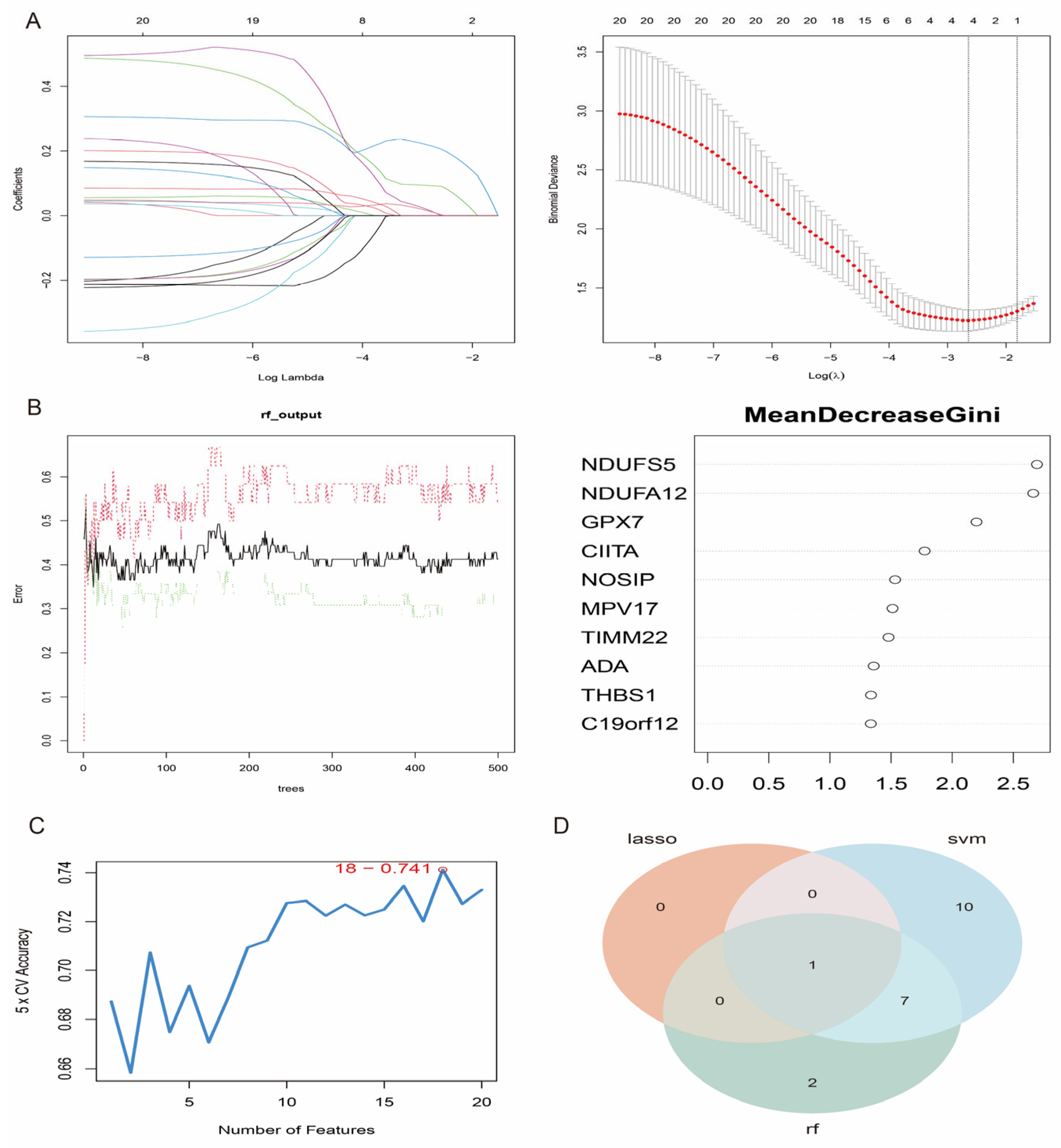
2.6. Machine Learning Algorithms for GPX7 Identification
2.7. Comparative Analysis of GPX7 High and Low Expression Groups
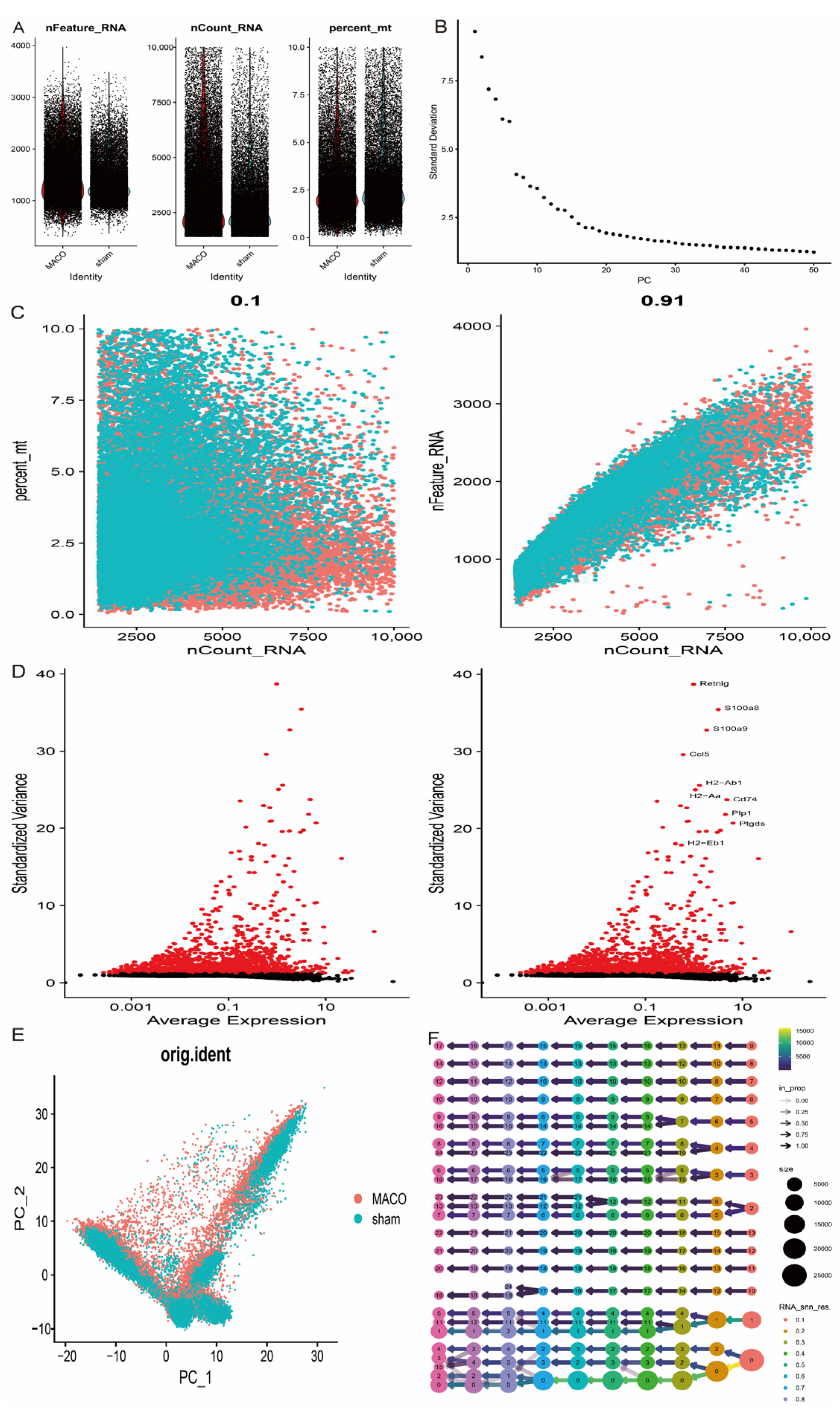
2.8. Single-Cell Data Analysis
2.9. Drug Prediction and Molecular Docking
2.10. pMCAO Model Construction
2.11. qRT-PCR Analysis
2.12. Statistical Analysis
3. Results
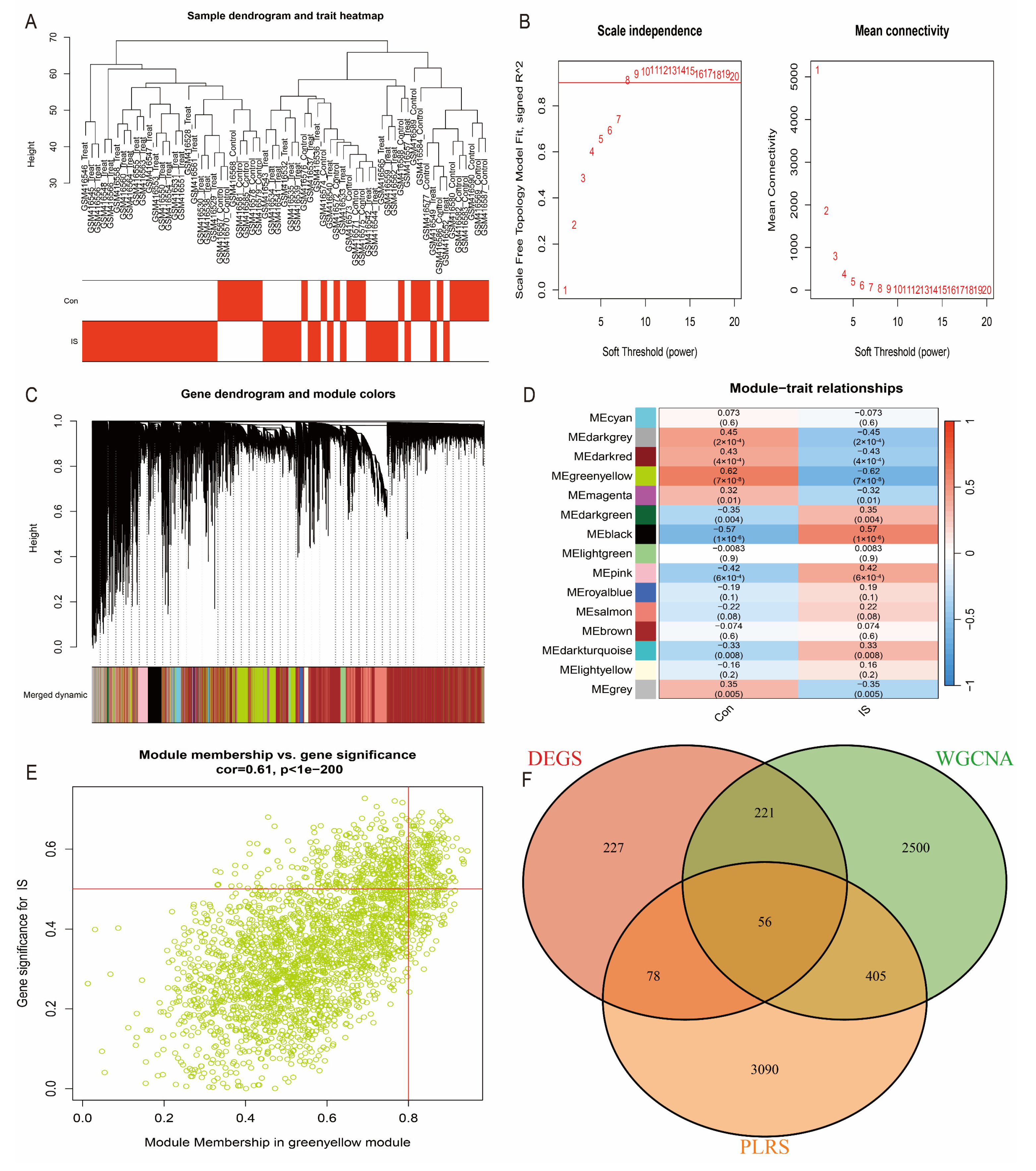
3.1. IS Can Be Stratified by Oxidative Stress-Related Genes
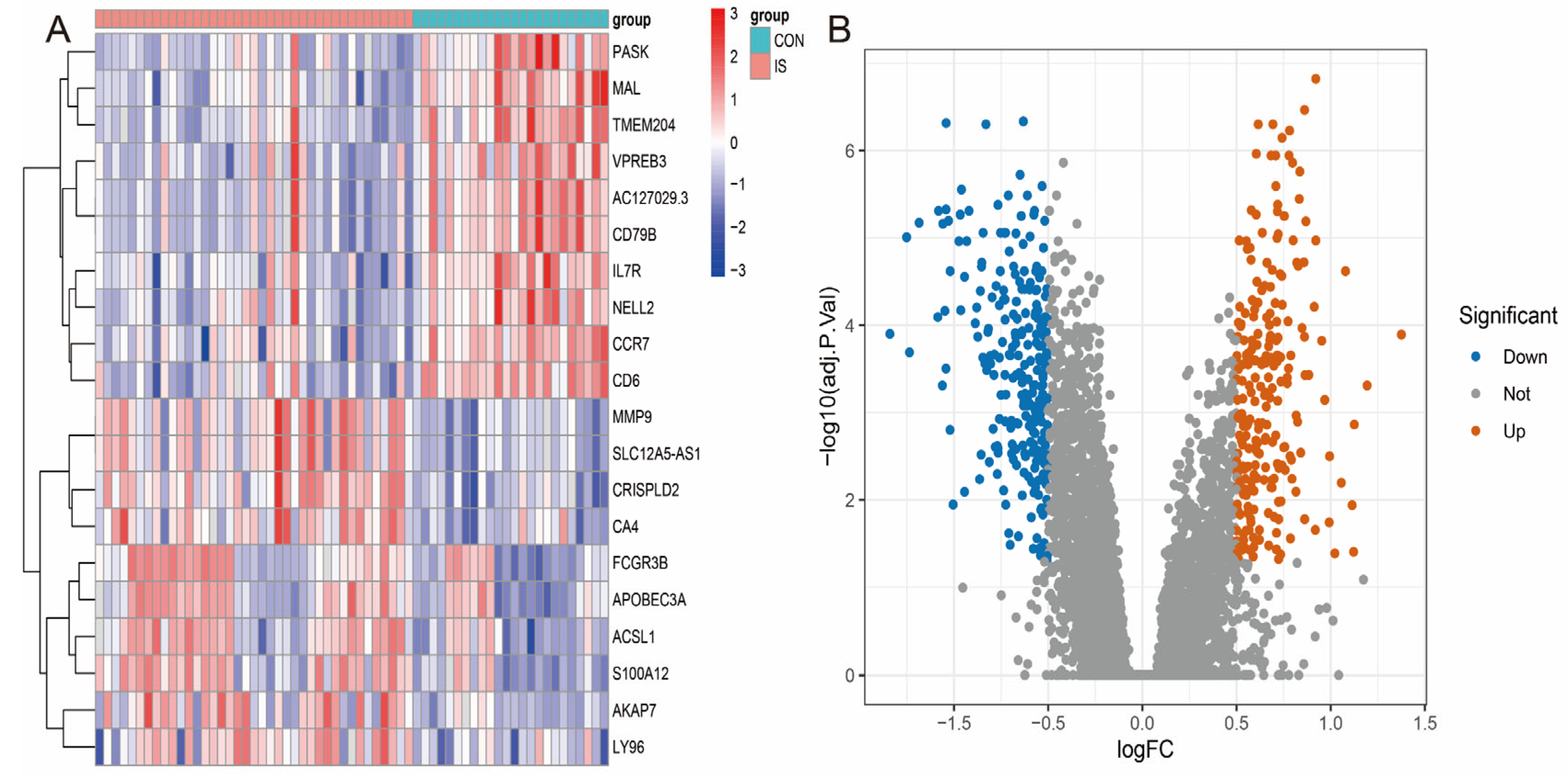
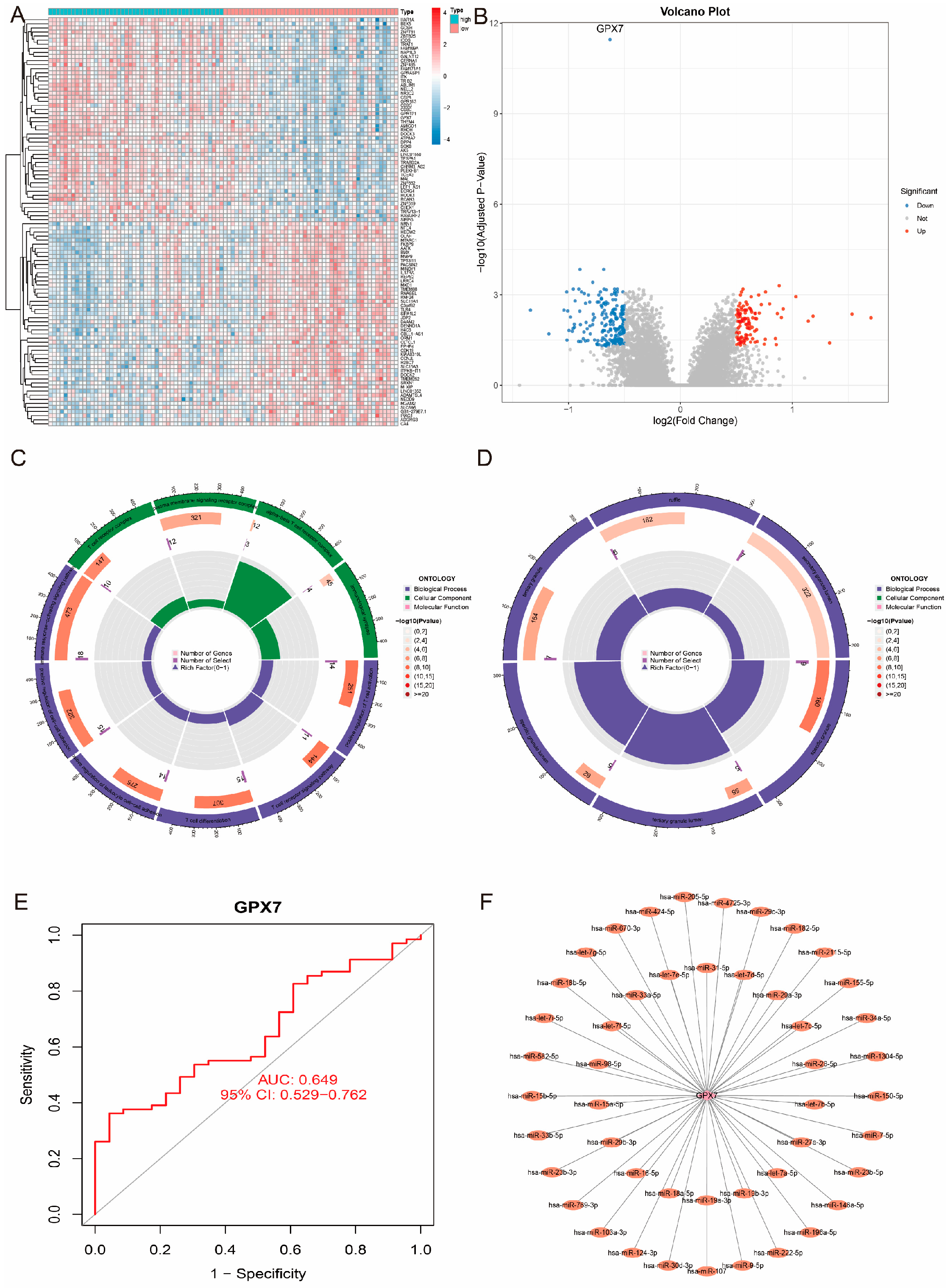
3.2. Differentially Expressed Genes (DEGs) in the GSE16561 Cohort
3.3. WGCNA Analysis to Identify Hub Genes
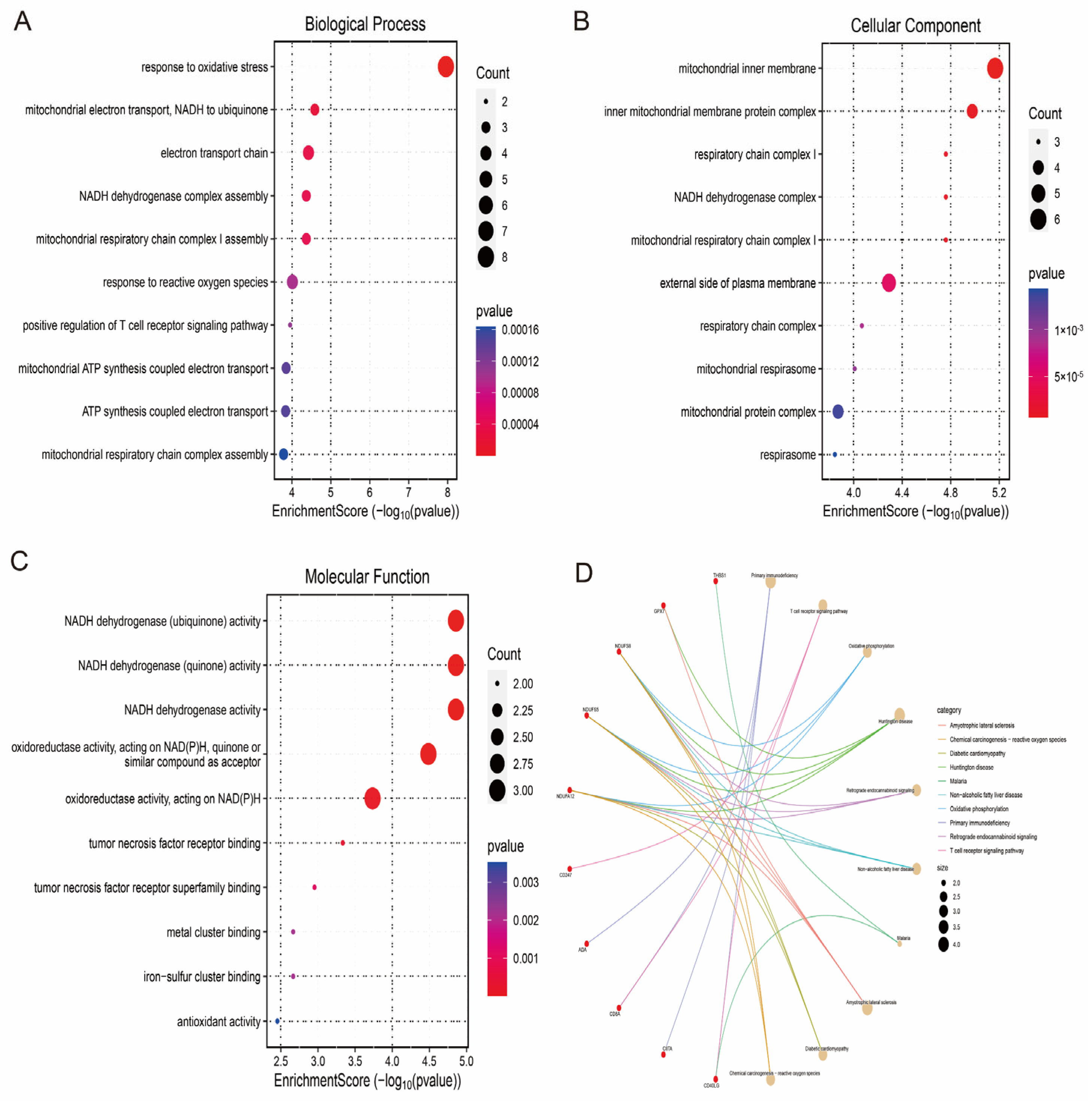
3.4. Enrichment Analysis Reveals the Functions of Hub Genes
3.5. Subtype Classification and Immune Infiltration Analysis
3.6. Feature Gene Selection
3.7. Functional Enrichment Analysis of GPX7-Related Genes and Their Predictive Ability
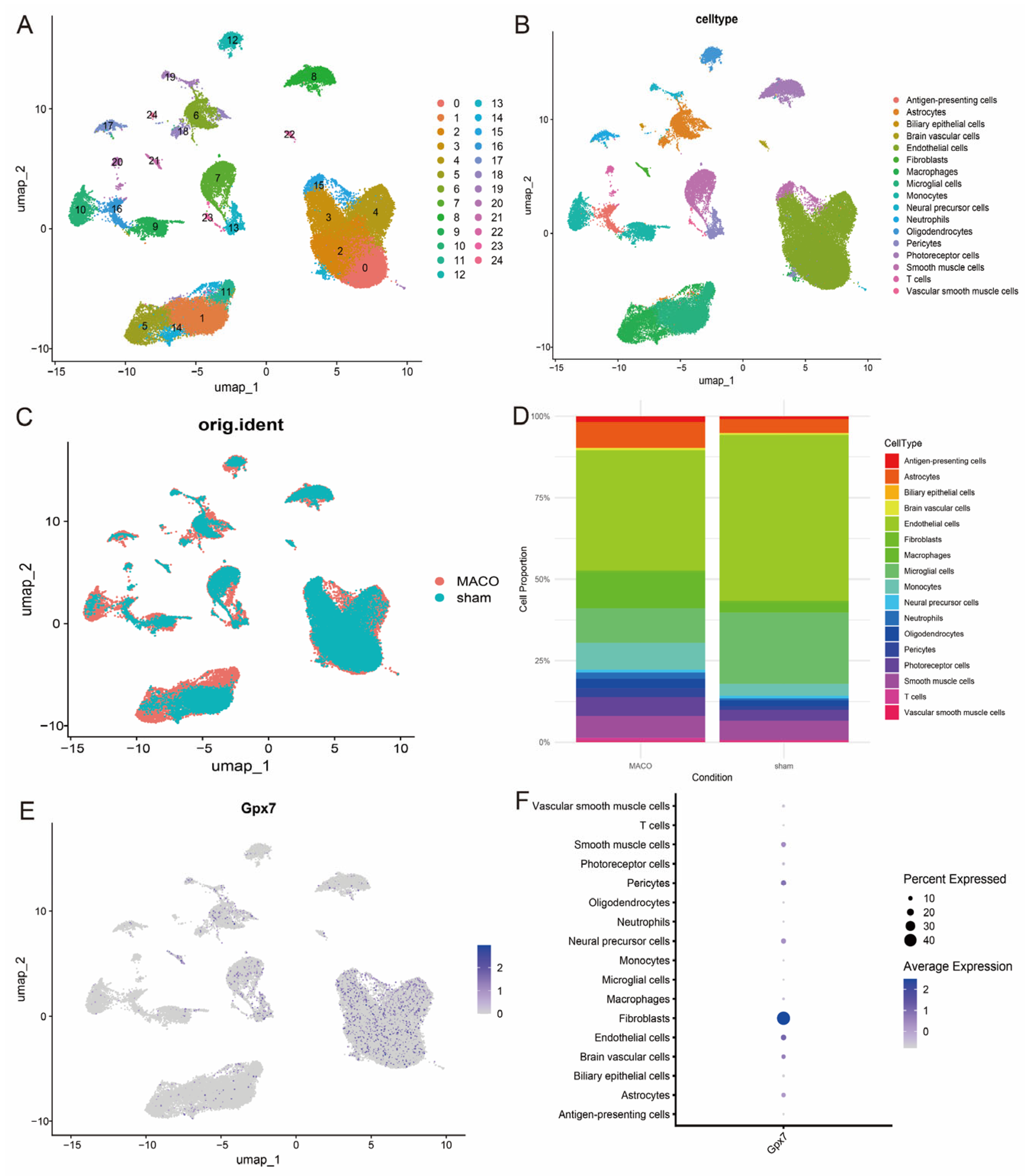
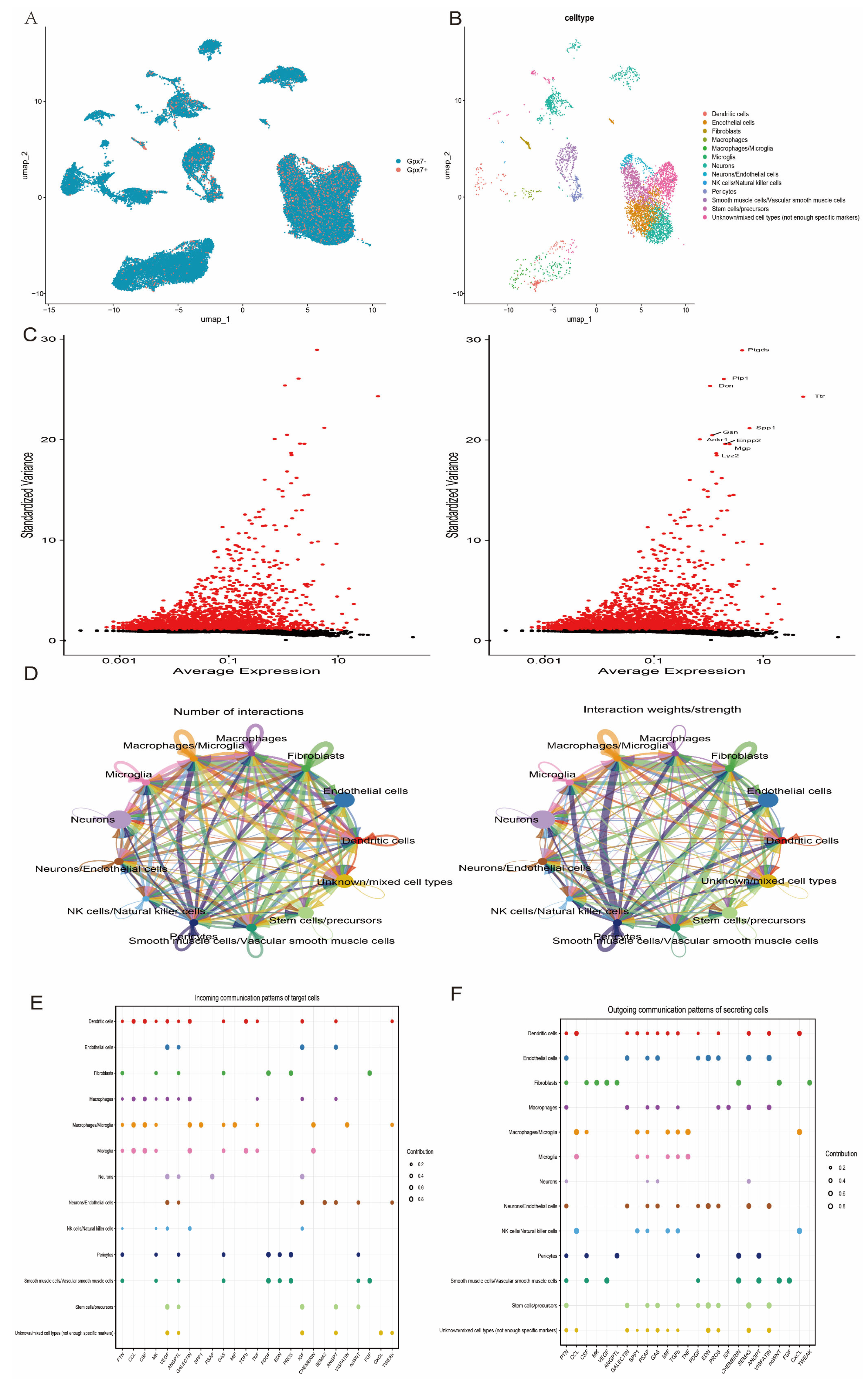
3.8. Single-Cell Transcriptome Data Analysis Reveals Cellular Heterogeneity
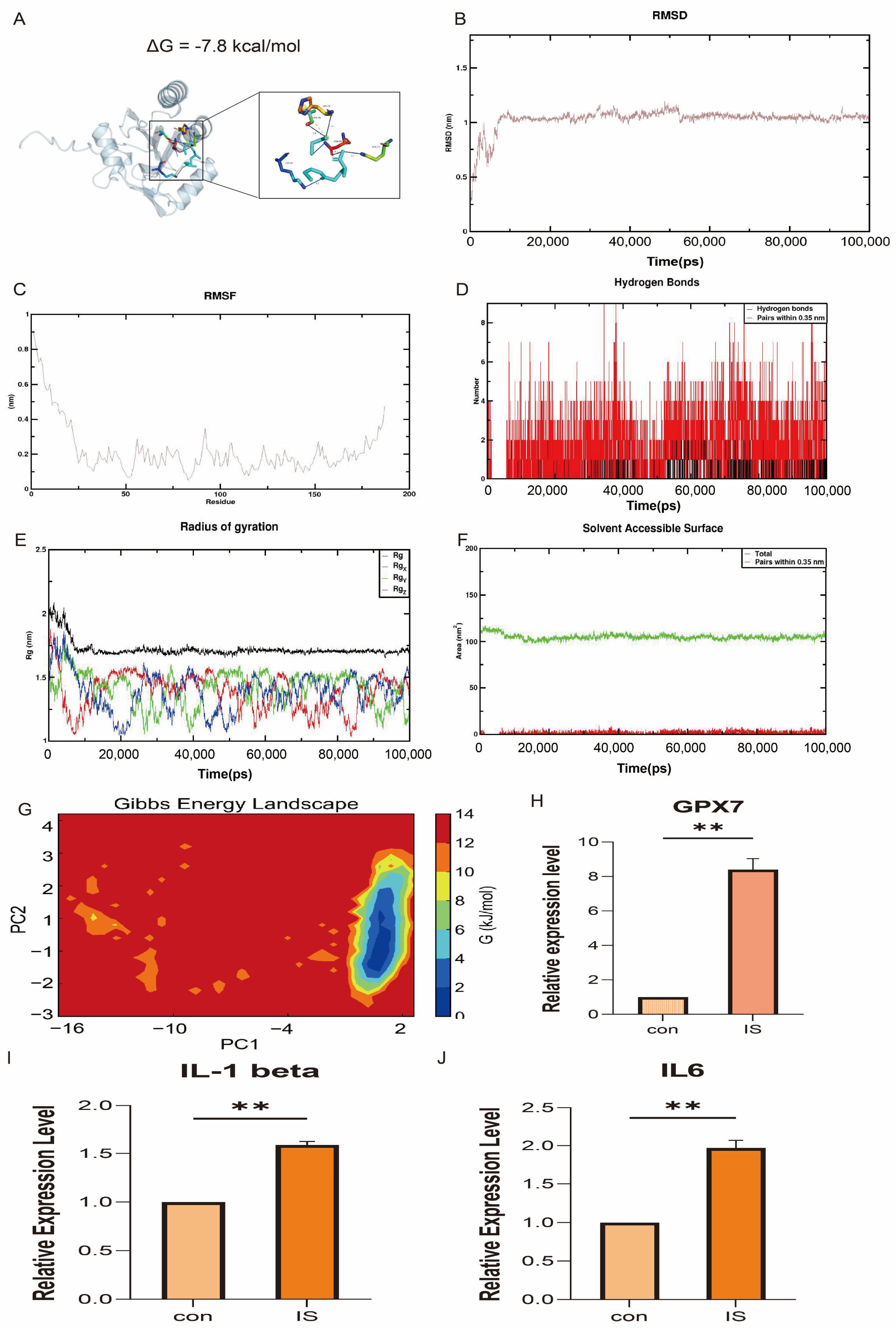
3.9. Molecular Docking and In Vivo Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, T.; Yuan, Y.; Wu, K.; Zhou, Y.; You, C.; Guan, J. Trends and patterns in the global burden of intracerebral hemorrhage: A comprehensive analysis from 1990 to 2019. Front. Neurol. 2023, 14, 1241158. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; Feld, J.; Rothwell, P.M.; Reinecke, H.; Koeppe, J. Admission Rates, Time Trends, Risk Factors, and Outcomes of Ischemic and Hemorrhagic Stroke From German Nationwide Data. Neurology 2022, 99, e2593–e2604. [Google Scholar] [CrossRef] [PubMed]
- Groff, H.; Yousfani, S.; Pantoja-Ruiz, C.; Douiri, A.; Bhalla, A.; Wolfe, C.; Marshall, I.J. A systematic review of the incidence and outcomes of ICD-11 defined stroke. J. Stroke Cerebrovasc. Dis. 2024, 33, 107784. [Google Scholar] [CrossRef] [PubMed]
- Rajashekar, D.; Wilms, M.; Macdonald, M.E.; Schimert, S.; Hill, M.D.; Demchuk, A.; Goyal, M.; Dukelow, S.P.; Forkert, N.D. Lesion-symptom mapping with NIHSS sub-scores in ischemic stroke patients. Stroke Vasc. Neurol. 2022, 7, 124–131. [Google Scholar] [CrossRef]
- Dammavalam, V.; Lin, S.; Nessa, S.; Daksla, N.; Stefanowski, K.; Costa, A.; Bergese, S. Neuroprotection during Thrombectomy for Acute Ischemic Stroke: A Review of Future Therapies. Int. J. Mol. Sci. 2024, 25, 891. [Google Scholar] [CrossRef]
- Wassélius, J.; Arnberg, F.; Von Euler, M.; Wester, P.; Ullberg, T. Endovascular thrombectomy for acute ischemic stroke. J. Intern. Med. 2022, 291, 303–316. [Google Scholar] [CrossRef]
- Guzik, A.; Bushnell, C. Stroke Epidemiology and Risk Factor Management. Contin. Lifelong Learn. Neurol. 2017, 23, 15–39. [Google Scholar] [CrossRef]
- Briyal, S.; Ranjan, A.K.; Gulati, A. Oxidative stress: A target to treat Alzheimer’s disease and stroke. Neurochem. Int. 2023, 165, 105509. [Google Scholar] [CrossRef]
- Evans, E.P.P.; Scholten, J.T.M.; Mzyk, A.; Reyes-San-Martin, C.; Llumbet, A.E.; Hamoh, T.; Arts, E.G.J.M.; Schirhagl, R.; Cantineau, A.E.P. Male subfertility and oxidative stress. Redox Biol. 2021, 46, 102071. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Zheng, H.; Ho, J.; Chan, M.T.V.; Wu, W.K.K. Protective roles of melatonin in central nervous system diseases by regulation of neural stem cells. Cell Prolif. 2017, 50, e12323. [Google Scholar] [CrossRef]
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar]
- Montenegro, J.D. Gene Co-Expression Network Analysis; Springer: New York, NY, USA, 2022; pp. 387–404. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar]
- Engebretsen, S.; Bohlin, J. Statistical predictions with glmnet. Clin. Epigenet. 2019, 11, 123. [Google Scholar] [CrossRef]
- Sanz, H.; Valim, C.; Vegas, E.; Oller, J.M.; Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 2018, 19, 432. [Google Scholar] [CrossRef]
- Ishwaran, H.; Kogalur, U.B. Consistency of random survival forests. Stat. Probab. Lett. 2010, 80, 1056–1064. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteom. Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Hou, W.; Ji, Z. Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis. Nat. Methods 2024, 21, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hilkens, N.A.; Casolla, B.; Leung, T.W.; De Leeuw, F.-E. Stroke. Lancet 2024, 403, 2820–2836. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 70. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Li, G.; Fang, Y.; Ma, Y.; Dawa, Y.; Wang, Q.; Gan, J.; Dang, J. Screening and Isolation of Potential Anti-Inflammatory Compounds from Saxifraga atrata via Affinity Ultrafiltration-HPLC and Multi-Target Molecular Docking Analyses. Nutrients 2022, 14, 2405. [Google Scholar] [CrossRef]
- Yu, W.; Ying, J.; Wang, X.; Liu, X.; Zhao, T.; Yoon, S.; Zheng, Q.; Fang, Y.; Yang, D.; Hua, F. The Involvement of Lactosylceramide in Central Nervous System Inflammation Related to Neurodegenerative Disease. Front. Aging Neurosci. 2021, 13, 691230. [Google Scholar] [CrossRef]
- Ludhiadch, A.; Sharma, R.; Muriki, A.; Munshi, A. Role of Calcium Homeostasis in Ischemic Stroke: A Review. CNS Neurol. Disord. Drug Targets 2022, 21, 52–61. [Google Scholar] [CrossRef]
- Yang, Z.; Shi, X.; Gao, Z.; Chu, L. MiR-155-5p in Extracellular Vesicles Derived from Choroid Plexus Epithelial Cells Promotes Autophagy and Inflammation to Aggravate Ischemic Brain Injury in Mice. Oxid. Med. Cell. Longev. 2022, 2022, 8603427. [Google Scholar] [CrossRef]
- Wegner, S.; Uhlemann, R.; Boujon, V.; Ersoy, B.; Endres, M.; Kronenberg, G.; Gertz, K. Endothelial Cell-Specific Transcriptome Reveals Signature of Chronic Stress Related to Worse Outcome After Mild Transient Brain Ischemia in Mice. Mol. Neurobiol. 2020, 57, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, O.H.; Degnan, S.M. Antioxidant enzymes that target hydrogen peroxide are conserved across the animal kingdom, from sponges to mammals. Sci. Rep. 2023, 13, 2510. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, S.; Sofia, E.F.; Hirai, N.; Okumura, M.; Kadokura, H.; Inaba, K. Characterization of the endoplasmic reticulum–resident peroxidases GPx7 and GPx8 shows the higher oxidative activity of GPx7 and its linkage to oxidative protein folding. J. Biol. Chem. 2020, 295, 12772–12785. [Google Scholar] [CrossRef] [PubMed]
- Buday, K.; Conrad, M. Emerging roles for non-selenium containing ER-resident glutathione peroxidases in cell signaling and disease. Biol. Chem. 2021, 402, 271–287. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef]
- Xu, J.; Guo, H.; Xing, Z.; Zhang, W.; He, J.; Cheng, J.; Cai, R. Mild Oxidative Stress Reduces NRF2 SUMOylation to Promote Kras/Lkb1/Keap1 Mutant Lung Adenocarcinoma Cell Migration and Invasion. Oxid. Med. Cell. Longev. 2020, 2020, 6240125. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, Y.; Zhang, L.; Wang, Y.; Wang, T.; Sun, Y.; Cao, J.; Qi, M.; Du, X.; Xia, Z.; et al. GPX7 Is Identified as a Novel Prognostic Indicator for Brain Lower Grade Glioma (LGG): Evidence from a Pan-Cancer Analysis; Research Square Platform LLC: Durham, NC, USA, 2021. [Google Scholar]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Pourzand, C.; Albieri-Borges, A.; Raczek, N.N. Shedding a New Light on Skin Aging, Iron- and Redox-Homeostasis and Emerging Natural Antioxidants. Antioxidants 2022, 11, 471. [Google Scholar] [CrossRef]
- Calabrese, G.; Morgan, B.; Riemer, J. Mitochondrial Glutathione: Regulation and Functions. Antioxid. Redox Signal. 2017, 27, 1162–1177. [Google Scholar] [CrossRef]
- Liu, L.; Chen, M.; Lin, K.; Xiang, X.; Zheng, Y.; Zhu, S. Inhibiting Caspase-12 Mediated Inflammasome Activation protects against Oxygen-Glucose Deprivation Injury in Primary Astrocytes. Int. J. Med. Sci. 2020, 17, 1936–1945. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zhang, S.; He, J.; Li, H.; Kang, J. Multi-Omics and Experimental Validation Identify GPX7 and Glutathione-Associated Oxidative Stress as Potential Biomarkers in Ischemic Stroke. Antioxidants 2025, 14, 665. https://doi.org/10.3390/antiox14060665
Li T, Zhang S, He J, Li H, Kang J. Multi-Omics and Experimental Validation Identify GPX7 and Glutathione-Associated Oxidative Stress as Potential Biomarkers in Ischemic Stroke. Antioxidants. 2025; 14(6):665. https://doi.org/10.3390/antiox14060665
Chicago/Turabian StyleLi, Tianzhi, Sijie Zhang, Jinshan He, Hongyan Li, and Jingsong Kang. 2025. "Multi-Omics and Experimental Validation Identify GPX7 and Glutathione-Associated Oxidative Stress as Potential Biomarkers in Ischemic Stroke" Antioxidants 14, no. 6: 665. https://doi.org/10.3390/antiox14060665
APA StyleLi, T., Zhang, S., He, J., Li, H., & Kang, J. (2025). Multi-Omics and Experimental Validation Identify GPX7 and Glutathione-Associated Oxidative Stress as Potential Biomarkers in Ischemic Stroke. Antioxidants, 14(6), 665. https://doi.org/10.3390/antiox14060665