
Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Oxidative Stress in Inflammation
2.1. Sources of Reactive Species
2.2. Oxidative Stress-Induced Signaling Pathways
2.3. Impact on Immune System and Cytokine Production
2.4. Role in Tissue Damage and Disease Progression
3. Reductive Stress in Inflammation
3.1. Definition and Mechanisms of Reductive Stress
3.2. Antioxidant Systems
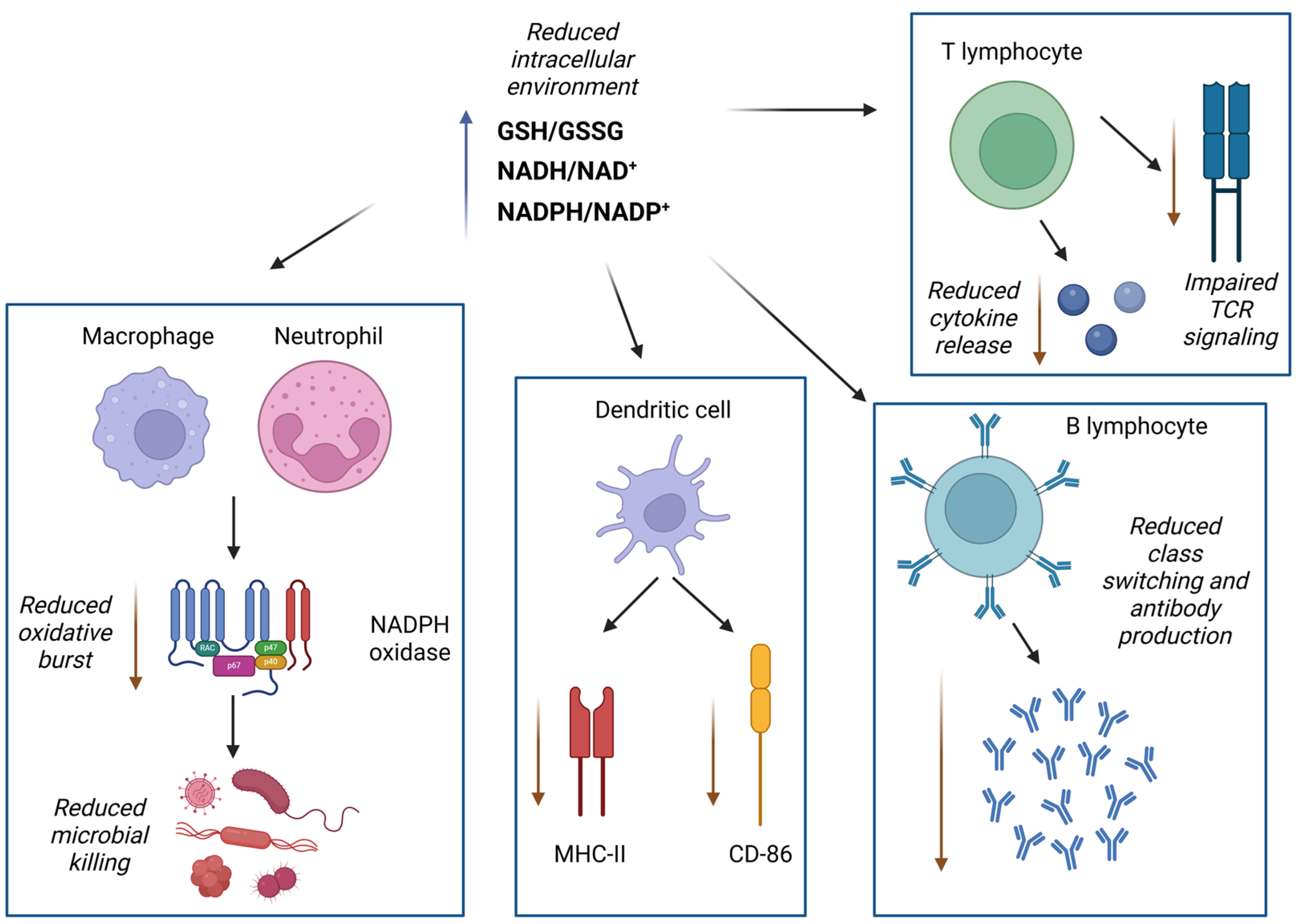
3.3. Effects of Excessive Reductive Conditions on Immune Function
3.4. Paradoxical Roles of Reductive Stress in Chronic Inflammation
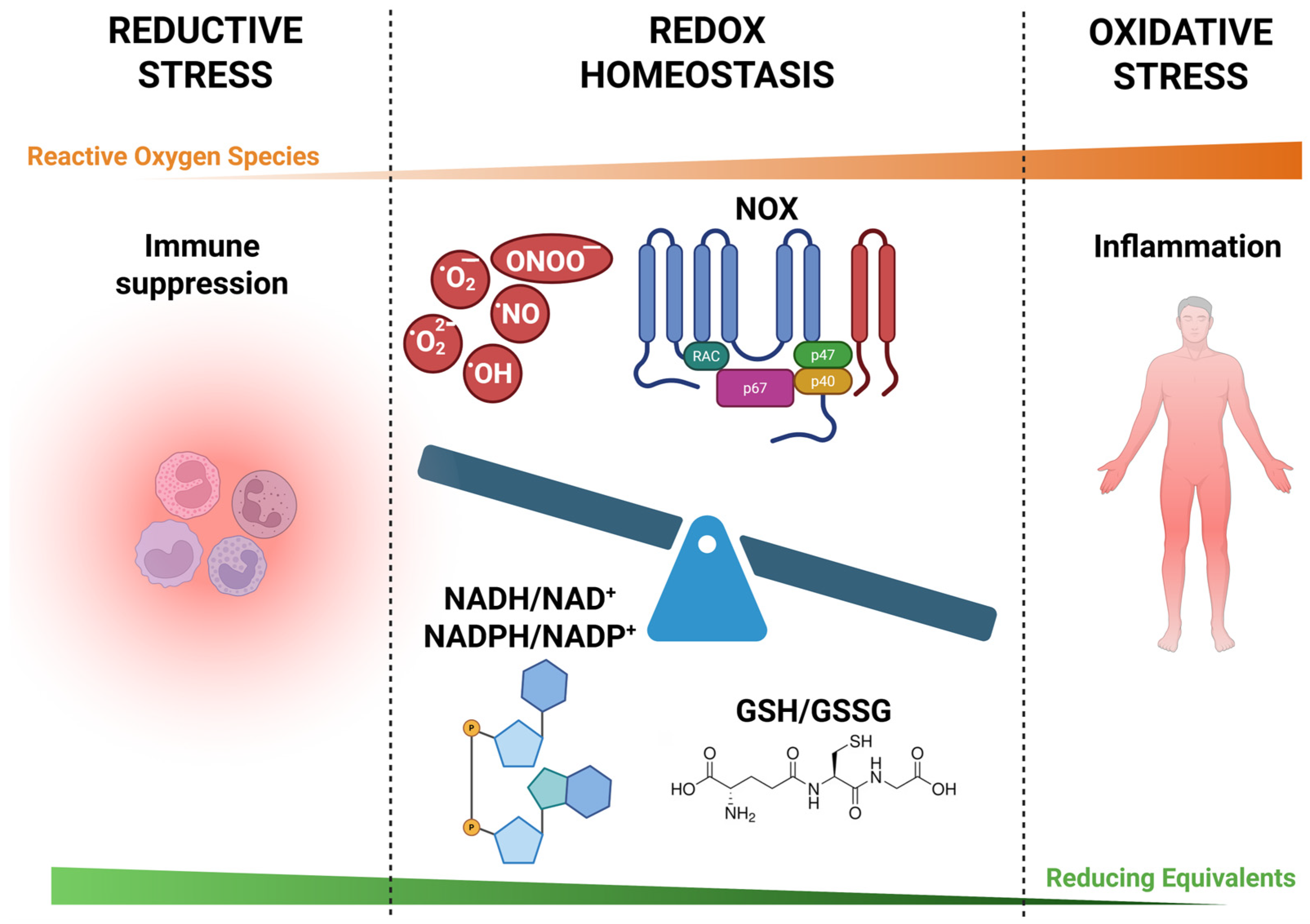
4. Interplay Between Oxidative and Reductive Stress
4.1. Dynamic Balance and Feedback Mechanisms
4.2. Conditions Leading to Redox Imbalance
- Environmental and lifestyle factors: Exposure to pollutants, heavy metals, tobacco smoke, radiation, and various xenobiotics can elevate ROS/RNS production, overwhelming antioxidant defense mechanisms and leading to OS [103]. Additionally, poor dietary habits, sedentarism, and chronic psychological stress contribute to systemic redox imbalance and heighten susceptibility to inflammation-related diseases [105];
- Inflammatory processes: The activation of immune cells, such as macrophages and neutrophils, during infections or chronic inflammation leads to excessive production of ROS/RNS, amplifying OS and tissue damage. This persistent redox imbalance is observed in conditions such as rheumatoid arthritis and inflammatory bowel disease [106];
- Genetic mutations and hereditary diseases: Inherited diseases including cystic fibrosis, familial amyotrophic lateral sclerosis (ALS), and certain mitochondrial encephalopathies are associated with chronic redox imbalance. Mutations in genes regulating antioxidant enzymes, mitochondrial function, or protein folding exacerbate ROS production and trigger inflammation through UPR and mitochondrial damage [107];
- Metabolic dysregulation: Conditions such as obesity, insulin resistance, and type 1 and type 2 diabetes are associated with heightened OS. Excess adipose tissue and hyperglycemia stimulate mitochondrial ROS production and NOX activity, impairing redox signaling and promoting systemic inflammation [108]. Mitochondria isolated from the carotid body of type 1 diabetic rats exhibited evidence of RS, supporting the role of mitochondrial redox imbalance in diabetic pathology and systemic inflammation [109]. In metabolic syndrome, this imbalance contributes to endothelial dysfunction, vascular inflammation, and progression to cardiovascular disease (CVD) [110];
- Aging: The aging process is associated with a decline in mitochondrial efficiency and antioxidant capacity, resulting in cumulative oxidative damage and the emergence of a pro-inflammatory state known as “inflammaging” [111]. Redox imbalance during aging contributes to tissue degeneration, neuroinflammation, and increased risk of chronic diseases such as Alzheimer’s and atherosclerosis [112];
- Endothelial dysfunction: An imbalance between NO bioavailability and OS in the endothelium contributes to impaired vasodilation, vascular inflammation, and thrombosis. This mechanism plays a central role in the development of hypertension and atherosclerosis, linking redox imbalance to cardiovascular events [113].
4.3. Consequences of Redox Dysregulation in Chronic Inflammatory Diseases
5. Redox Imbalance in Chronic Inflammatory Diseases
5.1. Redox Perturbations in Autoimmune Disorders
5.2. Redox Perturbations in Cardiovascular Diseases
5.3. Loss of Redox Balance in Neuroinflammatory Conditions
6. Therapeutic Strategies Targeting Redox Balance
6.1. Antioxidant Therapies and Their Limitations
6.2. Modulating Redox-Sensitive Signaling Pathways
6.3. Pharmacological and Dietary Approaches
6.4. Emerging Redox-Based Therapeutic Interventions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OS | Oxidative stress |
| RS | RS |
| GSH | Reduced glutathione |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| NF-κB | Nuclear factor-kappa B |
| GSSG | Oxidized glutathione |
| AP-1 | Activating protein-1 |
| HIF-1α | Hypoxia-inducible factor-1α |
| MMPs | Matrix metalloproteinases |
| NOS | Nitric oxide synthase |
| COX | Cyclooxygenase |
| NOX | NADPH oxidase |
| SOD | Superoxide dismutase |
| MPO | Myeloperoxidase |
| iNOS | Inducible nitric oxide synthase |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| Ero1 | ER oxidoreductin 1 |
| PDI | Protein disulfide isomerase |
| PERK | Protein kinase R-like endoplasmic reticulum kinase |
| ATF6 | Activating transcription factor 6 |
| IRE1α | Inositol-requiring enzyme 1α |
| UV | Ultraviolet |
| IKK | IκB kinase |
| MAPKs | Mitogen-activated protein kinases |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| AREs | Antioxidant response elements |
| HO-1 | Heme oxygenase-1 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| GCLC | glutamate–cysteine ligase catalytic subunit |
| IL | Interleukin |
| MHC | Major histocompatibility complex |
| Treg | Regulatory T cell |
| TGF-β | Tumor growth factor-β |
| MDA | Malondialdehyde |
| 4-HNE | 4-hydroxynonenal |
| 8-oxo-dG | 8-Oxo-2′-deoxyguanosine |
| IBD | Inflammatory bowel disease |
| FNIP1 | Folliculin-interacting protein 1 |
| BEX | Brain-expressed X-linked |
| Mtb | Mycobacterium tuberculosis |
| TR | Thioredoxin reductase |
| TCA | Tricarboxylic acid |
| GPX | Glutathione peroxidase |
| Trx | Thioredoxins |
| TrxR | Thioredoxin reductase |
| RA | Rheumatoid arthritis |
| SLE | Systemic lupus erythematosus |
| T1DM | Type 1 diabetes mellitus |
| AIH | Autoimmune hepatitis |
| AIT | Autoimmune thyroiditis |
| HT | Hashimoto’s thyroiditis |
| GD | Graves’ disease |
| CVDs | Cardiovascular diseases |
| CNS | Central nervous system |
| AD | Alzheimer’s disease |
| Aβ | Amyloid β |
| PD | Parkinson’s disease |
| MS | Multiple sclerosis |
| STAT3 | Signal transducer and activator of transcription 3 |
References
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Reductive Stress in Inflammation-Associated Diseases and the pro-Oxidant Effect of Antioxidant Agents. Int. J. Mol. Sci. 2017, 18, 2098. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal 2020, 32, 1330–1347. [Google Scholar] [CrossRef]
- Xiao, W.; Lee, L.Y.; Loscalzo, J. Metabolic Responses to Redox Stress in Vascular Cells. Antioxid. Redox Signal 2024, 41, 793–817. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Averill-Bates, D. Reactive Oxygen Species and Cell Signaling. Review. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119573. [Google Scholar] [CrossRef]
- Manoharan, R.R.; Prasad, A.; Pospíšil, P.; Kzhyshkowska, J. ROS Signaling in Innate Immunity via Oxidative Protein Modifications. Front. Immunol. 2024, 15, 1359600. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide Generation by Complex III: From Mechanistic Rationales to Functional Consequences. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Urbański, N.K.; Berȩsewicz, A. Generation of ·OH Initiated by Interaction of Fe2+ and Cu+ with Dioxygen; Comparison with the Fenton Chemistry. Acta Biochim. Pol. 2000, 47, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Bode, K.; Hauri-Hohl, M.; Jaquet, V.; Weyd, H. Unlocking the Power of NOX2: A Comprehensive Review on Its Role in Immune Regulation. Redox Biol. 2023, 64, 102795. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, Y.; Zhang, G.; Yang, Z.; Xu, W.; Chen, Q. The Applications and Mechanisms of Superoxide Dismutase in Medicine, Food, and Cosmetics. Antioxidants 2023, 12, 1675. [Google Scholar] [CrossRef]
- Frangie, C.; Daher, J. Role of Myeloperoxidase in Inflammation and Atherosclerosis (Review). Biomed. Rep. 2022, 16, 53. [Google Scholar] [CrossRef]
- Sun, J.; Druhan, L.J.; Zweier, J.L. Reactive Oxygen and Nitrogen Species Regulate Inducible Nitric Oxide Synthase Function Shifting the Balance of Nitric Oxide and Superoxide Production. Arch. Biochem. Biophys. 2010, 494, 130–137. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.R.; Chae, H.J. The Aftermath of the Interplay between the Endoplasmic Reticulum Stress Response and Redox Signaling. Exp. Mol. Med. 2021, 53, 151–157. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Mathes, E.; O’Dea, E.L.; Hoffmann, A.; Ghosh, G. NF-ΚB Dictates the Degradation Pathway of IκBα. EMBO J. 2008, 27, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-ΚB: A Key Role in Inflammatory Diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and Versatility of P38 Kinase Signalling in Health and Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Pant, T.; Uche, N.; Juric, M.; Zielonka, J.; Bai, X. Regulation of Immunomodulatory Networks by Nrf2-Activation in Immune Cells: Redox Control and Therapeutic Potential in Inflammatory Diseases. Redox Biol. 2024, 70, 103077. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Role of Reactive Species on Innate Immunity. Vaccines 2022, 10, 1735. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Pérez, S.; Rius-Pérez, S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants 2022, 11, 1394. [Google Scholar] [CrossRef]
- ten Broeke, T.; Wubbolts, R.; Stoorvogel, W. MHC Class II Antigen Presentation by Dendritic Cells Regulated through Endosomal Sorting. Cold Spring Harb. Perspect. Biol. 2013, 5, a016873. [Google Scholar] [CrossRef]
- Peng, H.Y.; Lucavs, J.; Ballard, D.; Das, J.K.; Kumar, A.; Wang, L.; Ren, Y.; Xiong, X.; Song, J. Metabolic Reprogramming and Reactive Oxygen Species in T Cell Immunity. Front. Immunol. 2021, 12, 652687. [Google Scholar] [CrossRef]
- Weiskopf, D.; Schwanninger, A.; Weinberger, B.; Almanzar, G.; Parson, W.; Buus, S.; Lindner, H.; Grubeck-Loebenstein, B. Oxidative Stress Can Alter the Antigenicity of Immunodominant Peptides. J. Leukoc. Biol. 2009, 87, 165–172. [Google Scholar] [CrossRef]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative Stress and Glutathione in TGF-β-Mediated Fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein Oxidation—Formation Mechanisms, Detection and Relevance as Biomarkers in Human Diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef] [PubMed]
- Gorini, F.; Scala, G.; Cooke, M.S.; Majello, B.; Amente, S. Towards a Comprehensive View of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine: Highlighting the Intertwined Roles of DNA Damage and Epigenetics in Genomic Instability. DNA Repair. 2021, 97, 103027. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Desai, L.P. Reciprocal Regulation of TGF-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Umar, M.I.; Imran, S.; Javaid, F.; Syed, S.K.; Riaz, R.; Hassan, W. TGF-Β1 Signaling Can Worsen NAFLD with Liver Fibrosis Backdrop. Exp. Mol. Pathol. 2022, 124, 104733. [Google Scholar] [CrossRef]
- Li, L.; Peng, P.; Ding, N.; Jia, W.; Huang, C.; Tang, Y. Oxidative Stress, Inflammation, Gut Dysbiosis: What Can Polyphenols Do in Inflammatory Bowel Disease? Antioxidants 2023, 12, 967. [Google Scholar] [CrossRef]
- Scioli, M.G.; Storti, G.; D’amico, F.; Guzmán, R.R.; Centofanti, F.; Doldo, E.; Miranda, E.M.C.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-Free Biology of Oxidative Stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Manzano-Pech, L.; Guarner-Lans, V.; Soto, M.E.; Díaz-Díaz, E.; Caballero-Chacón, S.; Díaz-Torres, R.; Rodríguez-Fierros, F.L.; Pérez-Torres, I. Excessive Consumption Hibiscus sabdariffa L. Increases Inflammation and Blood Pressure in Male Wistar Rats via High Antioxidant Capacity: The Preliminary Findings. Cells 2022, 11, 2774. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Pech, L.; Guarner-Lans, V.; Elena Soto, M.; Díaz-Díaz, E.; Pérez-Torres, I. Alteration of the Aortic Vascular Reactivity Associated to Excessive Consumption of Hibiscus sabdariffa Linnaeus: Preliminary Findings. Heliyon 2023, 9, e20020. [Google Scholar] [CrossRef]
- Manford, A.G.; Mena, E.L.; Shih, K.Y.; Gee, C.L.; McMinimy, R.; Martínez-González, B.; Sherriff, R.; Lew, B.; Zoltek, M.; Rodríguez-Pérez, F.; et al. Structural Basis and Regulation of the Reductive Stress Response. Cell 2021, 184, 5375–5390. [Google Scholar] [CrossRef]
- Manford, A.G.; Rodríguez-Pérez, F.; Shih, K.Y.; Shi, Z.; Berdan, C.A.; Choe, M.; Titov, D.V.; Nomura, D.K.; Rape, M. A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183, 46–61. [Google Scholar] [CrossRef]
- Jevtić, P.; Haakonsen, D.L.; Rapé, M. An E3 Ligase Guide to the Galaxy of Small-Molecule-Induced Protein Degradation. Cell Chem. Biol. 2021, 28, 1000–1013. [Google Scholar] [CrossRef]
- Ge, M.; Papagiannakopoulos, T.; Bar-Peled, L. Reductive Stress in Cancer: Coming out of the Shadows. Trends Cancer 2024, 10, 103–112. [Google Scholar] [CrossRef]
- Nasiri, M.J.; Venketaraman, V. Advances in Host–Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention. Int. J. Mol. Sci. 2025, 26, 1621. [Google Scholar] [CrossRef]
- Lin, K.; O’Brien, K.M.; Trujillo, C.; Wang, R.; Wallach, J.B.; Schnappinger, D.; Ehrt, S. Mycobacterium Tuberculosis Thioredoxin Reductase Is Essential for Thiol Redox Homeostasis but Plays a Minor Role in Antioxidant Defense. PLoS Pathog. 2016, 12, e1005675. [Google Scholar] [CrossRef] [PubMed]
- Mavi, P.S.; Singh, S.; Kumar, A. Reductive Stress: New Insights in Physiology and Drug Tolerance of Mycobacterium. Antioxid. Redox Signal 2020, 32, 1348–1366. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R. Skin Antioxidants: Their Role in Aging and in Oxidative Stress—New Approaches for Their Evaluation. Biomed. Pharmacother. 1999, 53, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.R.; Segal, A.W. The NADPH Oxidase of Professional Phagocytes—Prototype of the NOX Electron Transport Chain Systems. Biochim. Biophys. Acta Bioenerg. 2004, 1657, 1–22. [Google Scholar] [CrossRef]
- Brewer, A.C.; Mustafi, S.B.; Murray, T.V.A.; Rajasekaran, N.S.; Benjamin, I.J. Reductive Stress Linked to Small HSPs, G6PD, and Nrf2 Pathways in Heart Disease. Antioxid. Redox Signal 2013, 18, 1114–1127. [Google Scholar] [CrossRef]
- Hahn, M.E.; Timme-Laragy, A.R.; Karchner, S.I.; Stegeman, J.J. Nrf2 and Nrf2-Related Proteins in Development and Developmental Toxicity: Insights from Studies in Zebrafish (Danio rerio). Free Radic. Biol. Med. 2015, 88, 275–289. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Zhang, H.; Limphong, P.; Pieper, J.; Liu, Q.; Rodesch, C.K.; Christians, E.; Benjamin, I.J. Glutathione-dependent Reductive Stress Triggers Mitochondrial Oxidation and Cytotoxicity. FASEB J. 2012, 26, 1442–1451. [Google Scholar] [CrossRef]
- Cortassa, S.; O’Rourke, B.; Aon, M.A. Redox-Optimized ROS Balance and the Relationship between Mitochondrial Respiration and ROS. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 287–295. [Google Scholar] [CrossRef]
- Yan, L.J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 137979. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox Regulation of Cell Survival by the Thioredoxin Superfamily: An Implication of Redox Gene Therapy in the Heart. Antioxid. Redox Signal 2009, 11, 2741–2758. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N. Involvement of Thio-, Peroxi-, and Glutaredoxins in Cellular Redox-Dependent Processes. Biochemistry 2008, 73, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Owuor, E.D.; Kong, A.N.T. Antioxidants and Oxidants Regulated Signal Transduction Pathways. Biochem. Pharmacol. 2002, 64, 765–770. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Javadov, S.; Sommer, N. Cellular ROS and Antioxidants: Physiological and Pathological Role. Antioxidants 2024, 13, 602. [Google Scholar] [CrossRef]
- Møller, S.H.; Wang, L.; Ho, P.C. Metabolic Programming in Dendritic Cells Tailors Immune Responses and Homeostasis. Cell Mol. Immunol. 2022, 19, 370–383. [Google Scholar] [CrossRef]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T Cells and Reactive Oxygen Species. J. Biomed. Sci. 2015, 22, 85. [Google Scholar] [CrossRef]
- Weyand, C.M.; Shen, Y.; Goronzy, J.J. Redox-Sensitive Signaling in Inflammatory T Cells and in Autoimmune Disease. Free Radic. Biol. Med. 2018, 125, 36–43. [Google Scholar] [CrossRef]
- Boothby, M.; Rickert, R.C. Metabolic Regulation of the Immune Humoral Response. Immunity 2017, 46, 743–755. [Google Scholar] [CrossRef]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The Interplay between Cytokines, Inflammation, and Antioxidants: Mechanistic Insights and Therapeutic Potentials of Various Antioxidants and Anti-Cytokine Compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, N.; Gao, Z.; Gao, J.; Wang, X.; Xie, H.; Wang, C.-Y.; Zhang, S. Reductive Stress: The Key Pathway in Metabolic Disorders Induced by Overnutrition. J. Adv. Res. 2025; in press. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, N.R.; Nandan, D.; Nair, B.G.; Nair, V.A.; Venugopal, P.; Aradhya, R. Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases. Cells 2025, 14, 511. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, D.; Carter-Timofte, M.E.; van Grevenynghe, J.; Laguette, N.; Dinkova-Kostova, A.T.; Olagnier, D. Regulation of Innate Immunity by Nrf2. Curr. Opin. Immunol. 2022, 78, 102247. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B. Limiting Reductive Stress for Treating In-Stent Stenosis: The Heart of the Matter? J. Clin. Investig. 2014, 124, 5092–5094. [Google Scholar] [CrossRef]
- Dialynas, G.; Shrestha, O.K.; Ponce, J.M.; Zwerger, M.; Thiemann, D.A.; Young, G.H.; Moore, S.A.; Yu, L.; Lammerding, J.; Wallrath, L.L. Myopathic Lamin Mutations Cause Reductive Stress and Activate the Nrf2/Keap-1 Pathway. PLoS Genet. 2015, 11, e1005231. [Google Scholar] [CrossRef]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-Mediated Increases in l-2-Hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Cannon, J.R.; Drolet, R.; Mastroberardino, P.G. Lessons from the Rotenone Model of Parkinson’s Disease. Trends Pharmacol. Sci. 2010, 31, 141–142. [Google Scholar] [CrossRef]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of Insulin Resistance and Obesity in Mice Overexpressing Cellular Glutathione Peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring Oxidant Signaling Suppresses Proarthritogenic T Cell Effector Functions in Rheumatoid Arthritis. Sci. Transl. Med. 2016, 8, 331ra38. [Google Scholar] [CrossRef]
- Carne, N.A.; Bell, S.; Brown, A.P.; Määttä, A.; Flagler, M.J.; Benham, A.M. Reductive Stress Selectively Disrupts Collagen Homeostasis and Modifies Growth Factor-Independent Signaling through the MAPK/Akt Pathway in Human Dermal Fibroblasts. Mol. Cell Proteom. 2019, 18, 1123–1137. [Google Scholar] [CrossRef]
- Shanmugam, G.; Wang, D.; Gounder, S.S.; Fernandes, J.; Litovsky, S.H.; Whitehead, K.; Radhakrishnan, R.K.; Franklin, S.; Hoidal, J.R.; Kensler, T.W.; et al. Reductive Stress Causes Pathological Cardiac Remodeling and Diastolic Dysfunction. Antioxid. Redox Signal 2020, 32, 1293–1312. [Google Scholar] [CrossRef]
- Ma, K.; Miao, L.; Li, B.; Yu, W.; Liu, F.; Liu, K.; Li, Y.; Huang, C.; Yang, Z. Mechanism of Action of Nrf2 and Its Related Natural Regulators in Rheumatoid Arthritis. J. Orthop. Surg. Res. 2024, 19, 759. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, R.; Weng, S.; Xu, H.; Zhang, Y.; Chen, S.; Liu, S.; Ba, Y.; Zhou, Z.; Luo, P.; et al. Multifaceted Role of Redox Pattern in the Tumor Immune Microenvironment Regarding Autophagy and Apoptosis. Mol. Cancer 2023, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, G.; Bačić, G.; Bridal, L.; Schmidt, H.H.H.W.; Tavitian, B.; Viel, T.; Utsumi, H.; Yalçin, A.S.; De Spirito, M. Imaging Reactive Oxygen Species-Induced Modifications in Living Systems. Antioxid. Redox Signal 2016, 24, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Cipak Gasparovic, A.; Zarkovic, N.; Zarkovic, K.; Semen, K.; Kaminskyy, D.; Yelisyeyeva, O.; Bottari, S.P. Biomarkers of Oxidative and Nitro-Oxidative Stress: Conventional and Novel Approaches. Br. J. Pharmacol. 2017, 174, 1771–1783. [Google Scholar] [CrossRef]
- Ghezzi, P. Environmental Risk Factors and Their Footprints in Vivo—A Proposal for the Classification of Oxidative Stress Biomarkers. Redox Biol. 2020, 34, 101442. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for Measuring Reactive Oxygen Species and Oxidative Damage in Cells and in Vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox Regulation of Inflammation: Old Elements, a New Story. Med. Res. Rev. 2015, 35, 306–340. [Google Scholar] [CrossRef]
- Kračun, D.; Lopes, L.R.; Cifuentes-Pagano, E.; Pagano, P.J. NADPH Oxidases: Redox Regulation of Cell Homeostasis and Disease. Physiol. Rev. 2025, 105, 1291–1428. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxid. Med. Cell Longev. 2017, 2017, 8459402. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Kopf, M. Redox Regulation of Immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Ash, D.; Nagarkoti, S.; Belin De Chantemèle, E.J.; Fulton, D.J.R.; Fukai, T. Interplay between Reactive Oxygen/Reactive Nitrogen Species and Metabolism in Vascular Biology and Disease. Antioxid. Redox Signal 2021, 34, 1319–1354. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Phosphatidylinositol-3,4,5-Triphosphate and Cellular Signaling: Implications for Obesity and Diabetes. Cell Physiol. Biochem. 2015, 35, 1253–1275. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid. Redox Signal 2014, 20, 372–382. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin Resistance: Review of the Underlying Molecular Mechanisms. J. Cell Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Tejeda-Chavez, H.R.; Montero, S.; Saavedra-Molina, A.; Lemus, M.; Tejeda-Luna, J.B.; Roces de Alvarez-Buylla, E. Reductive Stress in Mitochondria Isolated from the Carotid Body of Type 1 Diabetic Male Wistar Rats. Physiol. Rep. 2024, 12, e70016. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative Stress and Metabolic Disorders: Pathogenesis and Therapeutic Strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune–Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Liu, G.H.; Qu, J. Mitochondrial Sirtuins, Metabolism, and Aging. J. Genet. Genom. 2022, 49, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P. Stay in Shape with BH4: Loss of Endothelial Tetrahydrobiopterin Promotes Aortic Aneurysm Development in Mice. Hypertension 2018, 72, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic Inflammation and the Hallmarks of Aging. Mol. Metab. 2023, 74, 101755. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.; Zamboni, P.; Mahajan, R. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative Stress and Stress-Activated Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative Stress and Pulmonary Fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef]
- Kalpakcioglu, B.; Şenel, K. The Interrelation of Glutathione Reductase, Catalase, Glutathione Peroxidase, Superoxide Dismutase, and Glucose-6-Phosphate in the Pathogenesis of Rheumatoid Arthritis. Clin. Rheumatol. 2008, 27, 141–145. [Google Scholar] [CrossRef]
- Vasanthi, P.; Nalini, G.; Rajasekhar, G. Status of Oxidative Stress in Rheumatoid Arthritis. Int. J. Rheum. Dis. 2009, 12, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakos, D.B.; Pitsavos, C.; Chrysohoou, C.; Skoumas, J.; Stefanadis, C. Status and Management of Blood Lipids in Greek Adults and Their Relation to Socio-Demographic, Lifestyle and Dietary Factors: The ATTICA Study: Blood Lipids Distribution in Greece. Atherosclerosis 2004, 173, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Roşian, Ş.H.; Boarescu, I.; Boarescu, P.-M. Antioxidant and Anti-Inflammatory Effects of Bioactive Compounds in Atherosclerosis. Int. J. Mol. Sci. 2025, 26, 1379. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Massaad, C.A.; Klann, E. Reactive Oxygen Species in the Regulation of Synaptic Plasticity and Memory. Antioxid. Redox Signal 2011, 14, 2013–2064. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial Oxidative Stress Causes Insulin Resistance without Disrupting Oxidative Phosphorylation. J. Biol. Chem. 2018, 293, 3715–3728. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Perl, A. Oxidative Stress in the Pathology and Treatment of Systemic Lupus Erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [Google Scholar] [CrossRef]
- Piganelli, J.D.; Delmastro, M.M. Oxidative Stress and Redox Modulation Potential in Type 1 Diabetes. Clin. Dev. Immunol. 2011, 2011, 593863. [Google Scholar]
- Yan, L. jun Redox Imbalance Stress in Diabetes Mellitus: Role of the Polyol Pathway. Animal Model. Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef]
- Pemberton, P.W.; Aboutwerat, A.; Smith, A.; Burrows, P.C.; McMahon, R.F.T.; Warnes, T.W. Oxidant Stress in Type I Autoimmune Hepatitis: The Link between Necroinflammation and Fibrogenesis? Biochim. Biophys. Acta Mol. Basis Dis. 2004, 1689, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.T.; Rigopoulou, E.I.; Koukoulis, G.K.; Dalekos, G.N.; Moulas, A.N. Oxidative Stress and Antioxidant Status in Patients with Autoimmune Liver Diseases. Redox Rep. 2015, 20, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kochman, J.; Jakubczyk, K.; Bargiel, P.; Janda-Milczarek, K. The Influence of Oxidative Stress on Thyroid Diseases. Antioxidants 2021, 10, 1442. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.H.; Griffiths, H.R. The Dual Role of Reactive Oxygen Species in Autoimmune and Inflammatory Diseases: Evidence from Preclinical Models. Free Radic. Biol. Med. 2018, 125, 63–71. [Google Scholar] [CrossRef]
- Kannan, K.P.; Girija, A.S.S. Exploring the ROS Reduction Strategies in Chronic Lupus Management. Front. Immunol. 2024, 15, 1346656. [Google Scholar] [CrossRef]
- Cuadrado, A.; Cazalla, E.; Bach, A.; Bathish, B.; Naidu, S.D.; DeNicola, G.M.; Dinkova-Kostova, A.T.; Fernández-Ginés, R.; Grochot-Przeczek, A.; Hayes, J.D.; et al. Health Position Paper and Redox Perspectives—Bench to Bedside Transition for Pharmacological Regulation of NRF2 in Noncommunicable Diseases. Redox Biol. 2025, 81, 103569. [Google Scholar] [CrossRef]
- Penna, C.; Pagliaro, P. Endothelial Dysfunction: Redox Imbalance, NLRP3 Inflammasome, and Inflammatory Responses in Cardiovascular Diseases. Antioxidants 2025, 14, 256. [Google Scholar] [CrossRef]
- Fei, J.; Demillard, L.J.; Ren, J. Reactive Oxygen Species in Cardiovascular Diseases: An Update. Explor. Med. 2022, 3, 188–204. [Google Scholar] [CrossRef]
- Szyller, J.; Antoniak, R.; Wadowska, K.; Bil-Lula, I.; Hrymniak, B.; Banasiak, W.; Jagielski, D. Redox Imbalance in Patients with Heart Failure and ICD/CRT-D Intervention. Can It Be an Underappreciated and Overlooked Arrhythmogenic Factor? A First Preliminary Clinical Study. Front. Physiol. 2023, 14, 1289587. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in Myocardial Ischemia Reperfusion and Remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Francisco, J.; Del Re, D.P. Inflammation in Myocardial Ischemia/Reperfusion Injury: Underlying Mechanisms and Therapeutic Potential. Antioxidants 2023, 12, 1944. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Responses to Reductive Stress in the Cardiovascular System. Free Radic. Biol. Med. 2017, 109, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.X.; Li, C.Y.; Tao, R.; Wang, X.P.; Yan, L.J. Reductive Stress-Induced Mitochondrial Dysfunction and Cardiomyopathy. Oxid. Med. Cell Longev. 2020, 2020, 5136957. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Du, L. An Update on Ox-LDL-Inducing Vascular Smooth Muscle Cell-Derived Foam Cells in Atherosclerosis. Front. Cell Dev. Biol. 2024, 12, 1481505. [Google Scholar] [CrossRef]
- Lorey, M.B.; Öörni, K.; Kovanen, P.T. Modified Lipoproteins Induce Arterial Wall Inflammation During Atherogenesis. Front. Cardiovasc. Med. 2022, 9, 841545. [Google Scholar] [CrossRef]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide Production by NAD(P)H Oxidase and Mitochondria Is Increased in Genetically Obese and Hyperglycemic Rat Heart and Aorta before the Development of Cardiac Dysfunction. The Role of Glucose-6-Phosphate Dehydrogenase-Derived NADPH. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H153–H162. [Google Scholar] [CrossRef]
- Wu, W.; Hendrix, A.; Nair, S.; Cui, T. Nrf2-Mediated Dichotomy in the Vascular System: Mechanistic and Therapeutic Perspective. Cells 2022, 11, 30–42. [Google Scholar] [CrossRef]
- Sairam, T.; Patel, A.N.; Subrahmanian, M.; Gopalan, R.; Pogwizd, S.M.; Ramalingam, S.; Sankaran, R.; Rajasekaran, N.S. Evidence for a Hyper-Reductive Redox in a Sub-Set of Heart Failure Patients. J. Transl. Med. 2018, 16, 130. [Google Scholar] [CrossRef]
- Wafi, A.M. Nrf2 and Autonomic Dysregulation in Chronic Heart Failure and Hypertension. Front. Physiol. 2023, 14, 1206527. [Google Scholar] [CrossRef]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative Stress in Multiple Sclerosis: Central and Peripheral Mode of Action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alape-Girón, A.; Flores-Díaz, M.; Echeverri-McCandless, A.; Parajeles-Vindas, A. Oxidative Stress Involvement in the Molecular Pathogenesis and Progression of Multiple Sclerosis: A Literature Review. Rev. Neurosci. 2024, 35, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, K.K.S.; Devarajan, A.; Karan, G.; Sundaram, S.; Wang, Q.; van Groen, T.; del Monte, F.; Rajasekaran, N.S. Reductive Stress Promotes Protein Aggregation and Impairs Neurogenesis. Redox Biol. 2020, 37, 101739. [Google Scholar] [CrossRef]
- Jagaraj, C.J.; Parakh, S.; Atkin, J.D. Emerging Evidence Highlighting the Importance of Redox Dysregulation in the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS). Front. Cell Neurosci. 2021, 14, 581950. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Cárcamo-Fonfría, A.; Caballero-Muñoz, M.d.M.; Agúndez, J.A.G. Oxidative Stress in Huntington’s Disease. Biomolecules 2025, 15, 527. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 664–674. [Google Scholar] [CrossRef]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Dodson, M.; Liang, Q.; Johnson, M.S.; Redmann, M.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Inhibition of Glycolysis Attenuates 4-Hydroxynonenal-Dependent Autophagy and Exacerbates Apoptosis in Differentiated SH-SY5Y Neuroblastoma Cells. Autophagy 2013, 9, 1996–2008. [Google Scholar] [CrossRef]
- Revi, N.; Rengan, A.K. Impact of Dietary Polyphenols on Neuroinflammation-Associated Disorders. Neurol. Sci. 2021, 42, 3101–3119. [Google Scholar] [CrossRef]
- Schaffer, S.; Asseburg, H.; Kuntz, S.; Muller, W.E.; Eckert, G.P. Effects of Polyphenols on Brain Ageing and Alzheimer’s Disease: Focus on Mitochondria. Mol. Neurobiol. 2012, 46, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Dinar, Y.; Elkhoudri, N.; Belahsen, R. Effect of Education on Nutrition and Diabetes Status in Type 2 Diabetics in El Jadida Province of Morocco. Med. J. Nutrition Metab. 2015, 8, 187–197. [Google Scholar] [CrossRef]
- Morris, G.; Gevezova, M.; Sarafian, V.; Maes, M. Redox Regulation of the Immune Response. Cell Mol. Immunol. 2022, 19, 1079–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, D.; Li, X.; Deng, Y.; Li, C. Redox Imbalance in Chronic Inflammatory Diseases. Biomed. Res. Int. 2022, 2022, 9813486. [Google Scholar] [CrossRef]
- Li, B.; Ming, H.; Qin, S.; Nice, E.C.; Dong, J.; Du, Z.; Huang, C. Redox Regulation: Mechanisms, Biology and Therapeutic Targets in Diseases. Signal Transduct. Target. Ther. 2025, 10, 72. [Google Scholar] [CrossRef]
- Chiurchiù, V.; MacCarrone, M. Chronic Inflammatory Disorders and Their Redox Control: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal 2011, 15, 2605–2641. [Google Scholar] [CrossRef]
- Kim, M.E.; Kim, D.H.; Lee, J.S. Transcription Factors as Targets of Natural Compounds in Age-Related Diseases and Cancer: Potential Therapeutic Applications. Int. J. Mol. Sci. 2022, 23, 13882. [Google Scholar] [CrossRef]
- Surh, Y.J.; Kundu, J.K.; Na, H.K.; Lee, J.S. Redox-Sensitive Transcription Factors as Prime Targets for Chemoprevention with Anti-Inflammatory and Antioxidative Phytochemicals. J. Nutr. 2005, 135, 2993–3001S. [Google Scholar] [CrossRef]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the Transcription Factor Nrf2 to Ameliorate Oxidative Stress and Inflammation in Chronic Kidney Disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef]
- Henning, T.; Weber, D. Redox Biomarkers in Dietary Interventions and Nutritional Observation Studies—From New Insights to Old Problems. Redox Biol. 2021, 41, 101922. [Google Scholar] [CrossRef]
- Meng, Q.; Su, C.-H. The Impact of Physical Exercise on Oxidative and Nitrosative Stress: Balancing the Benefits and Risks. Antioxidants 2024, 13, 573. [Google Scholar] [CrossRef]
- Tyuryaeva, I.; Lyublinskaya, O. Expected and Unexpected Effects of Pharmacological Antioxidants. Int. J. Mol. Sci. 2023, 24, 9303. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Hecker, L. NADPH Oxidases: Pathophysiology and Therapeutic Potential in Age-Associated Pulmonary Fibrosis. Redox Biol. 2020, 33, 101541. [Google Scholar] [CrossRef] [PubMed]
- Serra-Majem, L.; Roman, B.; Estruch, R. Scientific Evidence of Interventions Using the Mediterranean Diet: A Systematic Review. Nutr. Rev. 2006, 64, S27–S47. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, S.; Mackiewicz, M.; Yazdimamaghani, M. Recent Advances in the Redox-Responsive Drug Delivery Nanoplatforms: A Chemical Structure and Physical Property Perspective. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 118, 111536. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug Delivery Systems for RNA Therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.-J.; Won, Y.-S.; Kim, E.-K.; Park, S.-I.; Lee, S.J. Free Radicals and Their Impact on Health and Antioxidant Defenses: A Review. Cell Death Discov. 2025, 11, 19. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Shen, L.; Liu, C.C.; An, C.Y.; Ji, H.F. How Does Curcumin Work with Poor Bioavailability? Clues from Experimental and Theoretical Studies. Sci. Rep. 2016, 6, 20872. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, J.; Liu, Y.; Zhou, P.; Gu, Y. Mechanisms of Radiation-induced Tissue Damage and Response. MedComm 2024, 5, e725. [Google Scholar] [CrossRef]
- Surh, Y.J.; Na, H.K. NF-ΚB and Nrf2 as Prime Molecular Targets for Chemoprevention and Cytoprotection with Anti-Inflammatory and Antioxidant Phytochemicals. Genes. Nutr. 2008, 2, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, F.; Catanzaro, M.; Buoso, E.; Basagni, F.; Di Marino, D.; Raniolo, S.; Amadio, M.; Frost, E.H.; Corsini, E.; Racchi, M.; et al. Targeting Cytokine Release Through the Differential Modulation of Nrf2 and NF-ΚB Pathways by Electrophilic/Non-Electrophilic Compounds. Front. Pharmacol. 2020, 11, 1256. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-ΚB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Daverey, A.; Agrawal, S.K. Pre and Post Treatment with Curcumin and Resveratrol Protects Astrocytes after Oxidative Stress. Brain Res. 2018, 1692, 45–55. [Google Scholar] [CrossRef]
- Banaszak, M.; Górna, I.; Woźniak, D.; Przysławski, J.; Drzymała-Czyż, S. The Impact of Curcumin, Resveratrol, and Cinnamon on Modulating Oxidative Stress and Antioxidant Activity in Type 2 Diabetes: Moving beyond an Anti-Hyperglycaemic Evaluation. Antioxidants 2024, 13, 510. [Google Scholar] [CrossRef]
- Kostenko, V.; Akimov, O.; Gutnik, O.; Kostenko, H.; Kostenko, V.; Romantseva, T.; Morhun, Y.; Nazarenko, S.; Taran, O. Modulation of Redox-Sensitive Transcription Factors with Polyphenols as Pathogenetically Grounded Approach in Therapy of Systemic Inflammatory Response. Heliyon 2023, 9, e15551. [Google Scholar] [CrossRef]
- Acosta-Martinez, M.; Cabail, M.Z. The PI3K/Akt Pathway in Meta-Inflammation. Int. J. Mol. Sci. 2022, 23, 15330. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-ΚB in Biology and Targeted Therapy: New Insights and Translational Implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Bresciani, G.; Manai, F.; Davinelli, S.; Tucci, P.; Saso, L.; Amadio, M. Novel Potential Pharmacological Applications of Dimethyl Fumarate—An Overview and Update. Front. Pharmacol. 2023, 14, 1264842. [Google Scholar] [CrossRef]
- Berger, A.A.; Sottosanti, E.R.; Winnick, A.; Izygon, J.; Berardino, K.; Cornett, E.M.; Kaye, A.D.; Varrassi, G.; Viswanath, O.; Urits, I. Monomethyl Fumarate (Mmf, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (Ms). Neurol. Int. 2021, 13, 207–223. [Google Scholar] [CrossRef]
- Jonasson, E.; Sejbaek, T. Diroximel Fumarate in the Treatment of Multiple Sclerosis. Neurodegener. Dis. Manag. 2020, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, A.; Coronado-Albi, M.J.; Muñoz-Viana, R.; García-Merino, A.; Sánchez-López, A.J. Neuroprotective and Anti-Inflammatory Effects of Dimethyl Fumarate, Monomethyl Fumarate, and Cannabidiol in Neurons and Microglia. Int. J. Mol. Sci. 2024, 25, 13082. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Perlman, S.; Schadt, K. Omaveloxolone for the Treatment of Friedreich Ataxia: Clinical Trial Results and Practical Considerations. Expert. Rev. Neurother. 2024, 24, 251–258. [Google Scholar] [CrossRef]
- Pilotto, F.; Chellapandi, D.M.; Puccio, H. Omaveloxolone: A Groundbreaking Milestone as the First FDA-Approved Drug for Friedreich Ataxia. Trends Mol. Med. 2024, 30, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; De La Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Laurindo, L.F.; Santos, A.R. de O. dos; Carvalho, A.C.A. de; Bechara, M.D.; Guiguer, E.L.; Goulart, R. de A.; Vargas Sinatora, R.; Araújo, A.C.; Barbalho, S.M. Phytochemicals and Regulation of NF-KB in Inflammatory Bowel Diseases: An Overview of In Vitro and In Vivo Effects. Metabolites 2023, 13, 96. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Togni, L.; Santarelli, A.; Olivieri, F.; Marzioni, D.; Rippo, M.R. Modulation of NRF2/KEAP1 Signaling by Phytotherapeutics in Periodontitis. Antioxidants 2024, 13, 1270. [Google Scholar] [CrossRef]
- Kell, D.B. Iron Behaving Badly: Inappropriate Iron Chelation as a Major Contributor to the Aetiology of Vascular and Other Progressive Inflammatory and Degenerative Diseases. BMC Med. Genom. 2009, 2, 2. [Google Scholar]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and Natural Iron Chelators: Therapeutic Potential and Clinical Use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef]
- Bellanti, F.; Lo Buglio, A.; Dobrakowski, M.; Kasperczyk, A.; Kasperczyk, S.; Serviddio, G.; Vendemiale, G. Adherence to Mediterranean Diet and Biomarkers of Redox Balance and Inflammation in Old Patients Hospitalized in Internal Medicine. Nutrients 2024, 16, 3359. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Koelman, L.; Rodrigues, C.E. Dietary Patterns and Biomarkers of Oxidative Stress and Inflammation: A Systematic Review of Observational and Intervention Studies. Redox Biol. 2021, 42, 101869. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Zhao, N.; Wang, D.; Shams-White, M.; Karlsen, M.; Cassidy, A.; Ferruzzi, M.; Jacques, P.F.; Johnson, E.J.; Wallace, T.C. Dose-Response Relation between Tea Consumption and Risk of Cardiovascular Disease and All-Cause Mortality: A Systematic Review and Meta-Analysis of Population-Based Studies. Adv. Nutr. 2020, 11, 790–814. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent Developments in Delivery, Bioavailability, Absorption and Metabolism of Curcumin: The Golden Pigment from Golden Spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Liang, X.; Ao, Z.; Tang, X.; Li, C.; Yan, K.; Yu, X.; Wan, Y.; Li, Y.; Li, C.; et al. Stimulus-Responsive Drug Delivery Nanoplatforms for Inflammatory Bowel Disease Therapy. Acta Biomater. 2024, 188, 27–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, M.; Wang, T. Reactive Oxygen Species (ROS)-Responsive Biomaterials for Treating Myocardial Ischemia-Reperfusion Injury. Front. Bioeng. Biotechnol. 2024, 12, 1469393. [Google Scholar] [CrossRef]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-Associated Virus as a Delivery Vector for Gene Therapy of Human Diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Category | Oxidative Stress Markers | Reductive Stress Markers |
|---|---|---|
| Reactive species | Superoxide (O2•−) Hydroxyl radicals (•OH) | Excess NADH and NADPH |
| Oxidation to cell membrane lipids | HNE MDA | Altered lipid peroxidation ratio |
| Protein damage | Carbonylated proteins Oxidized thiols | Increased free thiol groups and disrupted disulfide bonds |
| DNA damage | 8-OHdG Nitrosylated DNA | Reduced DNA oxidation markers |
| Antioxidant levels | Decreased SOD, catalase, and glutathione | Overactive glutathione system |
| Disease | Oxidative Stress Role | Reductive Stress Role |
|---|---|---|
| Rheumatoid arthritis | Increased ROS leads to joint inflammation [120] | Reduced immune activation affects response [121] |
| Atherosclerosis | LDL oxidation triggers plaque formation [122] | Excess NADPH alters cholesterol metabolism [123] |
| Neurodegeneration | ROS damage neurons (Parkinson’s, Alzheimer’s) [87,124] | Excess reducing equivalents alters synaptic plasticity [125] |
| Diabetes | Mitochondrial ROS disrupt insulin signaling [126] | Increased NADH contributes to insulin resistance [70] |
| Therapeutic Category | Examples | Mechanism of Action | Targeted Redox Imbalance |
|---|---|---|---|
| Antioxidants [172] | Vitamin C, NAC, and resveratrol | ROS scavenging and NF-κB inhibition | Oxidative stress |
| Pharmacological agents [31,173] | Nrf2 activators NOX inhibitors | Modulate redox-sensitive transcription factors | Both |
| Dietary approaches [160,174] | Mediterranean diet Polyphenols | Enhancing endogenous antioxidant defenses | Both |
| Nanomedicine [175] | ROS-responsive nanoparticles | Targeted drug release in inflamed tissues | Oxidative stress |
| Gene therapy [176] | siRNA against NOX enzymes | Downregulating excess ROS production | Oxidative stress |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellanti, F.; Coda, A.R.D.; Trecca, M.I.; Lo Buglio, A.; Serviddio, G.; Vendemiale, G. Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress. Antioxidants 2025, 14, 656. https://doi.org/10.3390/antiox14060656
Bellanti F, Coda ARD, Trecca MI, Lo Buglio A, Serviddio G, Vendemiale G. Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress. Antioxidants. 2025; 14(6):656. https://doi.org/10.3390/antiox14060656
Chicago/Turabian StyleBellanti, Francesco, Anna Rita Daniela Coda, Maria Incoronata Trecca, Aurelio Lo Buglio, Gaetano Serviddio, and Gianluigi Vendemiale. 2025. "Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress" Antioxidants 14, no. 6: 656. https://doi.org/10.3390/antiox14060656
APA StyleBellanti, F., Coda, A. R. D., Trecca, M. I., Lo Buglio, A., Serviddio, G., & Vendemiale, G. (2025). Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress. Antioxidants, 14(6), 656. https://doi.org/10.3390/antiox14060656