1. Introduction
The retinal pigment epithelium (RPE) lies between the neuronal retina and the choriocapillaris. It forms a crucial part of the outer retinal blood barrier. The epithelium is vital for ocular health due to performing functions such as the phagocytosis of photoreceptor outer segments, the maintenance of the visual cycle, and the regulation of the energy balance of the retina [
1]. RPE cells are terminally differentiated and post-mitotic, a state that enables them to maintain their specialized roles in managing oxidative stress and retinal homeostasis over a lifetime but also renders them vulnerable to cumulative damage from reactive oxygen species.
The RPE also is a hub of ocular lipid metabolism and regulates the uptake of isoprenoid lipids, such as cholesterol, carotenoids, and retinoids, which are essential for photoreceptor function and survival [
2,
3,
4]. Disruptions in lipid transport can lead to lipid accumulation at the basolateral membrane of the RPE or lipid deficiencies in photoreceptors, contributing to oxidative stress and inflammation [
5]. This impairment is linked to ocular diseases such as age-related macular degeneration (AMD) and neurodegeneration [
6,
7]. Therefore, understanding lipid transport mechanisms at the RPE is critical for uncovering the molecular basis of associated diseases and identifying potential therapeutic targets.
Scavenger receptor class B type 1 (SR-B1) is expressed in the RPE and plays a crucial role in lipoprotein metabolism [
8]. SR-B1 facilitates the selective uptake of lipids, including high-density lipoprotein (HDL)-derived cholesterol and dietary carotenoids such as lutein and zeaxanthin [
9,
10], by loading them into the plasma membrane (PM). Recent studies have revealed that Aster proteins, encoded by the
GRAMD1A,
B, and
C genes, operate downstream of SR-B1, aiding in the transportation of lipids through non-vesicular transport mechanisms [
11,
12].
Aster proteins exhibit a tripartite structure featuring a StART-like lipid-binding domain flanked by an N-terminal GRAM domain and a C-terminal membrane anchor [
12]. The crystal structure of the lipid-binding domain of Aster-A, with bound cholesterol, has been solved [
12], and subsequent studies have demonstrated its capability to accommodate rigid C40 carotenoids [
13,
14]. Binding assays with recombinant Aster-A and B have confirmed that carotenoids can compete with cholesterol for binding to their StART-like domains [
14], indicating that Aster proteins display transport activity with several lipid classes. Aster proteins form contact points between the endoplasmic reticulum (ER) and PM, as well as between the ER and mitochondria [
15], thereby facilitating the non-vesicular transport of cholesterol and carotenoids between different cellular compartments.
GRAMD1A-C genes are expressed in several cell types and organs [
16]. A critical role of
GRAMD1B, encoding the Aster-B protein, has been established in cholesterol absorption in the enterocytes of the small intestine [
17]. Moreover, Aster-B plays a critical role in steroidogenesis in the adrenal glands [
12]. The
GRAMD1B gene is also highly expressed in several cell types of the human retina and the RPE [
14]. However, the ocular role of Aster-B has not been studied in detail.
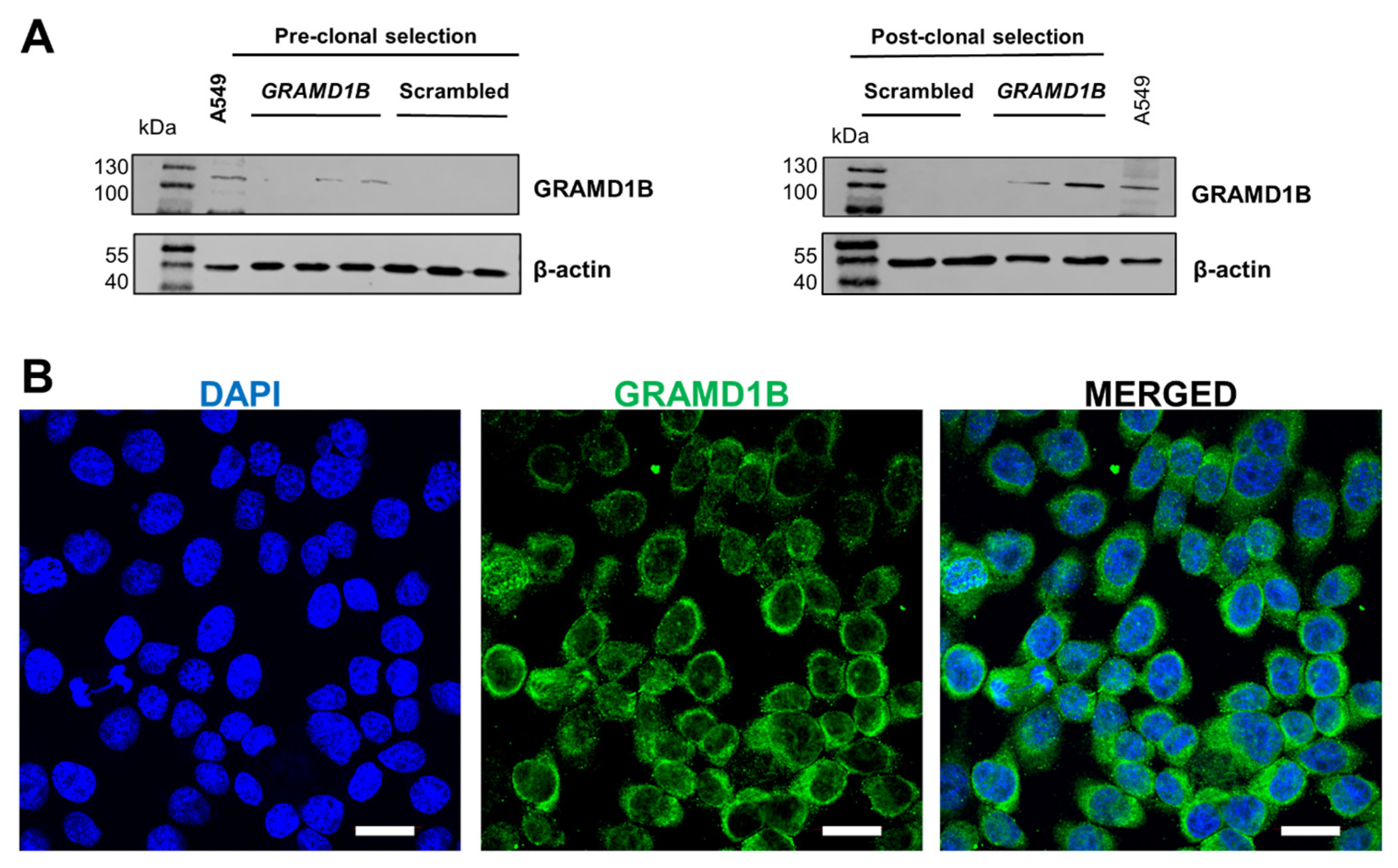
To investigate the role of Aster-B in ocular lipid metabolism, we utilized ARPE-19 cells, a human-derived cell line that retains many characteristics of primary RPE cells and is widely used in ocular disease research. Since ARPE-19 cells lack endogenous expression of the
GRAMD1B gene [
13], we employed CRISPR/dCas9 gene activation to induce its expression. To mimic the disease states of the RPE, we exposed cells to oxidative stress induced by hydrogen peroxide. Our study demonstrated that Aster-B facilitated lipid transport in ARPE-19 cells and influenced the cellular response to oxidative stress. These results uncover a novel role for Aster-B in oxidative stress defense, linked to lipid metabolism in ocular cells.
2. Materials and Methods
2.1. Reagents
H2O2, DCFH-DA, anti-COX IV antibody (Sigma, St. Loius, MO, USA), CyQUANT MTT Cell Viability Assay kit (V131540, Thermo Fisher, Waltham, MA, USA),TF Activation Profiling Plate Array (FA-1005) and Nuclear Extraction kit (SK-001) (Signosis, Santa Cruz, CA, USA). Control CRISPR Activation Plasmid (sc-437275), GRAMD1B CRISPR Activation Plasmid (h) (sc-408929-ACT), plasmid transfection medium (sc-108062), Ultra Cruz transfection reagent (sc-395739), hygromycin B (sc-29067), puromycin (sc-108071), and blasticidin S HCl (sc-204655A) (Santa Cruz Biotechnology, Dallas, TX, USA). Anti-GRAMD1B Polyclonal antibody (24905-1-AP, Proteintech, Rosemont, IL, USA), anti-Caspase-3 #9662, anti-NRF2 #20733S, anti-HO-1 #43966S, anti-P53 #9282 (Cell Signaling Technology, Danvers, MA, USA), Alexa Fluor 488-conjugated goat anti-rabbit IgG (A11008) HRP-conjugated goat anti-mouse and anti-rabbit IgG (H + L) (Invitrogen, Carlsbad, CA, USA), DAPI mounting medium (Southern biotech, Birmingham, AL, USA), HRP-conjugated secondary antibodies (Abcam, Waltham, MA, USA), Mitochondrial isolation kit, M-Per, Protease Inhibitor, Pierce ECL Western blotting substrate, BCA assay kit (Thermo Scientific, Waltham, MA, USA), PDVF membrane (Bio-Rad, Hercules, CA, USA), TaqMan primers (Thermo fisher, Waltham, MA, USA), TaqMan fast universal PCR master mix (Applied Biosystems, Foster City, CA, USA).
2.2. Cell Culture
The human ARPE-19 cell line was obtained from the American Type Culture Collection (ATCC), USA. ARPE-19 GRAMD1B-null ARPE-19 (−) and GRAMD1B-stable ARPE-19 (+) cells were cultured in DMEM/F-12 (1:1) medium (Gibco), supplemented with 2.5 mM L-glutamine, 15 mM HEPES buffer, 10% fetal bovine serum (FBS) (Gibco), and 1% (v/v) antibiotic antimycotic (Gibco) at 37 °C and 5% CO2. Typically, cells were seeded in a 24-well plate, a 6-well plate, or a 10 cm dish at a concentration of 150 K and 400 K per well and 2 × 107 cells per dish and grown overnight at 37 °C.
2.3. CRISPR/dCas9 Activation of Endogenous GRAMD1B Gene Expression
Transfection with plasmid DNA and the selection of GRAMD1B-stable clones were performed according to the manufacturer’s instructions. Initially, the optimal plasmid DNA transfection reagent ratio (manufacturer) was determined experimentally and optimized to minimize cell toxicity. For this purpose, the appropriate ratio of plasmid DNA to transfection reagent (manufacturer) used per well was established to determine which amount provided the highest transfection efficiency. The following solutions were prepared. Solution A: For each transfection, 0.5–2 µg of plasmid DNA and plasmid transfection medium were mixed to a final volume of 150 µL. The mix was incubated for 5 min at room temperature. Solution B: For each transfection, 5–15 µL of Ultra Cruz transfection reagent was diluted with plasmid transfection medium to a final volume of 150 µL and incubated for 5 min at room temperature. Then, solution A was added dropwise to solution B using a pipette. The combined solutions were vortexed immediately and incubated for 20 min at room temperature. Prior to transfection, the culture medium was replaced with fresh antibiotic-free growth medium, and a 300 µL plasmid transfection medium (combined solutions A and B) was added dropwise to the cells and gently mixed by swirling the plate.
For clonal selection, the human ARPE-19 cells seeded in a 6-well plate at a 40 to 60% confluency were transfected with scrambled plasmid DNA or GRAMD1B plasmid DNA at a concentration of 1.5 (µg/well) and cultured in normal medium for 60 h at 37 °C. The normal medium was replaced with 3 mL of the selection medium containing all hygromycin, blasticidin, and puromycin at concentrations of 200, 7.5, and 7.5 µg/mL of medium, respectively, following two rounds of antibiotic selection for a total duration of five days. The antibiotic-resistant scrambled and GRAMD1B stable clones were pooled and expanded in the normal medium for five days. Cells were then immediately used for further studies or frozen in liquid nitrogen for storage until future use.
2.4. Staining for Reactive Oxygen Species
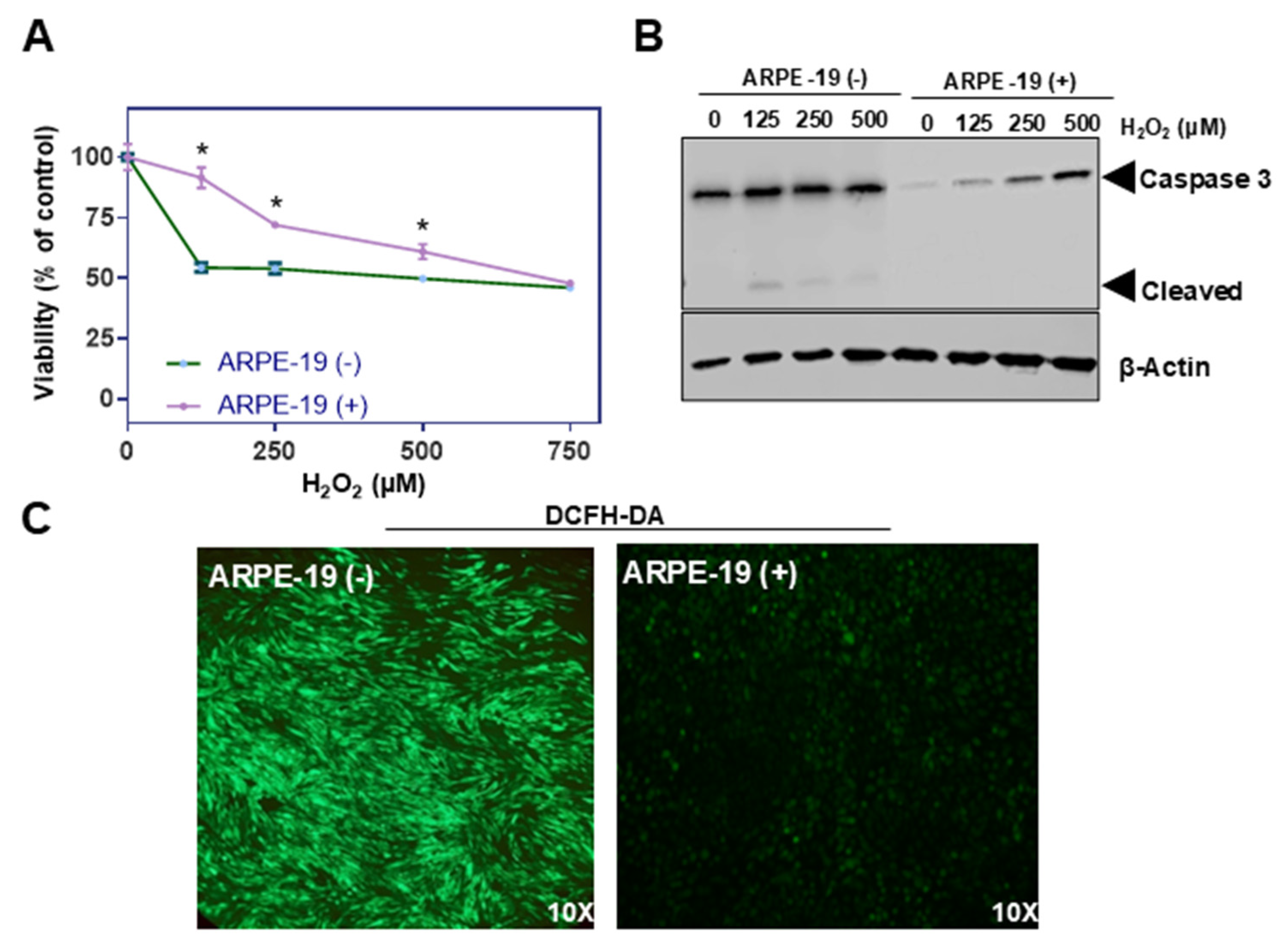
ROS levels were determined using a fluorescent probe, 2,7-dichlorofluorescin diacetate (DCFH-DA, Sigma). Briefly, ARPE-19 cells were seeded at a density of 2 × 105 cells per well in a 24-well plate and grown overnight at 37 °C. Then, cells were treated, respectively, with a defined dose of hydrogen peroxide for 24 h. Untreated cells served as controls. After washing cells with phosphate-buffer saline (PBS), medium containing 10 μM of DCF-DA was added for 30 min at 37 °C. After treatment, cells were washed once with DMEM and twice with PBS. After washing, 500 μL of PBS was added to each well, and the staining of the cells was observed and photographed using the green fluorescent protein (GFP) channel of a fluorescence microscope (Leica DMI6000B, Leica Microsystems, Wetzlar, Germany) equipped with a digital camera.
2.5. Treatment of Cells with Carotenoids and Hydrogen Peroxide
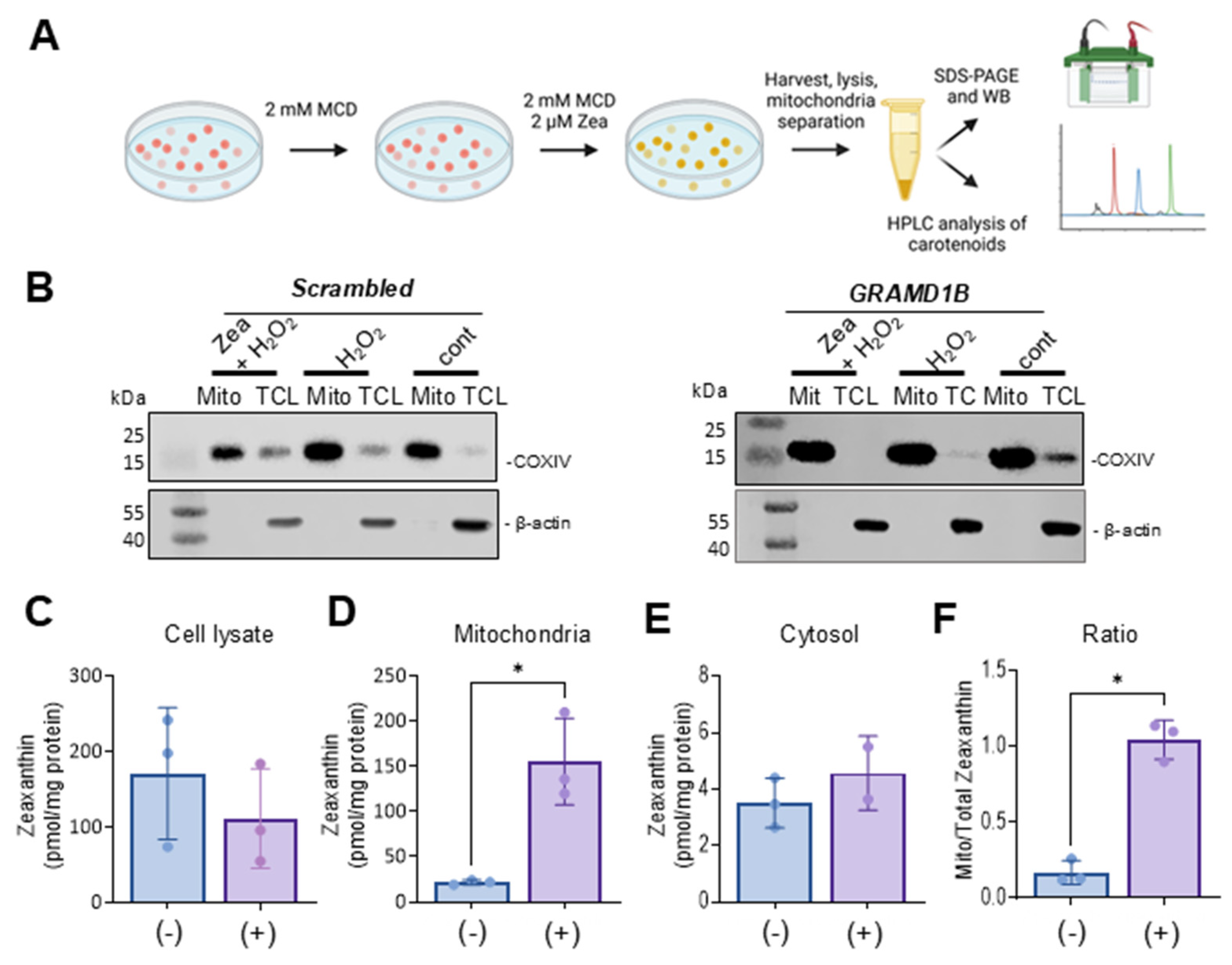
ARPE-19 cell lines were seeded in a 6-well plate or in a 10 cm dish at a concentration of 300 K and 2 × 107 cells/well or dish and grown until they reached ~90% confluency. Then, the medium was changed to a serum-free medium with 2 mM methyl-β-cyclodextrin (MCD) (Thermo Scientific) for 2 h at 37 °C in 5% CO2. Serum-free medium was then mixed with 2 mM MCD and 2 μM of zeaxanthin dissolved in acetone (<1% (v/v) final concentration). The cells were incubated for 24 h with the zeaxanthin/MCD mix to achieve cellular carotenoid uptake and transport. Cells were then washed and incubated in the presence of various concentrations of H2O2 for 24 h. Cells were then washed with PBS three consecutive times before they were collected by scraping and centrifugation. The collected cell pellets were either immediately subject to analyses or stored at −80 °C until further use.
2.6. Total Cell Lysate, Mitochondrial, Cytosolic, and Nuclear Extract Isolation
The total, mitochondrial, cytosolic, and nuclear fractions of the ARPE-19 cells were prepared according to the manufacturer’s instructions using the M-PER (Thermo Scientific), mitochondrial (Thermo Scientific), and Nuclear Extraction (Signosis) kits, respectively. For the isolation of total cell lysates, cell pellets were suspended in a 200 to 400 μL M-PER buffer containing a Protease Inhibitor cocktail (Thermo Scientific) and homogenized gently for 5 min. The samples were centrifuged at 15,000 rpm for 15 min to pellet the cell debris. The supernatant containing total cell lysates was used for further studies. For the isolation of mitochondrial and cytosolic fractions, 800 μL of mitochondria isolation reagent A was added to the cell pellets, vortexed for 5 s at medium speed and incubated on ice for two minutes. Then, 10 μL of mitochondria isolation reagent B was added, and the mix was vortexed at maximum speed for 5 s. The mix was incubated on ice for 5 min, and vortexing at maximum speed was repeated every minute. Then, 800 μL of mitochondria isolation reagent C was added, and the tubes were inverted several times to mix the solution. The extracts were then centrifuged at 700× g for 10 min at 4 °C. The pellet was discarded, and the supernatant was centrifuged at 12,000× g for 15 min at 4 °C. The supernatant containing the cytosolic fraction was saved, and the pellet containing the mitochondria was washed once with 500 μL of mitochondria isolation reagent C and centrifuged at 12,000× g for 5 min. For the isolation of nuclear extracts, 250 μL of buffer 1 was added to the cell pellet (1 × 106 cells/well), and the tubes were rocked at 200 rpm for 10 min on a shaking platform. The tubes were centrifuged at 12,000 rpm for 5 min at 4 °C, and the supernatant was discarded thoroughly and completely. In total, 50 μL of freshly prepared Buffer II was added to the pellet, and this allowed the pellet to gently float in the Buffer II working reagents. The tubes were placed on ice and shaken at 200 rpm for two hours. Then, the samples were centrifuged at 12,000 rpm for 5 min at 4 °C. The supernatants containing the nuclear extract were transferred to a new tube and stored on ice until further analysis. A BCA assay (Thermo scientific) was used to determine the protein concentration of the different subcellular fractions. Typically, the protein concentrations in different cellular fractions ranged as follows: total cell lysates (15 to 20 µg/µL); mitochondria (8 to 10 µg/µL); cytosol (1 to 2.5 µg/µL); nuclear extracts (2 to 3 µg/µL).
2.7. SDS-PAGE and Immunoblotting
Total soluble protein lysates from ARPE19-null and -stable cell pellets treated as described above were extracted using M-PER (Mammalian Protein Extraction Reagent) (Thermo scientific), containing Protease Inhibitor (Thermo scientific). The protein concentration was determined by the BCA (Thermo scientific) assays. In total, 30 to 60 μg of protein per lane was denatured in 2 × gel-loading buffer (4% SDS, 20% glycerol, 120 mM Tris-HCl (pH 6.8), and 0.01% bromophenol blue, with 10%‚ β-mercaptoethanol), heated to 95 °C for 5 min, and resolved by SDS-PAGE. Proteins were transferred to the PVDF membrane for 2 h and blocked for one hour at room temperature. The blots were incubated overnight at 4 °C with the primary antibody with 5% BSA in TBST. The antibodies used included GRAMD1B (Proteintech), COXIV, Caspase-3, PARP, and Histone H3 (Cell Signaling Technology) at a dilution of 1:1000. For protein loading controls, anti-β-actin (Cell Signaling) was used at a dilution of 1:2000. The blots were washed with TBST and incubated with secondary anti-rabbit IgG antibody or anti-mouse IgG antibody (Abcam) at a dilution of 1:5000 in 5% milk in TBST for one hour at room temperature. The blots were washed with 1X TBST thrice. The PierceTM ECL Western blotting substrate (Thermo scientific) was added to the blots for 1–3 min and was scanned with the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE, USA) for chemiluminescence detection.
2.8. Carotenoid Extraction and HPLC Analysis
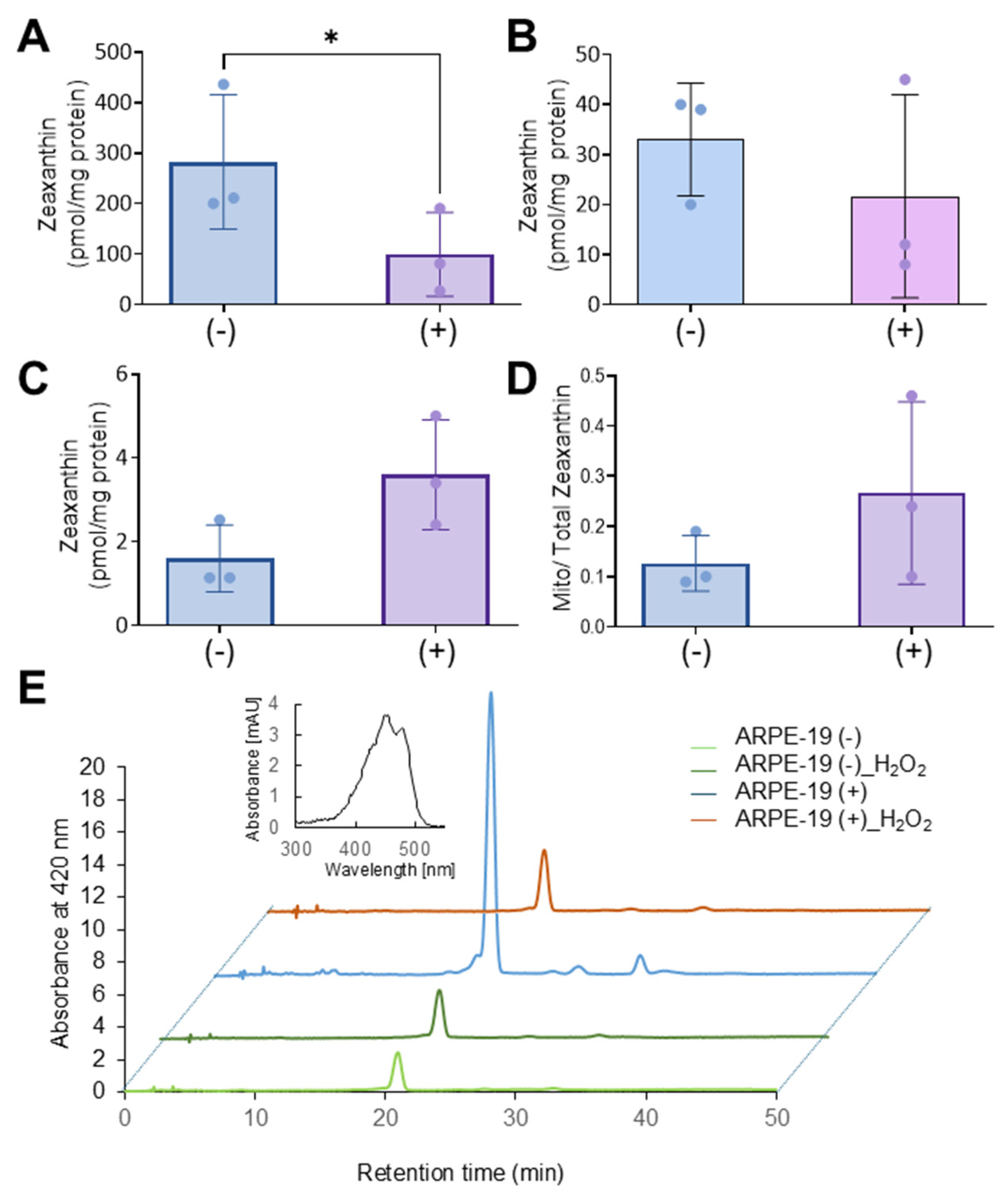
Zeaxanthin (β,β-carotene-3R,3R’-diol) was a gift from DSM (Sisseln, Switzerland). Zeaxanthin was extracted from total cell lysate, mitochondrial, and cytoplasm fractions, using previously published protocols [
13], and subjected to HPLC analysis on a silica column using hexanes: ethyl acetate (70:30
v/
v) as the mobile phase. In a typical zeaxanthin extraction, 200 µL of total cell lysate, 100 µL of the mitochondrial fraction, and 750 µL of cytosolic fractions were mixed with the same amount of methanol. Then, 400 µL of acetone, 400 µL of diethyl ether, and 100 µL of petroleum ether were added. The mixture was vortexed two times for 15 s. Phase separation was achieved by centrifugation for one minute at room temperature (2000×
g). The organic phase was transferred to a new tube and vacuum-dried using a speed vacufuge (Eppendorf). The extracted zeaxanthin was reconstituted in 150 uL of hexane/ethyl acetate (70:30
v/
v) solvent. HPLC analysis was carried out with an Agilent 1260 Infinity Quaternary HPLC system (Santa Clara, CA, USA) equipped with a pump (G1312C), with an integrated degasser (G1322A), a thermostated column compartment (G1316A), an autosampler (G1329B), a diode array detector (G1315D), and online analysis software (Agilent OpenLab CDS Chemstation, A.01.02). The analyses were carried out at 25 °C using a normal-phase Zorbax Sil (5 μm, 4.6 × 150 mm) column (Agilent Technologies, Santa Clara, CA, USA). Chromatographic separation was achieved by isocratic flow at 1.4 mL per minute. Zeaxanthin and its metabolic products were identified based on the absorption spectrum of their peaks. The integration area for each peak was calculated, and the picomole amount of zeaxanthin was determined based on scaling the column with known amounts of zeaxanthin standard and normalized to the mg protein amount of the samples.
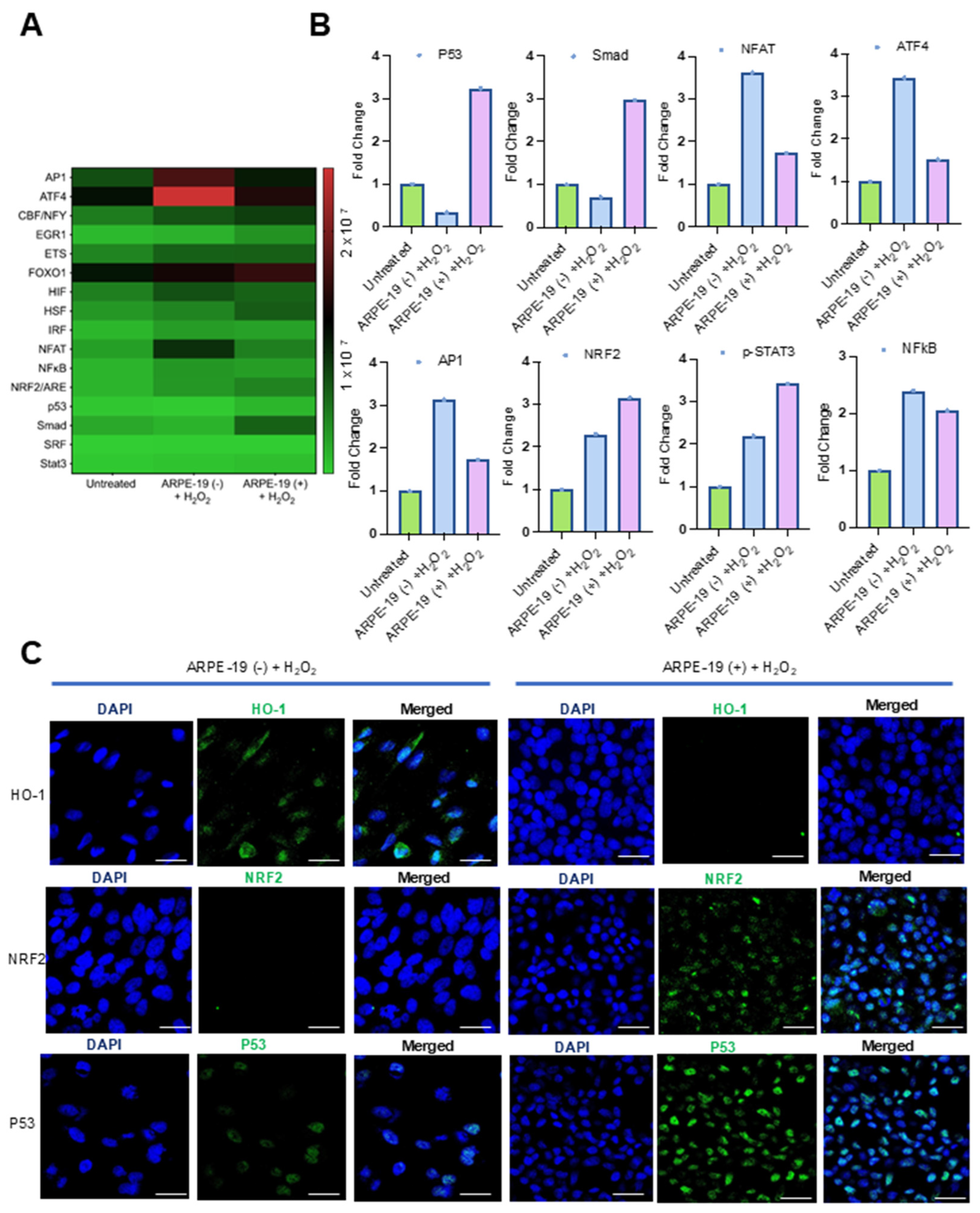
2.9. Oxidative Stress Transcription Factor Activation Profiling
The oxidative stress transcription factor activation profiling assay (Signosis) was used for monitoring the activation of oxidative stress-related transcription factors (TFs) following the manufacturer’s instructions. Briefly, the nuclear extracts of ARPE-19 cells were mixed with a series of biotin-labeled DNA probes based on the consensus sequences of transcription factor DNA-binding sites and incubated at RT for 30 min. After the incubation, the bound TFs were separated from the free TFs through spin column purification. Next, the bound probe was eluted and analyzed through hybridization with a hybridization plate where each well was precoated with complementary DNA sequences of the TF probe. The assay allowed the simultaneous analysis of the activation of 16 oxidative stress-related TFs. In total, 100 µL of denatured probes were diluted in 10 mL of warmed hybridization buffer, and 95 µL of the mixture was added into the corresponding 16 wells, and the plate was incubated at 42 °C overnight. The wells were washed thrice with the hybridization wash buffer and blocked for 15 min with the blocking buffer. After 15 min of blocking, 20 μL of Streptavidin-HRP Conjugate in 10 mL of blocking buffer (1:500 dilution) was added to each well and incubated for 45 min at room temperature with gentle shaking. After 45 min, a 200 μL detection wash buffer was added to each well, and the plate was incubated for 5 min with gentle shaking on a plate-shaker at room temperature. The wells were washed thrice for a total of 15 min. After the last wash, 1 mL of substrate A and 1 mL of substrate B were mixed with 8 mL of substrate dilution buffer and 95 μL of substrate solution in each well and incubated for 1 min. The captured DNA probe was detected with the diluted Streptavidin-HRP Conjugate. The plate was placed in the microplate luminometer and read within 5 min. Luminescence is reported as relative light units (RLUs).
2.10. Cell Viability Assays
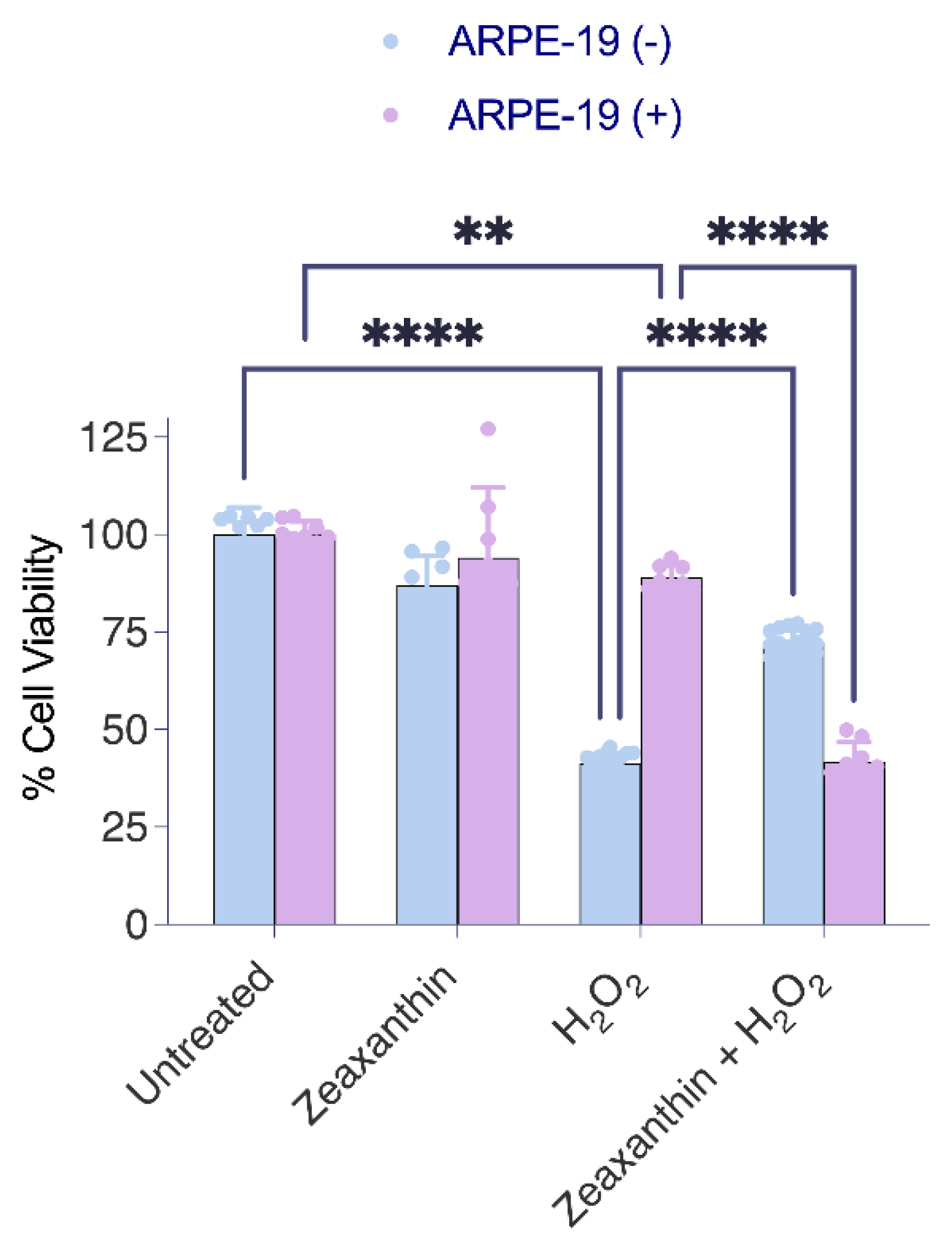
Cell viability was measured by an MTT assay (CyQUANT MTT, Thermo scientific). Briefly, ARPE-19 cells were seeded in 96 96-well plates at a density of 4 × 103 cells per well and grown overnight at 37 °C. Cells were left either untreated or treated with H2O2 in the presence or absence of zeaxanthin for 24 h in serum-free medium. Following treatment, the cells were washed with PBS 3 times and were incubated with a 10 μL MTT solution for 4 h. This was followed by 100 μL of the SDS-HCL solution for 4 h at 37 °C. The amount of formazan dye was measured by detecting the absorbance at a wavelength of 570 nm with a microplate reader (Versamax Microplate Reader, Molecular devices). Eight readings per sample were taken to reduce random errors, and the average was calculated.
2.11. qRT-PCR Analysis of Marker Genes
Total RNA was isolated from ARPE19 cells using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) and the RNeasy Mini Kit (Qiagen, Germantown, MD, USA). RNA concentration and purity were determined with a nano-drop spectrophotometer (Thermo Scientific). Isolated RNA was converted to cDNA using the High-Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Gene expression analysis was carried out by real-time quantitative PCR using an Applied Biosystems real-time PCR instrument with Taq Man probes (Thermo Fisher Scientific). Primers used for analysis were human GAPDH (Hs99999905) and BCO2 (Hs01568558). We used 20 ng of cDNA in a 10 μL reaction, and the amplification was carried out using TaqMan polymerase Fast Universal PCR Master Mix (2×) No Amp Erase, UNG (Applied Biosystems), following the manufacturer’s protocol. Gene expression levels were normalized to the expression of the housekeeping gene GAPDH using the ∆∆Ct method.
2.12. Immunocytochemistry and Confocal Imaging
ARPE19 (−) and ARPE19 (+) cells were grown on Labtek chamber slides (Nunc Thermofisher, Walthamn, MA, USA) for 24 h at 37 °C in a 5% CO2 incubator. Cells were washed with 1 × phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) for 15 min at room temperature. Cells were washed and permeabilized with 0.2% Triton X-100 (Sigma) for 15 min. Cells were washed and blocked with 5% goat serum in PBST (Tween 20, 0.05%) for 1 h at 37 °C and incubated with primary antibodies GRAMD-1B (2 μg/mL), NRF2, p53, and HO-1 (0.4 μg/mL) at 4 °C overnight. After a wash with PBST buffer, cells were incubated with secondary antibody Alexa Fluor rabbit 488 and Alexa Fluor mouse 555 (4 μg/mL) at room temperature for 2 h. DAPI Fluoromount G (Southern biotech) was used to stain the nucleus. Images were taken with an Olympus FV1200 laser scanning confocal microscope (Olympus America, Waltham, MA, USA) using a 405 nm diode laser for the blue channel, 473 nm diode laser for the green channel with a UPLXAPO100XO oil OFN26.5, NA1.45 objective.
2.13. Statistical Analysis
Data shown are the mean ± SD and are representative of more than two or three independent experiments. Analysis was performed using an unpaired two-tailed t test and one-way ANOVA using GraphPad Prism 10.2 software, and results are considered significant at * p < 0.05, ** p < 0.005, and **** p < 0.0001.
4. Discussion
GRAMD1B, encoding the lipid carrier protein Aster-B, is emerging as a critical regulator of cholesterol homeostasis and isoprenoid lipid transport [
11,
12,
17,
25], including carotenoids [
13,
14]. This protein is expressed in the human neuronal retina and retinal pigment epithelium (RPE) [
14], where lipid metabolism plays a pivotal role in cellular health [
2]. Dysregulated lipid metabolism in the RPE, characterized by lipid-rich drusen accumulation and oxidative damage, is a hallmark of chronic ocular diseases such as age-related macular degeneration (AMD) [
26]. To investigate
GRAMD1B’s role, we developed an ARPE-19 cell model with CRISPR/dCas9-induced
GRAMD1B expression and assessed its impact on lipid transport and oxidative stress responses.
Our findings provide evidence that Aster-B expression confers protective effects against oxidative stress, as ARPE-19 (+) cells expressing
GRAMD1B exhibited reduced ROS levels and improved survival under hydrogen peroxide-induced stress. These effects appear to stem from Aster-B in maintaining membrane cholesterol homeostasis, highlighting its capacity to modulate oxidative stress independently of carotenoid transport. Additionally, lysosomes are essential for the degradation and recycling of lipid-rich photoreceptor outer segments. However, with aging or stress, lysosomal function can become compromised, leading to the accumulation of lipofuscin, an autofluorescent aggregate composed of oxidized lipids and proteins. Lipofuscin components, particularly bisretinoids such as A2E, have been shown to interfere with cholesterol metabolism by impairing cholesterol efflux from the lysosomal compartment, resulting in intracellular cholesterol accumulation [
27]. This disruption in cholesterol homeostasis can contribute to lipid dysregulation, and Aster proteins may help to prevent this accumulation by moving cholesterol between membranes [
28]. Cholesterol’s influence on cellular stress pathways, including the Nrf2 pathway and mitochondrial membrane integrity, aligns with our results [
29].
By facilitating non-vesicular cholesterol transport [
15], Aster-B likely supports cellular health, reducing ARPE-19 cells’ vulnerability to oxidative damage. Such a scenario is particularly relevant in AMD, where mitochondrial dysfunction and lipid dysregulation are key pathological features [
5].
We used a commercial TF panel to detect oxidative stress pathways that are modulated by Aster-B. We observed a putative interplay between Aster-B and the tumor suppressor protein p53. It has been reported that cholesterol impacts p53 activity through mechanisms such as the oxysterol-mediated activation of apoptotic pathways and the modulation of membrane fluidity, which influence p53 localization and activity [
30]. Aster-B’s role in cholesterol transport may indirectly affect p53-mediated stress responses, including oxidative stress and ER stress. Additionally, p53’s regulation of cholesterol metabolism genes, such as sterol regulatory element-binding proteins (SREBPs), may contribute to a potential feedback loop between Aster-B and p53 in maintaining lipid homeostasis and cellular health [
31].
Nrf2 signaling was activated in both ARPE-19 cell lines. Interestingly, the Nrf2 target gene HO-1 was only detectable in ARPE-19 (−) but not ARPE-19 (+) cells (
Figure 3 and
Supplementary Figure S2). Aster-B expression likely modulates the oxidative stress response by altering intracellular lipid homeostasis and redox signaling, thereby impacting Nrf2-driven transcriptional activity in a complex manner. While Nrf2 protein levels are elevated in ARPE-19 (+) cells, the efficient transcriptional activation of downstream targets like HO-1 may require not only Nrf2 nuclear accumulation but also a favorable chromatin environment, intact redox-sensitive co-factors, and appropriate cellular context. Aster-B-mediated alterations in lipid and cholesterol distribution during oxidative stress could disrupt these regulatory mechanisms, preventing HO-1 induction despite increased Nrf2 levels. More research is needed to elucidate the biochemical and molecular details of this interaction.
Carotenoids, including lutein and zeaxanthin, add complexity to the oxidative stress response in human eyes. Known for their antioxidant properties, these pigments filter blue light and neutralize ROS, offering protection to ocular tissues [
32,
33,
34,
35]. Our study showed that Aster-B facilitates the intracellular movement of carotenoids in ARPE-19 cells. Interestingly, Aster-B expression had a pronounced effect on the response to hydrogen peroxide treatment. In ARPE-19 (+) cells, carotenoids surprisingly exacerbate the response to oxidative stress, as indicated by reduced survival rates as compared to control cells. By contrast, carotenoid treatment improved survival in ARPE-19 (−) cells when compared to controls. Such improved resistance to abiotic stress has been previously reported in carotenoid-treated ARPE-19 cells and might be related to the physical properties of the pigments [
22,
36]. The observations in ARPE-19 (+) cells suggest that excessive carotenoid accumulation in mitochondria may aggravate oxidative stress and apoptosis, revealing a potential trade-off in their protective roles. Interestingly, the adverse effects of carotenoids in ARPE-19 (+) cells in the presence of hydrogen peroxide were associated with reduced zeaxanthin levels in cells. The determination of BCO2 mRNA levels, encoding a carotenoid catabolizing enzyme, ruled out that this reduction went along with enzymatic carotenoid turnover. Therefore, we assume that the unspecific oxidation of carotenoids contributed to the reduced zeaxanthin levels in ARPE-19 (+) cells and the increased cell mortality in our experiments.
We and others previously have shown that a mitochondrial accumulation of carotenoids can be associated with cell death in the absence of BCO2 [
23,
37,
38,
39]. Notably, the RPE and many cell types of the neuronal retina express mitochondrial BCO2 [
40,
41,
42], likely to protect them from detrimental carotenoid accumulation in mitochondria. Thus, our findings suggest that carotenoids act context- and location-dependently and provide either protection against ROS or exacerbate stress responses.
5. Conclusions
Our investigation highlights Aster-B’s structural and functional versatility. By forming contact sites between the PM, endoplasmic reticulum, and mitochondria, the lipid carrier facilitates the movement of lipids across cellular membranes, integrating cholesterol and carotenoid metabolism with stress responses. This structural organization positions Aster-B as a central player in maintaining cellular lipid balance and mitigating oxidative stress.
The clinical relevance of these findings is underscored by the success of carotenoid-based interventions, such as the Age-Related Eye Disease Study (AREDS), which demonstrated the protective effects of lutein and zeaxanthin against AMD progression [
9,
10]. Elevated high-density lipoprotein cholesterol levels and an altered composition in AMD patients further suggest that Aster-B’s role in cholesterol transport could influence disease outcomes [
43]. Understanding how Aster proteins integrate with cholesterol-mediated pathways, such as NF-κB and Nrf2, and its interactions with carotenoids, could pave the way for targeted therapeutic strategies.
Therefore, future research should focus on delineating the mechanisms by which Aster-B coordinates cholesterol and carotenoid metabolism under stress conditions. Investigating its regulatory interactions with p53 and cholesterol-mediated signaling pathways, such as NF-κB and Nrf2, could provide deeper insights into its role in oxidative stress defense. Furthermore, optimizing the balance between carotenoids’ antioxidant properties and their potential pro-oxidant effects in an Aster-B-dependent context may improve therapeutic interventions. These efforts could significantly enhance our understanding of lipid dysregulation and oxidative stress in retinal diseases like AMD, offering promising avenues for disease management and treatment.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}