
The Role of Oxidative Stress-Induced Senescence in the Pathogenesis of Preeclampsia


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Definition and Types of Preeclampsia
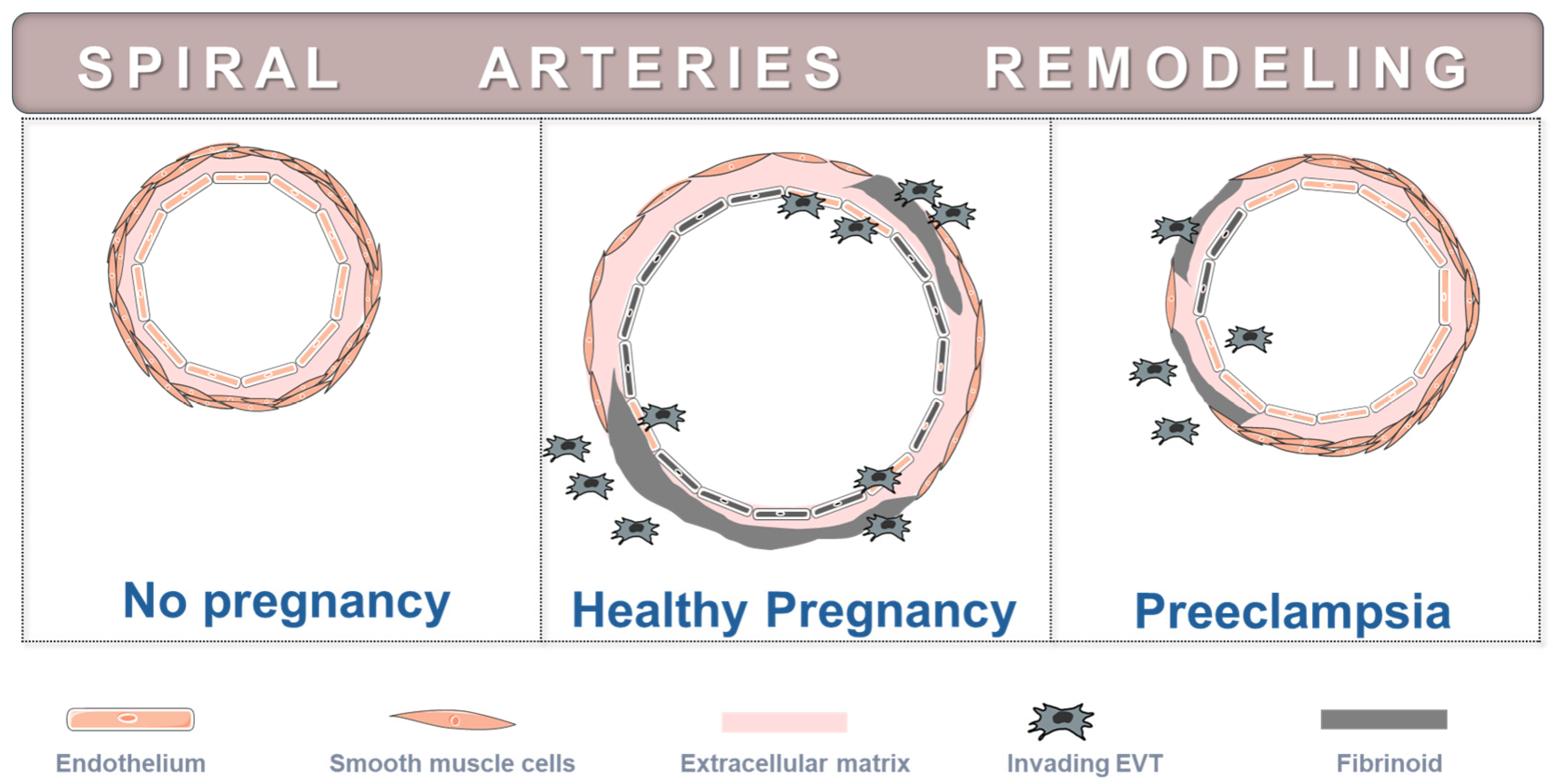
3. Placental Development and Placenta Morphology in Preeclampsia
4. Pathogenetic Implications of Oxidative Stress in Preeclampsia
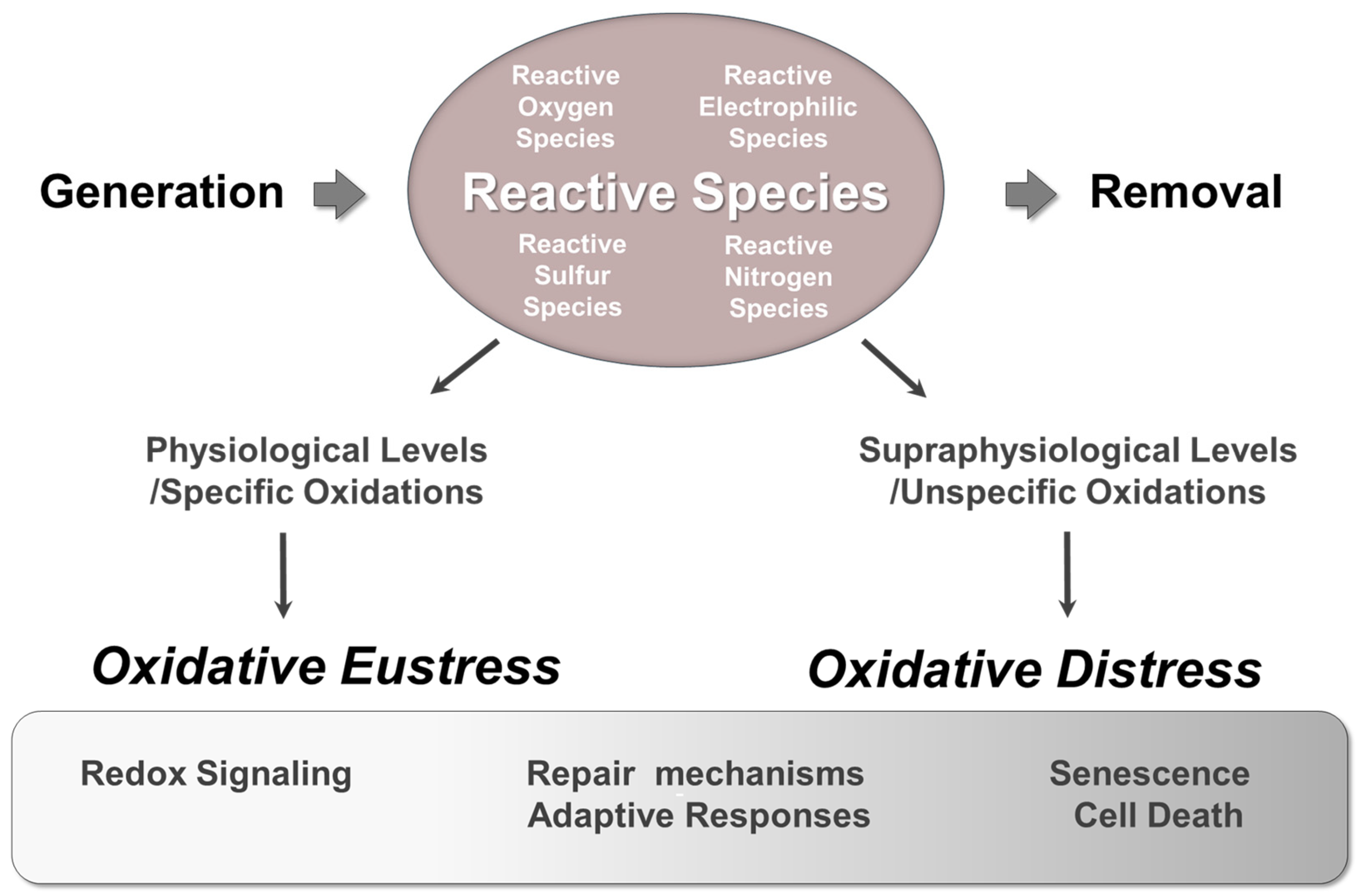
4.1. The Dual Role of Reactive Species in Aerobes: Physiological Functions Versus Pathophysiological Consequences
4.2. Ischemia and Reperfusion-Induced Injury
4.3. Reactive Oxygen Species as Contributors to Ischemia and Reperfusion Injury
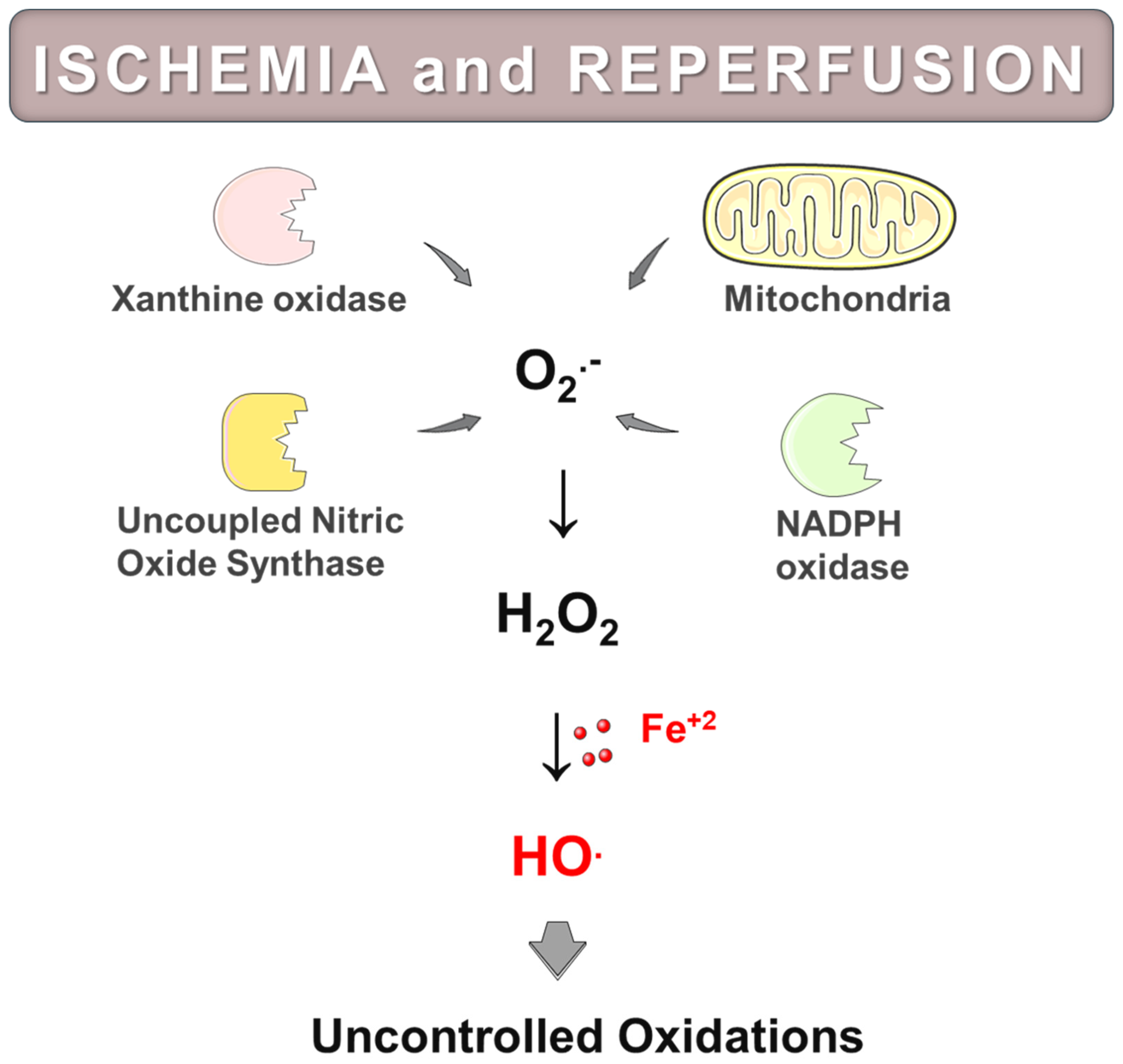
4.4. Sources and Biochemical Mechanisms of Reactive Oxygen Species Generation in Post-Ischemic Tissues
4.5. Damaging Effects of Oxidative Stress in Preeclampsia
5. Pathogenetic Implications of Oxidative Stress-Induced Placental Senescence
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dimitriadis, E.; Rolnik, D.L.; Zhou, W.; Estrada-Gutierrez, G.; Koga, K.; Francisco, R.P.V.; Whitehead, C.; Hyett, J.; da Silva Costa, F.; Nicolaides, K.; et al. Pre-Eclampsia. Nat. Rev. Dis. Primers 2023, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis, and Management Recommendations for International Practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia—Pathophysiology and Clinical Presentations. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef] [PubMed]
- Erez, O.; Romero, R.; Jung, E.; Chaemsaithong, P.; Bosco, M.; Suksai, M.; Gotsch, F. Preeclampsia and Eclampsia: The Conceptual Evolution of a Syndrome. Am. J. Obstet. Gynecol. 2022, 226, S786–S803. [Google Scholar] [CrossRef] [PubMed]
- Gusella, A.; Martignoni, G.; Giacometti, C. Behind the Curtain of Abnormal Placentation in Pre-Eclampsia: From Molecular Mechanisms to Histological Hallmarks. Int. J. Mol. Sci. 2024, 25, 7886. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Swiader, A.; Salvayre, R.; Guerby, P. Oxidative Stress, Lipid Peroxidation and Premature Placental Senescence in Preeclampsia. Arch. Biochem. Biophys. 2022, 730, 109416. [Google Scholar] [CrossRef]
- Burton, G.J.; Cindrova-Davies, T.; Yung, H.W.; Jauniaux, E. Oxygen and Development of the Human Placenta. Reproduction 2021, 161, F53–F65. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast Stress in Preeclampsia: The Convergence Point for Multiple Pathways. Am. J. Obstet. Gynecol. 2022, 226, S907–S927. [Google Scholar] [CrossRef]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef]
- Manna, S.; McCarthy, C.; McCarthy, F.P. Placental Ageing in Adverse Pregnancy Outcomes: Telomere Shortening, Cell Senescence, and Mitochondrial Dysfunction. Oxidative Med. Cell. Longev. 2019, 2019, 1496. [Google Scholar] [CrossRef]
- Sultana, Z.; Maiti, K.; Dedman, L.; Smith, R. Is There a Role for Placental Senescence in the Genesis of Obstetric Complications and Fetal Growth Restriction? Am. J. Obstet. Gynecol. 2018, 218, S762–S773. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Atkinson, D.; Campion-Smith, T.; Olovsson, M.; Charnock-Jones, D.S.; Burton, G.J. Differential Activation of Placental Unfolded Protein Response Pathways Implies Heterogeneity in Causation of Early- and Late-Onset Pre-Eclampsia. J. Pathol. 2014, 234, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Colleoni, F.; Dommett, E.; Cindrova-Davies, T.; Kingdom, J.; Murray, A.J.; Burton, G.J. Noncanonical Mitochondrial Unfolded Protein Response Impairs Placental Oxidative Phosphorylation in Early-Onset Preeclampsia. Proc. Natl. Acad. Sci. USA 2019, 116, 18109–18118. [Google Scholar] [CrossRef]
- Burton, G.J.; Yung, H.W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta 2009, 30, 43–48. [Google Scholar] [CrossRef]
- Cox, L.S.; Redman, C. The Role of Cellular Senescence in Ageing of the Placenta. Placenta 2017, 52, 139–145. [Google Scholar] [CrossRef]
- Farladansky-Gershnabel, S.; Gal, H.; Kidron, D.; Krizhanovsky, V.; Amiel, A.; Sukenik-Halevy, R.; Biron-Shental, T. Telomere Homeostasis and Senescence Markers Are Differently Expressed in Placentas from Pregnancies with Early- Versus Late-Onset Preeclampsia. Reprod. Sci. 2019, 26, 1203–1209. [Google Scholar] [CrossRef]
- Scaife, P.J.; Simpson, A.; Kurlak, L.O.; Briggs, L.V.; Gardner, D.S.; Pipkin, F.B.; Jones, C.J.P.; Mistry, H.D. Increased Placental Cell Senescence and Oxidative Stress in Women with Pre-Eclampsia and Normotensive Post-Term Pregnancies. Int. J. Mol. Sci. 2021, 22, 7295. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Fogarty, N.M.E.; Jones, C.J.P.; Kingdom, J.; Burton, G.J. Evidence of Oxidative Stress-Induced Senescence in Mature, Post-Mature and Pathological Human Placentas. Placenta 2018, 68, 15–22. [Google Scholar] [CrossRef]
- Huppertz, B. Traditional and New Routes of Trophoblast Invasion and Their Implications for Pregnancy Diseases. Int. J. Mol. Sci. 2020, 21, 289. [Google Scholar] [CrossRef]
- Mizuuchi, M.; Cindrova-Davies, T.; Olovsson, M.; Charnock-Jones, D.S.; Burton, G.J.; Yung, H.W. Placental Endoplasmic Reticulum Stress Negatively Regulates Transcription of Placental Growth Factor via ATF4 and ATF6β: Implications for the Pathophysiology of Human Pregnancy Complications. J. Pathol. 2016, 238, 550–561. [Google Scholar] [CrossRef]
- Burton, G.J.; Yung, H.W.; Murray, A.J. Mitochondrial—Endoplasmic Reticulum Interactions in the Trophoblast: Stress and Senescence. Placenta 2017, 52, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Romero, R.; Yeo, L.; Gomez-Lopez, N.; Chaemsaithong, P.; Jaovisidha, A.; Gotsch, F.; Erez, O. The Etiology of Preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S844–S866. [Google Scholar] [CrossRef] [PubMed]
- Many, A.; Hubel, C.A.; Fisher, S.J.; Roberts, J.M.; Zhou, Y. Invasive Cytotrophoblasts Manifest Evidence of Oxidative Stress in Preeclampsia. Am. J. Pathol. 2000, 156, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, S.; Li, L. Advanced Oxidative Protein Products Drive Trophoblast Cells Into Senescence by Inhibiting the Autophagy: The Potential Implication of Preeclampsia. Front. Cell Dev. Biol. 2022, 10, 810282. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. The Cytotrophoblastic Shell and Complications of Pregnancy. Placenta 2017, 60, 134–139. [Google Scholar] [CrossRef]
- Londero, A.P.; Orsaria, M.; Marzinotto, S.; Grassi, T.; Fruscalzo, A.; Calcagno, A.; Bertozzi, S.; Nardini, N.; Stella, E.; Lellé, R.J.; et al. Placental Aging and Oxidation Damage in a Tissue Micro-Array Model: An Immunohistochemistry Study. Histochem. Cell. Biol. 2016, 146, 191–204. [Google Scholar] [CrossRef]
- Takagi, Y.; Nikaido, T.; Toki, T.; Kita, N.; Kanai, M.; Ashida, T.; Ohira, S.; Konishi, I. Levels of Oxidative Stress and Redox-Related Molecules in the Placenta in Preeclampsia and Fetal Growth Restriction. Virchows Arch. 2004, 444, 49–55. [Google Scholar] [CrossRef]
- Kimura, C.; Watanabe, K.; Iwasaki, A.; Mori, T.; Matsushita, H.; Shinohara, K.; Wakatsuki, A. The Severity of Hypoxic Changes and Oxidative DNA Damage in the Placenta of Early-Onset Preeclamptic Women and Fetal Growth Restriction. J. Matern. Fetal Neonatal Med. 2013, 26, 491–496. [Google Scholar] [CrossRef]
- Cui, X.L.; Brockman, D.; Campos, B.; Myatt, L. Expression of NADPH Oxidase Isoform 1 (Nox1) in Human Placenta: Involvement in Preeclampsia. Placenta 2006, 27, 422–431. [Google Scholar] [CrossRef]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-Eclampsia: Pathophysiology and Clinical Implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef]
- Fujimaki, A.; Watanabe, K.; Mori, T.; Kimura, C.; Shinohara, K.; Wakatsuki, A. Placental Oxidative DNA Damage and Its Repair in Preeclamptic Women with Fetal Growth Restriction. Placenta 2011, 32, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Kohlrausch, F.B.; Keefe, D.L. Telomere Erosion as a Placental Clock: From Placental Pathologies to Adverse Pregnancy Outcomes. Placenta 2020, 97, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sugulle, M.; Fiskå, B.S.; Jacobsen, D.P.; Fjeldstad, H.E.; Staff, A.C. Placental Senescence and the Two-Stage Model of Preeclampsia. Am. J. Reprod. Immunol. 2024, 92, e13904. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell. Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of Redox Regulation in Biology. Nat. Rev. Mol. Cell. Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Evangelou, K.; Belogiannis, K.; Papaspyropoulos, A.; Petty, R.; Gorgoulis, V.G. Escape from Senescence: Molecular Basis and Therapeutic Ramifications. J. Pathol. 2023, 260, 649–665. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Zambella, E.; Peruffo, B.; Guarano, A.; Inversetti, A.; Di Simone, N. The Hidden Relationship between Intestinal Microbiota and Immunological Modifications in Preeclampsia Pathogenesis. Int. J. Mol. Sci. 2024, 25, 10099. [Google Scholar] [CrossRef] [PubMed]
- Herrock, O.; Deer, E.; LaMarca, B. Setting a Stage: Inflammation during Preeclampsia and Postpartum. Front. Physiol. 2023, 14, 1130116. [Google Scholar] [CrossRef] [PubMed]
- Cristodoro, M.; Messa, M.; Tossetta, G.; Marzioni, D.; Dell’Avanzo, M.; Inversetti, A.; Di Simone, N. First Trimester Placental Biomarkers for Pregnancy Outcomes. Int. J. Mol. Sci. 2024, 25, 6136. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Giannubilo, S.R.; Marzioni, D.; Tossetta, G. HTRA Family Proteins in Pregnancy Outcome. Tissue Cell 2021, 72, 101549. [Google Scholar] [CrossRef]
- Dasilva-Arnold, S.; James, J.L.; Al-Khan, A.; Zamudio, S.; Illsley, N.P. Differentiation of First Trimester Cytotrophoblast to Extravillous Trophoblast Involves an Epithelial-Mesenchymal Transition. Placenta 2015, 36, 1412–1418. [Google Scholar] [CrossRef]
- Natenzon, A.; McFadden, P.; DaSilva-Arnold, S.C.; Zamudio, S.; Illsley, N.P. Diminished Trophoblast Differentiation in Early Onset Preeclampsia. Placenta 2022, 120, 25–31. [Google Scholar] [CrossRef]
- Farah, O.; Nguyen, C.; Tekkatte, C.; Parast, M.M. Trophoblast Lineage-Specific Differentiation and Associated Alterations in Preeclampsia and Fetal Growth Restriction. Placenta 2020, 102, 4–9. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Placental Oxidative Stress: From Miscarriage to Preeclampsia. J. Soc. Gynecol. Investig. 2004, 11, 342–352. [Google Scholar] [CrossRef]
- Jauniaux, E.; Hempstock, J.; Greenwold, N.; Burton, G.J. Trophoblastic Oxidative Stress in Relation to Temporal and Regional Differences in Maternal Placental Blood Flow in Normal and Abnormal Early Pregnancies. Am. J. Pathol. 2003, 162, 115–125. [Google Scholar] [CrossRef]
- Jauniaux, E.; Pahal, G.; Gervy, C.; Gulbis, B. Blood Biochemistry and Endocrinology in the Human Fetus between 11 and 17 Weeks of Gestation. Reprod. Biomed. Online 2000, 1, 38–44. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Tu, B.P.; Tang, Y. Eight kinetically stable but thermodynamically activated molecules that power cell metabolism. Chem. Rev. 2018, 118, 1460–1494. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox Metabolism: ROS as Specific Molecular Regulators of Cell Signaling and Function. Mol. Cell. 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E. Nitric Oxide Signaling in Health and Disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 6, 715–748. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and Reperfusion-from Mechanism to Translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2017, 7, 113–170. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Korantzopoulos, P. Oxidative Stress in Hepatic Ischemia-Reperfusion Injury: The Role of Antioxidants and Iron Chelating Compounds. Curr. Pharm. Des. 2006, 12, 2875–2890. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C.P. Rheological and Physiological Consequences of Conversion of the Maternal Spiral Arteries for Uteroplacental Blood Flow during Human Pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef]
- Hung, T.-H.; Skepper, J.N.; Burton, G.J. In Vitro Ischemia-Reperfusion Injury in Term Human Placenta as a Model for Oxidative Stress in Pathological Pregnancies. Am. J. Pathol. 2001, 159, 1031–1043. [Google Scholar] [CrossRef]
- Jauniaux, E.; Poston, L.; Burton, G.J. Placental-Related Diseases of Pregnancy: Involvement of Oxidative Stress and Implications in Human Evolution. Hum. Reprod. Update 2006, 12, 747–755. [Google Scholar] [CrossRef]
- Hung, T.H.; Burton, G.J. Hypoxia and Reoxygenation: A Possible Mechanism for Placental Oxidative Stress in Preeclampsia. Taiwan J. Obstet. Gynecol. 2006, 45, 189–200. [Google Scholar] [CrossRef]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D.Y. The Cellular and Molecular Origin of Reactive Oxygen Species Generation during Myocardial Ischemia and Reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Zweierl, J.L.; Brodericko, R.; Kuppusamy, P.; Thompson-Gorman, S.; Luttyy, G.A. Determination of the Mechanism of Free Radical in Human Aortic Endothelial Cells Exposed to Anoxia and Reoxygenation. J. Biol. Chem. 1994, 269, 24156–24162. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct Measurement of Free Radical Generation Following Reperfusion of Ischemic Myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef]
- Garlick, P.B.; Davies, M.J.; Hearse, D.J.; Slater, T.F. Direct Detection of Free Radicals in the Reperfused Rat Heart Using Electron Spin Resonance Spectroscopy. Circ. Res. 1987, 61, 757–760. [Google Scholar] [CrossRef]
- Kadkhodaee, M.; Hanson, G.R.; Towner, R.A.; Endre, Z.H. Detection of Hydroxyl and Carbon-Centred Radicals by EPR Spectroscopy after Ischaemia and Reperfusion of the Rat Kidney. Free Radic. Res. 1996, 25, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.W.; Chen, G.; Janzen, E.G. Detection of Free Radicals in Reperfused Dog Skin Flaps Using Electron Paramagnetic Resonance Spectroscopy: A Pilot Study. Microsurgery 1999, 19, 171–175. [Google Scholar] [CrossRef]
- Egemnazarov, B.; Sydykov, A.; Schermuly, R.T.; Weissmann, N.; Stasch, J.-P.; Sarybaev, A.S.; Seeger, W.; Grimminger, F.; Ghofrani, H.A. Novel Soluble Guanylyl Cyclase Stimulator BAY 41-2272 Attenuates Ischemia-Reperfusion-Induced Lung Injury. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L462–L469. [Google Scholar] [CrossRef]
- Togashi, H.; Shinzawa, H.; Matsuo, T.; Takeda, Y.; Takahashi, T.; Aoyama, M.; Oikawa, K.; Kamada, H. Analysis of hepatic oxidative stress status by electron spin resonance spectroscopy and imaging. Free Radic. Biol. Med. 2000, 28, 846–853. [Google Scholar] [CrossRef]
- Kono, H.; Woods, C.G.; Maki, A.; Connor, H.D.; Mason, R.P.; Rusyn, I.; Fujii, H. Electron Spin Resonance and Spin Trapping Technique Provide Direct Evidence That Edaravone Prevents Acute Ischemia-Reperfusion Injury of the Liver by Limiting Free Radical-Mediated Tissue Damage. Free Radic. Res. 2006, 40, 579–588. [Google Scholar] [CrossRef]
- Burke, A.; Fitzgerald, G.A.; Lucey, M.R. A prospective analysis of oxidative stress and liver transplantation. Transplantation 2002, 74, 217–221. [Google Scholar] [CrossRef]
- Biasi, F.; Bosco, M.; Lanfranco, G.; Massano, G.; Donadio, P.P.; Vaj, M.; Andorno, E.; Rizzeti, M.; Salizzoni, M.; Poli, G. Oxidative damage in human liver transplantation. Free Radic. Biol. Med. 1995, 19, 311–317. [Google Scholar] [CrossRef]
- Mantelou, A.G.; Barbouti, A.; Goussia, A.; Zacharioudaki, A.; Papoudou-Bai, A.; Vlachou, C.; Kokkoris, S.; Papalois, A.; Galaris, D.; Glantzounis, G.K. Combined Administration of Membrane-Permeable and Impermeable Iron-Chelating Drugs Attenuates Ischemia/Reperfusion-Induced Hepatic Injury. Free Radic. Biol. Med. 2022, 193, 227–237. [Google Scholar] [CrossRef]
- Kinouchi, H.; Epsteintt, C.J.; Mizui, T.; Carlsont, E.; Chen, S.F.; Chan, P.H. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc. Natl. Acad. Sci. USA 1991, 88, 11158–11162. [Google Scholar] [CrossRef]
- Horie, Y.; Wolf, R.; Flores, S.C.; Mccord, J.M.; Epstein, C.J.; Granger, D.N. Transgenic mice with increased copper/zinc-superoxide dismutase activity are resistant to hepatic leukostasis and capillary no-reflow after gut ischemia/reperfusion. Circ. Res. 1998, 83, 691–696. [Google Scholar] [CrossRef]
- Chen, E.; Bittner, H.; Davis, R.; Folz, R.; Van Trigt, P. Extracellular Superoxide Dismutase Transgene Overexpression Preserves Postischemic Myocardial Function in Isolated Murine Hearts. Circulation 1996, 94, II412–II417. [Google Scholar] [PubMed]
- Li, G.; Chen, Y.; Saari, J.T.; Kangl, Y.J.; Kang, Y.J. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am. J. Physiol. 1997, 273, H1090–H1095. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, N.; Weisbrot-Lefkowitz, M.; Reuhl, K.; Inouye, M.; Mirochnitchenko, O. Modulation of Chemokine Expression during Ischemia/Reperfusion in Transgenic Mice Overproducing Human Glutathione Peroxidases. J. Immunol. 1999, 163, 5666–5677. [Google Scholar] [CrossRef] [PubMed]
- Meneshian, A.; Bulkley, G.B. The Physiology of Endothelial Xanthine Oxidase: From Urate Catabolism to Reperfusion Injury to Inflammatory Signal Transduction. Microcirculation 2002, 9, 161–175. [Google Scholar] [CrossRef]
- Vega, V.; Mardones, L.; Maldonado, M.; Nicovani, S.; Manríquez, V.; Roa, J.; Ward, P. Xanthine Oxidase Released from Reperfused Hind Limbs Mediate Kupffer Cell Activation, Neutrophil Sequestration, and Hepatic Oxidative Stress in Rats Subjected to Tourniquet Shock. Shock 2000, 14, 565–571. [Google Scholar] [CrossRef]
- Matsumura, F.; Yamaguchi, Y.; Goto, M.; Ichiguchi, O.; Akizuki, E.; Matsuda, T.; Okabe, K.; Liang, J.; Ohshiro, H.; Iwamoto, T.; et al. Xanthine oxidase inhibition attenuates kupffer cell production of neutrophil chemoattractant following ischemia-reperfusion in rat liver. Hepatology 1998, 28, 1578–1587. [Google Scholar] [CrossRef]
- Otamiri, T. Oxygen Radicals, Lipid Peroxidation, and Neutrophil Infiltration after Small-Intestinal Ischemia and Reperfusion. Surgery 1989, 10, 593–597. [Google Scholar]
- Downey, J.M.; Miura, T.; Eddy, L.J.; Chambers, D.E.; Mellert, T.; Hearse, D.J.; Yellon, D.M.; Downey, M.; Miura, T.; Eddy, L.J.; et al. Xanthine oxidase is not a source of free radicals in the ischemic rabbit heart. J. Mol. Cell. Cardiol. 1987, 19, 1053–1060. [Google Scholar] [CrossRef]
- Kennedy, T.P.; Rao, N.V.; Hopkins, C.; Pennington, L.; Tolley, E.; Hoidal, J.R. Role of Reactive Oxygen Species in Reperfusion Injury of the Rabbit Lung. J. Clin. Investig. 1989, 83, 1326–1335. [Google Scholar] [CrossRef]
- Eddy, L.; Stewart, J.; Jones, H.; Engerson, T.; McCord, J.; Downey, J. Free Radical-Producing Enzyme, Xanthine Oxidase, Is Undetectable in Human Hearts. Am. J. Physiol. 1987, 253, H709–H711. [Google Scholar] [CrossRef]
- Many, A.; Hubel, C.A.; Roberts, J.M. Hyperuricemia and xanthine oxidase in preeclampsia, revisited. Am. J. Obstet. Gynecol. 1996, 174, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Krause, K.H.; Clark, R.A. NOX Enzymes as Novel Targets for Drug Development. Semin. Immunopathol. 2008, 30, 339–363. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.M.; Wingler, K.; Hermans, J.J.R.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H.H.W. NADPH Oxidases as a Source of Oxidative Stress and Molecular Target in Ischemia/Reperfusion Injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Kahles, T.; Brandes, R.P. Which NADPH Oxidase Isoform Is Relevant for Ischemic Stroke? The Case for Nox 2. Antioxid. Redox. Signal. 2013, 18, 1400–1417. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Filippi, M.D. Neutrophil Transendothelial Migration: Updates and New Perspectives. Blood 2019, 133, 2149–2158. [Google Scholar] [CrossRef]
- Myatt, L.; Cui, X. Oxidative Stress in the Placenta. Histochem. Cell Biol. 2004, 122, 369–382. [Google Scholar] [CrossRef]
- Chen, J.; Gao, Q.; Jiang, L.; Feng, X.; Zhu, X.; Fan, X.; Mao, C.; Xu, Z. The NOX2-Derived Reactive Oxygen Species Damaged Endothelial Nitric Oxide System via Suppressed BKCa/SKCa in Preeclampsia. Hypertens. Res. 2017, 40, 457–464. [Google Scholar] [CrossRef]
- Matsubara, S.; Sato, I. Enzyme Histochemically Detectable NAD(P)H Oxidase in Human Placental Trophoblasts: Normal, Preeclamptic, and Fetal Growth Restriction-Complicated Pregnancy. Histochem. Cell Biol. 2001, 116, 1–7. [Google Scholar] [CrossRef]
- Raijmakers, M.T.M.; Peters, W.H.M.; Steegers, E.A.P.; Poston, L. NAD(P)H Oxidase Associated Superoxide Production in Human Placenta from Normotensive and Pre-Eclamptic Women. Placenta 2004, 25 (Suppl. A), S85–S89. [Google Scholar] [CrossRef]
- Lim, R.; Acharya, R.; Delpachitra, P.; Hobson, S.; Sobey, C.G.; Drummond, G.R.; Wallace, E.M. Activin and NADPH-Oxidase in Preeclampsia: Insights from in Vitro and Murine Studies. Am. J. Obstet. Gynecol. 2015, 212, 86.e1–86.e12. [Google Scholar] [CrossRef] [PubMed]
- Roe, N.D.; Ren, J. Nitric Oxide Synthase Uncoupling: A Therapeutic Target in Cardiovascular Diseases. Vascul. Pharmacol. 2012, 57, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Park, H.S.; Lee, H.Y.; Ha, E.H.; Suh, S.H.; Oh, S.K.; Yoo, H.S. Reduced L-Arginine Level and Decreased Placental ENOS Activity in Preeclampsia. Placenta 2006, 27, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Tashie, W.; Fondjo, L.A.; Owiredu, W.K.B.A.; Ephraim, R.K.D.; Asare, L.; Adu-Gyamfi, E.A.; Seidu, L. Altered Bioavailability of Nitric Oxide and L-Arginine Is a Key Determinant of Endothelial Dysfunction in Preeclampsia. BioMed Res. Int. 2020, 2020, 3251956. [Google Scholar] [CrossRef]
- Guerby, P.; Swiader, A.; Augé, N.; Parant, O.; Vayssière, C.; Uchida, K.; Salvayre, R.; Negre-Salvayre, A. High Glutathionylation of Placental Endothelial Nitric Oxide Synthase in Preeclampsia. Redox Biol. 2019, 22, 101126. [Google Scholar] [CrossRef]
- Guerby, P.; Swiader, A.; Tasta, O.; Pont, F.; Rodriguez, F.; Parant, O.; Vayssière, C.; Shibata, T.; Uchida, K.; Salvayre, R.; et al. Modification of Endothelial Nitric Oxide Synthase by 4-Oxo-2(E)-Nonenal(ONE) in Preeclamptic Placentas. Free Radic. Biol. Med. 2019, 141, 416–425. [Google Scholar] [CrossRef]
- Pell, V.R.; Chouchani, E.T.; Murphy, M.P.; Brookes, P.S.; Krieg, T. Moving Forwards by Blocking Back-Flow the Yin and Yang of MI Therapy. Circ. Res. 2016, 118, 898–906. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-Specific Targets of Mitochondrial Reactive Oxygen Species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial Ischemia-Reperfusion Injury: A Neglected Therapeutic Target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Sorby-Adams, A.; Prime, T.A.; Miljkovic, J.L.; Prag, H.A.; Krieg, T.; Murphy, M.P. A Model of Mitochondrial Superoxide Production during Ischaemia-Reperfusion Injury for Therapeutic Development and Mechanistic Understanding. Redox Biol. 2024, 72, 103161. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Holland, O.J.; Cuffe, J.S.M.; Dekker Nitert, M.; Callaway, L.; Kwan Cheung, K.A.; Radenkovic, F.; Perkins, A.V. Placental Mitochondrial Adaptations in Preeclampsia Associated with Progression to Term Delivery. Cell Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef]
- Shi, Z.; Long, W.; Zhao, C.; Guo, X.; Shen, R.; Ding, H. Comparative Proteomics Analysis Suggests That Placental Mitochondria Are Involved in the Development of Pre-Eclampsia. PLoS ONE 2013, 8, e64351. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for Measuring Reactive Oxygen Species and Oxidative Damage in Cells and in Vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The Chemistry of Cell Signaling by Reactive Oxygen and Nitrogen Species and 4-Hydroxynonenal. Arch. Biochem. Biophys. 2008, 477, 183–195. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced Lipid Peroxidation End Products in Oxidative Damage to Proteins. Potential Role in Diseases and Therapeutic Prospects for the Inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef]
- Benedetti, A.; Comportia, M.; Esterbauerb, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta. 1980, 620, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Madazli, R.; Benian, A.; Aydin, S.; Uzun, H.; Tolun, N. The Plasma and Placental Levels of Malondialdehyde, Glutathione and Superoxide Dismutase in Pre-Eclampsia. J. Obstet. Gynaecol. 2002, 22, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Afrose, D.; Chen, H.; Ranashinghe, A.; Liu, C.-c.; Henessy, A.; Hansbro, P.M.; McClements, L. The Diagnostic Potential of Oxidative Stress Biomarkers for Preeclampsia: Systematic Review and Meta-Analysis. Biol. Sex Differ. 2022, 13, 26. [Google Scholar] [CrossRef]
- Sahay, A.S.; Sundrani, D.P.; Wagh, G.N.; Mehendale, S.S.; Joshi, S.R. Regional Differences in the Placental Levels of Oxidative Stress Markers in Pre-Eclampsia. Int. J. Gynaecol. Obstet. 2015, 129, 213–218. [Google Scholar] [CrossRef]
- Aydin, S.; Benian, A.; Madazli, R.; Uludaǧ, S.; Uzun, H.; Kaya, S. Plasma Malondialdehyde, Superoxide Dismutase, SE-Selectin, Fibronectin, Endothelin-1 and Nitric Oxide Levels in Women with Preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 113, 21–25. [Google Scholar] [CrossRef]
- Kaur, G.; Mishra, S.; Sehgal, A.; Prasad, R. Alterations in Lipid Peroxidation and Antioxidant Status in Pregnancy with Preeclampsia. Mol. Cell. Biochem. 2008, 313, 37–44. [Google Scholar] [CrossRef]
- Tasta, O.; Swiader, A.; Grazide, M.H.; Rouahi, M.; Parant, O.; Vayssière, C.; Bujold, E.; Salvayre, R.; Guerby, P.; Negre-Salvayre, A. A Role for 4-Hydroxy-2-Nonenal in Premature Placental Senescence in Preeclampsia and Intrauterine Growth Restriction. Free Radic. Biol. Med. 2021, 164, 303–314. [Google Scholar] [CrossRef]
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef]
- Karacay, Ö.; Sepici-Dincel, A.; Karcaaltincaba, D.; Sahin, D.; Yalvaç, S.; Akyol, M.; Kandemir, Ö.; Altan, N. A Quantitative Evaluation of Total Antioxidant Status and Oxidative Stress Markers in Preeclampsia and Gestational Diabetic Patients in 24-36 Weeks of Gestation. Diabetes Res. Clin. Pract. 2010, 89, 231–238. [Google Scholar] [CrossRef]
- Huang, Q.T.; Zhong, M.; Tian, J.W.; Hou, F.F. Higher Plasma AOPP Is Associated with Increased Proteinuria Excretion and Decreased Glomerular Filtration Rate in Pre-Eclamptic Women. Pregnancy Hypertens. 2013, 3, 16–20. [Google Scholar] [CrossRef]
- Noyan, T.; Güler, A.; Şekeroǧlu, M.R.; Kamaci, M. Serum Advanced Oxidation Protein Products, Myeloperoxidase and Ascorbic Acid in Pre-Eclampsia and Eclampsia. Aust. N. Z. J. Obstet. Gynaecol. 2006, 46, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kυks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell. Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A.; Toulmay, A.; Balla, T. The Functional Universe of Membrane Contact Sites. Nat. Rev. Mol. Cell. Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Resende, R.; Fernandes, T.; Pereira, A.; Marques, A.; Pereira, C. Endoplasmic Reticulum-Mitochondria Contacts Modulate Reactive Oxygen Species-Mediated Signaling and Oxidative Stress in Brain Disorders: The Key Role of Sigma-1 Receptor. Antiox. Red. Signal. 2022, 37, 758–780. [Google Scholar] [CrossRef]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medraño-Fernandez, I. Human Aquaporin-11 Guarantees Efficient Transport of H2O2 across the Endoplasmic Reticulum Membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef]
- Sorrentino, I.; Galli, M.; Medraño-Fernandez, I.; Sitia, R. Transfer of H2O2 from Mitochondria to the Endoplasmic Reticulum via Aquaporin-11. Redox Biol. 2022, 55, 102410. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial Control of Cellular Life, Stress, and Death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox Crosstalk at Endoplasmic Reticulum (ER) Membrane Contact Sites (MCS) Uses Toxic Waste to Deliver Messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef]
- Higuchi, S.; Miyamoto, T.; Kobara, H.; Yamada, S.; Asaka, R.; Kikuchi, N.; Kashima, H.; Ohira, S.; Shiozawa, T. Trophoblast Type-Specific Expression of Senescence Markers in the Human Placenta. Placenta 2019, 85, 56–62. [Google Scholar] [CrossRef]
- Gauster, M.; Moser, G.; Wernitznig, S.; Kupper, N.; Huppertz, B. Early Human Trophoblast Development: From Morphology to Function. Cell. Mol. Life Sci. 2022, 79, 345. [Google Scholar] [CrossRef] [PubMed]
- Goldman-Wohl, D.; Yagel, S. United We Stand Not Dividing: The Syncytiotrophoblast and Cell Senescence. Placenta 2014, 35, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell Fusion Induced by ERVWE1 or Measles Virus Causes Cellular Senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef]
- Farfαn-Labonne, B.; Leff-Gelman, P.; Pellσn-Dνaz, G.; Camacho-Arroyo, I. Cellular Senescence in Normal and Adverse Pregnancy. Reprod. Biol. 2023, 23, 100734. [Google Scholar] [CrossRef]
- Biron-Shental, T.; Sukenik-Halevy, R.; Sharon, Y.; Goldberg-Bittman, L.; Kidron, D.; Fejgin, M.D.; Amiel, A. Short Telomeres May Play a Role in Placental Dysfunction in Preeclampsia and Intrauterine Growth Restriction. Am. J. Obstet. Gynecol. 2010, 202, 381.e1–381.e7. [Google Scholar] [CrossRef]
- Sukenik-Halevy, R.; Amiel, A.; Kidron, D.; Liberman, M.; Ganor-Paz, Y.; Biron-Shental, T. Telomere homeostasis in trophoblasts and in cord blood cells from pregnancies complicated with preeclampsia. Am. J. Obstet. Gynecol. 2016, 214, 283. [Google Scholar] [CrossRef]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell Signalling by Reactive Lipid Species: New Concepts and Molecular Mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) Modified Proteins in Metabolic Diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef]
- Chapple, S.J.; Cheng, X.; Mann, G.E. Effects of 4-Hydroxynonenal on Vascular Endothelial and Smooth Muscle Cell Redox Signaling and Function in Health and Disease. Redox Biol. 2013, 1, 319–331. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are They Causal Players in Cellular Senescence? Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 1373–1379. [Google Scholar] [CrossRef]
- Holland, O.; Dekker Nitert, M.; Gallo, L.A.; Vejzovic, M.; Fisher, J.J.; Perkins, A.V. Review: Placental Mitochondrial Function and Structure in Gestational Disorders. Placenta 2017, 54, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The Mitochondrial Unfolded Protein Response, a Conserved Stress Response Pathway with Implications in Health and Disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, I.; Dhar, R.; Singh, S.; Sharma, J.B.; Nag, T.C.; Mridha, A.R.; Jaiswal, P.; Biswas, S.; Karmakar, S. Oxidative Stress-Induced Impairment of Trophoblast Function Causes Preeclampsia through the Unfolded Protein Response Pathway. Sci. Rep. 2021, 11, 18415. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The Unfolded Protein Response and Cellular Senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Cell Physiol. 2015, 308, 415–425. [Google Scholar] [CrossRef]
- Liu, J.; Huang, K.; Cai, G.Y.; Chen, X.M.; Yang, J.R.; Lin, L.R.; Yang, J.; Huo, B.G.; Zhan, J.; He, Y.N. Receptor for Advanced Glycation End-Products Promotes Premature Senescence of Proximal Tubular Epithelial Cells via Activation of Endoplasmic Reticulum Stress-Dependent P21 Signaling. Cell Signal. 2014, 26, 110–121. [Google Scholar] [CrossRef]
- Calvert, S.J.; Jones, C.J.P.; Sibley, C.P.; Aplin, J.D.; Heazell, A.E.P. Analysis of Syncytial Nuclear Aggregates in Preeclampsia Shows Increased Sectioning Artefacts and Decreased Inter-Villous Bridges Compared to Healthy Placentas. Placenta 2013, 34, 1251–1254. [Google Scholar] [CrossRef]
- Fogarty, N.M.E.; Ferguson-Smith, A.C.; Burton, G.J. Syncytial Knots (Tenney-Parker Changes) in the Human Placenta: Evidence of Loss of Transcriptional Activity and Oxidative Damage. Am. J. Pathol. 2013, 183, 144–152. [Google Scholar] [CrossRef]
- Heazell, A.E.P.; Moll, S.J.; Jones, C.J.P.; Baker, P.N.; Crocker, I.P. Formation of Syncytial Knots Is Increased by Hyperoxia, Hypoxia and Reactive Oxygen Species. Placenta 2007, 28 (Suppl. A), S33–S40. [Google Scholar] [CrossRef]
- Donthi, D.; Malik, P.; Mohamed, A.; Kousar, A.; Subramanian, R.A.; Manikyam, U.K. An Objective Histopathological Scoring System for Placental Pathology in Pre-Eclampsia and Eclampsia. Cureus 2020, 12, e11104. [Google Scholar] [CrossRef]
- Barbouti, A.; Goulas, V. Dietary Antioxidants in the Mediterranean Diet. Antioxidants 2021, 10, 1213. [Google Scholar] [CrossRef]
- Barbouti, A.; Lagopati, N.; Veroutis, D.; Goulas, V.; Evangelou, K.; Kanavaros, P.; Gorgoulis, V.G.; Galaris, D. Implication of Dietary Iron-Chelating Bioactive Compounds in Molecular Mechanisms of Oxidative Stress-Induced Cell Ageing. Antioxidants 2021, 10, 491. [Google Scholar] [CrossRef] [PubMed]
- Barbouti, A.; Briasoulis, E.; Galaris, D. Protective Effects of Olive Oil Components Against Hydrogen Peroxide-Induced DNA Damage. In Olives and Olive Oil in Health and Disease Prevention; Elsevier: Amsterdam, The Netherlands, 2010; pp. 1103–1109. [Google Scholar]
- Nousis, L.; Kanavaros, P.; Barbouti, A. Oxidative Stress-Induced Cellular Senescence: Is Labile Iron the Connecting Link? Antioxidants 2023, 12, 1250. [Google Scholar] [CrossRef]
- Barbouti, A.; Amorgianiotis, C.; Kolettas, E.; Kanavaros, P.; Galaris, D. Hydrogen Peroxide Inhibits Caspase-Dependent Apoptosis by Inactivating Procaspase-9 in an Iron-Dependent Manner. Free. Radic. Biol. Med. 2007, 43, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Tenopoulou, M.; Doulias, P.-T.; Barbouti, A.; Brunk, U.; Galaris, D. Role of Compartmentalized Redox-Active Iron in Hydrogen Peroxide-Induced DNA Damage and Apoptosis. Biochem. J. 2005, 387 Pt 3, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Barbouti, A.; Doulias, P.-T.; Nousis, L.; Tenopoulou, M.; Galaris, D. DNA Damage and Apoptosis in Hydrogen Peroxide-Exposed Jurkat Cells: Bolus Addition versus Continuous Generation of H2O2. Free. Radic. Biol. Med. 2002, 33, 691–702. [Google Scholar] [CrossRef]
- Barbouti, A.; Doulias, P.-T.; Zhu, B.-Z.; Frei, B.; Galaris, D. Intracellular Iron, but Not Copper, Plays a Critical Role in Hydrogen Peroxide-Induced DNA Damage. Free. Radic. Biol. Med. 2001, 31, 490–498. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbouti, A.; Varvarousis, D.N.; Kanavaros, P. The Role of Oxidative Stress-Induced Senescence in the Pathogenesis of Preeclampsia. Antioxidants 2025, 14, 529. https://doi.org/10.3390/antiox14050529
Barbouti A, Varvarousis DN, Kanavaros P. The Role of Oxidative Stress-Induced Senescence in the Pathogenesis of Preeclampsia. Antioxidants. 2025; 14(5):529. https://doi.org/10.3390/antiox14050529
Chicago/Turabian StyleBarbouti, Alexandra, Dimitrios N. Varvarousis, and Panagiotis Kanavaros. 2025. "The Role of Oxidative Stress-Induced Senescence in the Pathogenesis of Preeclampsia" Antioxidants 14, no. 5: 529. https://doi.org/10.3390/antiox14050529
APA StyleBarbouti, A., Varvarousis, D. N., & Kanavaros, P. (2025). The Role of Oxidative Stress-Induced Senescence in the Pathogenesis of Preeclampsia. Antioxidants, 14(5), 529. https://doi.org/10.3390/antiox14050529