
Activation of the Nrf2 Pathway by Sulforaphane Improves Hypoglycaemia-Induced Cognitive Impairment in a Rodent Model of Type 1 Diabetes

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Experimental Groups
2.3. Recurrent Hypoglycaemia
2.4. Administration of Sulforaphane (SFN)
2.5. Behavioural Procedures
2.6. Biochemical Analyses
2.7. Lipid Peroxidation and Protein Carbonylation
2.8. Inflammatory Protein Array
2.9. RNA Extraction and PCR
2.10. Statistical Analysis
2.11. Data and Resource Availability
3. Results
3.1. Sulforaphane Had No Impact on Body Weight or Glycaemic Control
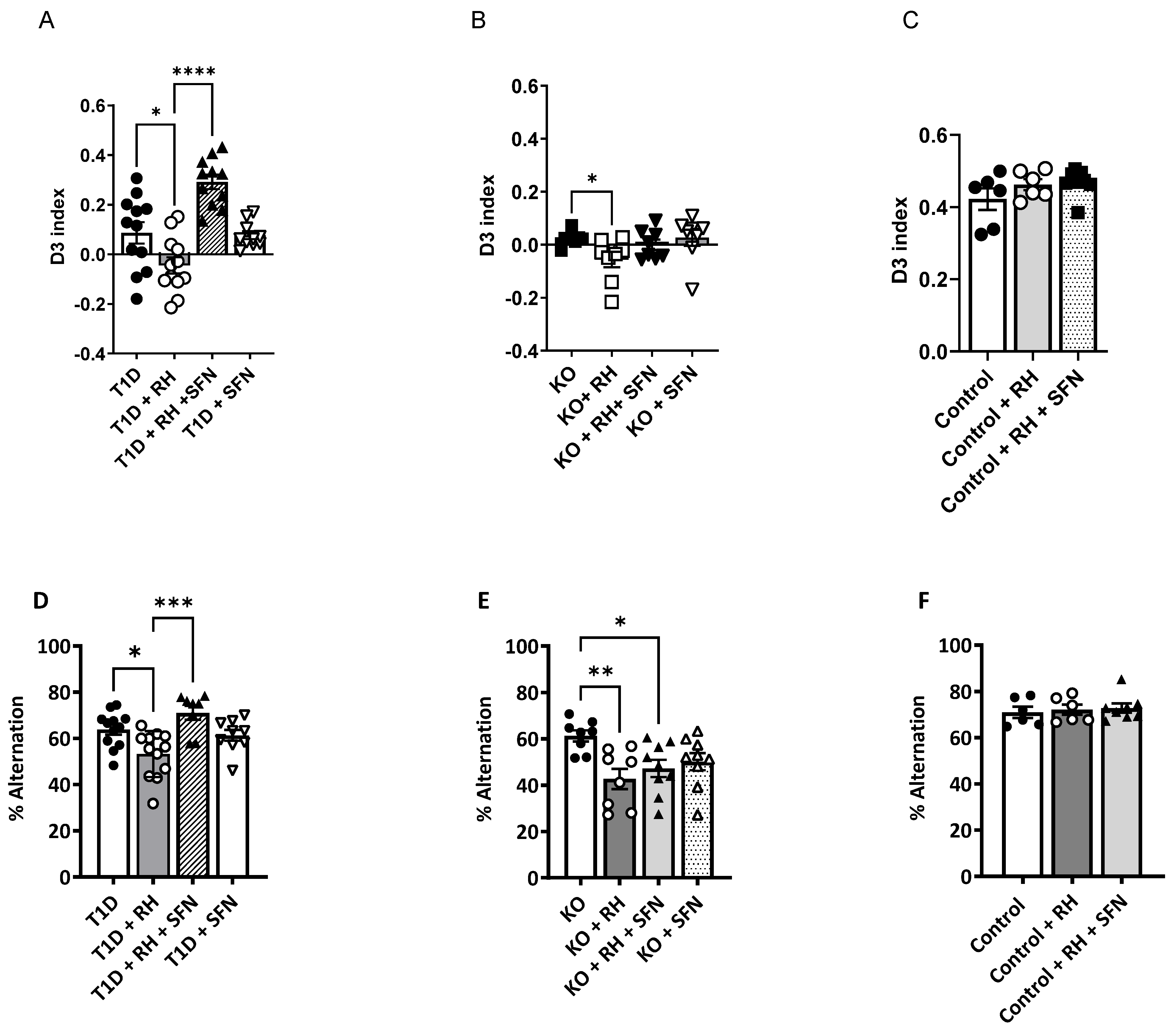
3.2. Sulforaphane Improves Spatial and Long-Term Working Memory in Diabetic Animals
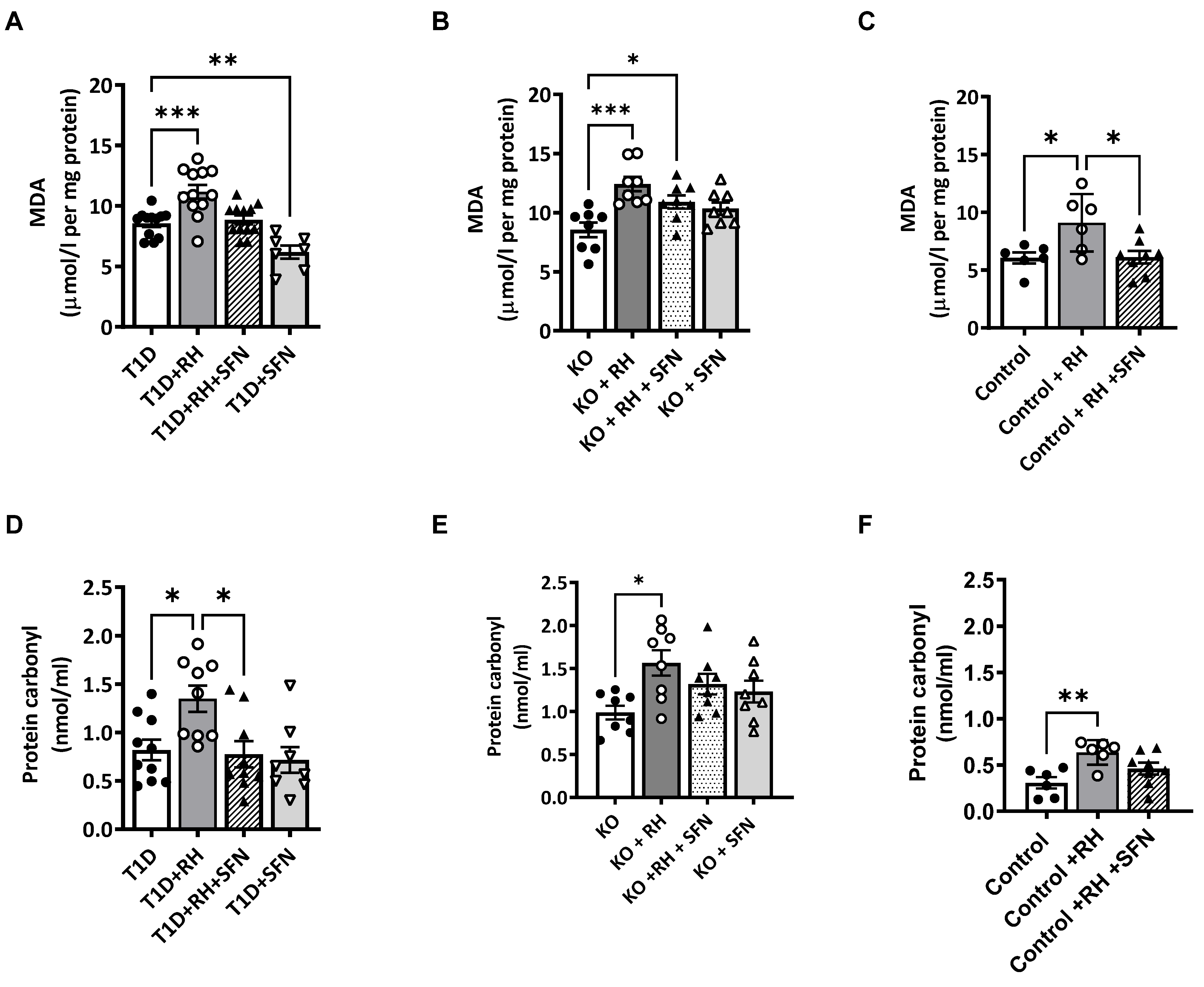
3.3. Sulforaphane-Mediated Induction of the Nrf2 Pathway Induces Antioxidant Defence Associated with STZ-T1D and RH
3.4. Sulforaphane Decreases ROS-Induced Cell Damage Within the Hippocampus
3.5. Sulforaphane Decreases Inflammation in the Hippocampus of STZ-T1D and RH Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCrimmon, R.J.; Sherwin, R.S. Hypoglycemia in type 1 diabetes. Diabetes 2010, 59, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.R.; Cryer, P.E. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes 1991, 40, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.M.; Rombouts, S.A.; Simsek, S.; Kuijer, J.P.; Kostense, P.J.; Barkhof, F.; Scheltens, P.; Snoek, F.J.; Heine, R.J. Microvascular disease in type 1 diabetes alters brain activation: A functional magnetic resonance imaging study. Diabetes 2006, 55, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef]
- Austin, E.J.; Deary, I.J. Effects of repeated hypoglycemia on cognitive function: A psychometrically validated reanalysis of the Diabetes Control and Complications Trial data. Diabetes Care 1999, 22, 1273–1277. [Google Scholar] [CrossRef]
- Hershey, T.; Perantie, D.C.; Warren, S.L.; Zimmerman, E.C.; Sadler, M.; White, N.H. Frequency and timing of severe hypoglycemia affects spatial memory in children with type 1 diabetes. Diabetes Care 2005, 28, 2372–2377. [Google Scholar] [CrossRef]
- Jacobson, A.M.; Ryan, C.M.; Braffett, B.H.; Gubitosi-Klug, R.A.; Lorenzi, G.M.; Luchsinger, J.A.; Trapani, V.R.; Bebu, I.; Chaytor, N.; Hitt, S.M.; et al. Cognitive performance declines in older adults with type 1 diabetes: Results from 32 years of follow-up in the DCCT and EDIC Study. Lancet Diabetes Endocrinol. 2021, 9, 436–445. [Google Scholar] [CrossRef]
- McNeilly, A.D.; Gallagher, J.R.; Dinkova-Kostova, A.T.; Hayes, J.D.; Sharkey, J.; Ashford, M.L.; McCrimmon, R.J. Nrf2-Mediated Neuroprotection Against Recurrent Hypoglycemia Is Insufficient to Prevent Cognitive Impairment in a Rodent Model of Type 1 Diabetes. Diabetes 2016, 65, 3151–3160. [Google Scholar] [CrossRef]
- Hong, F.; Freeman, M.L.; Liebler, D.C. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005, 18, 1917–1926. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef]
- Prestera, T.; Holtzclaw, W.D.; Zhang, Y.; Talalay, P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc. Natl. Acad. Sci. USA 1993, 90, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Rojo, A.I.; Garcia-Yague, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernandez-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal 2011, 14, 2347–2360. [Google Scholar] [CrossRef]
- Benedict, A.L.; Mountney, A.; Hurtado, A.; Bryan, K.E.; Schnaar, R.L.; Dinkova-Kostova, A.T.; Talalay, P. Neuroprotective effects of sulforaphane after contusive spinal cord injury. J. Neurotrauma 2012, 29, 2576–2586. [Google Scholar] [CrossRef]
- Langston, R.F.; Wood, E.R. Associative recognition and the hippocampus: Differential effects of hippocampal lesions on object-place, object-context and object-place-context memory. Hippocampus 2010, 20, 1139–1153. [Google Scholar] [CrossRef]
- McNay, E.C.; Sherwin, R.S. Effect of recurrent hypoglycemia on spatial cognition and cognitive metabolism in normal and diabetic rats. Diabetes 2004, 53, 418–425. [Google Scholar] [CrossRef]
- McNay, E.C.; Fries, T.M.; Gold, P.E. Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc. Natl. Acad. Sci. USA 2000, 97, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Uchiyama, M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal. Biochem. 1978, 86, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Why the need for qPCR publication guidelines?--The case for MIQE. Methods 2010, 50, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, 126, 55718. [Google Scholar] [CrossRef]
- Goldberg, R.B. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J. Clin. Endocrinol. Metab. 2009, 94, 3171–3182. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Ceriello, A.; Novials, A.; Ortega, E.; La Sala, L.; Pujadas, G.; Testa, R.; Bonfigli, A.R.; Esposito, K.; Giugliano, D. Evidence that hyperglycemia after recovery from hypoglycemia worsens endothelial function and increases oxidative stress and inflammation in healthy control subjects and subjects with type 1 diabetes. Diabetes 2012, 61, 2993–2997. [Google Scholar] [CrossRef]
- Cardoso, S.; Santos, R.X.; Correia, S.C.; Carvalho, C.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Insulin-induced recurrent hypoglycemia exacerbates diabetic brain mitochondrial dysfunction and oxidative imbalance. Neurobiol. Dis. 2013, 49, 1–12. [Google Scholar] [CrossRef]
- McNeilly, A.D.; Gallagher, J.R.; Evans, M.L.; de Galan, B.E.; Pedersen-Bjergaard, U.; Thorens, B.; Dinkova-Kostova, A.T.; Huang, J.T.; Ashford, M.L.J.; McCrimmon, R.J.; et al. Chronic hyperglycaemia increases the vulnerability of the hippocampus to oxidative damage induced during post-hypoglycaemic hyperglycaemia in a mouse model of chemically induced type 1 diabetes. Diabetologia 2023, 66, 1340–1352. [Google Scholar] [CrossRef]
- Benson, M.; Hossain, J.; Darmaun, D. Improved glycemic control either alone, or combined with antioxidant supplementation, fails to restore blood glutathione or markers of oxidative stress in adolescents with poorly controlled type 1 diabetes. Nutr. Res. 2023, 117, 83–90. [Google Scholar] [CrossRef]
- Liu, H.; Dinkova-Kostova, A.T.; Talalay, P. Coordinate regulation of enzyme markers for inflammation and for protection against oxidants and electrophiles. Proc. Natl. Acad. Sci. USA 2008, 105, 15926–15931. [Google Scholar] [CrossRef] [PubMed]
- Dick, R.A.; Kwak, M.K.; Sutter, T.R.; Kensler, T.W. Antioxidative function and substrate specificity of NAD(P)H-dependent alkenal/one oxidoreductase. A new role for leukotriene B4 12-hydroxydehydrogenase/15-oxoprostaglandin 13-reductase. J. Biol. Chem. 2001, 276, 40803–40810. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef]
- Healy, Z.R.; Liu, H.; Holtzclaw, W.D.; Talalay, P. Inactivation of tautomerase activity of macrophage migration inhibitory factor by sulforaphane: A potential biomarker for anti-inflammatory intervention. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1516–1523. [Google Scholar] [CrossRef]
- Frier, B.M. The incidence and impact of hypoglycemia in type 1 and type 2 diabetes. Int. Diabetes Monit. 2009, 21, 210–218. [Google Scholar]
- Hu, R.; Hebbar, V.; Kim, B.R.; Chen, C.; Winnik, B.; Buckley, B.; Soteropoulos, P.; Tolias, P.; Hart, R.P.; Kong, A.N. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J. Pharmacol. Exp. Ther. 2004, 310, 263–271. [Google Scholar] [CrossRef]
- Bergstrom, P.; Andersson, H.C.; Gao, Y.; Karlsson, J.O.; Nodin, C.; Anderson, M.F.; Nilsson, M.; Hammarsten, O. Repeated transient sulforaphane stimulation in astrocytes leads to prolonged Nrf2-mediated gene expression and protection from superoxide-induced damage. Neuropharmacology 2011, 60, 343–353. [Google Scholar] [CrossRef]
- Talalay, P.; Fahey, J.W.; Healy, Z.R.; Wehage, S.L.; Benedict, A.L.; Min, C.; Dinkova-Kostova, A.T. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 17500–17505. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Burness, C.B.; Deeks, E.D. Dimethyl fumarate: A review of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs 2014, 28, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Omaveloxolone (Skyclarys(TM)) for patients with Friedreich’s ataxia. Trends Pharmacol. Sci. 2023, 44, 394–395. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Control + RH | Control + RH + SFN | T1D | T1D + RH | T1D + RH + SFN | T1D + SFN | Nrf2−/− | Nrf2−/− + RH | Nrf2−/− + RH + SFN | Nrf2−/− + SFN | |
| Nrf2 | 1.0 ± 0.07 | 0.96 ± 0.08 | 1.49 ± 0.15 * | 1.0 ± 0.05 | 1.46 ± 0.02 **# | 1.82 ± 0.07 ** | 1.46 ± 0.07 *# | ND | ND | ND | ND |
| Nqo1 | 1.0 ± 0.06 | 0.95 ± 0.21 | 1.51 ± 0.13 * | 1.0 ± 0.06 | 1.31 ± 0.09 *# | 1.87 ± 0.15 ** | 1.59 ± 0.16 ** | 1.0 ± 0.06 | 1.05 ± 0.04 | 0.92 ± 0.05 | 1.03 ± 0.04 |
| Hmox-1 | 1.0 ± 0.09 | 0.96 ± 0.09 | 0.97 ± 0.06 | 1.0 ± 0.06 | 1.34 ± 0.09 | 1.32 ± 0.08 | 1.32 ± 0.08 | 1.0 ± 0.10 | 1.07 ± 0.12 | 1.07 ± 0.14 | 1.14 ± 0.09 |
| Sod2 | 1.0 ± 0.14 | 0.94 ± 0.06 | 1.01 ± 0.05 | 1.0 ± 0.14 | 0.81 ± 0.04 | 0.87 ± 0.06 | 0.82 ± 0.04 | 1.0 ± 0.10 | 0.94 ± 0.15 | 0.80 ± 0.05 | 1.10 ± 0.09 |
| Gclc | 1.0 ± 0.05 | 0.98 ± 0.08 | 1.48 ± 0.15 * | 1.0 ± 0.05 | 1.43 ± 0.11 * | 1.50 ± 0.15 ** | 1.49 ± 0.09 * | 1.0 ± 0.05 | 1.05 ± 0.04 | 0.93 ± 0.05 | 1.04 ± 0.02 |
| Gclm | 1.0 ± 0.03 | 0.99 ± 0.04 | 1.31 ± 0.07 * | 1.0 ± 0.10 | 1.43 ± 0.12 * | 1.62 ± 0.11 ** | 1.31 ± 0.10 | 1.0 ± 0.05 | 1.04 ± 0.06 | 0.94 ± 0.07 | 0.97 ± 0.04 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| T1D | T1D + RH | T1D + RH + SFN | T1D + SFN | Nrf2−/− | Nrf2−/−+ RH | Nrf2−/−+ RH + SFN | Nrf2−/−+ SFN | |
| Pro-inflammatory | ||||||||
| IFNg | 4.77 ± 0.94 | 4.15 ± 0.66 | 4.31 ± 0.78 | 4.15 ± 0.58 | 5.17 ± 0.44 | 4.67 ± 0.65 | 6.61 ± 0.41 | 6.57 ± 0.69 |
| IL-12p70 | 117 ± 10.2 | 128 ± 13.2 | 127 ± 14.4 | 131 ± 10.1 | 163 ± 33.2 | 158 ± 7.27 | 147 ± 14.5 | 74.9 ± 14.1 |
| IL-1β | 2.35 ± 0.51 | 8.61 ± 1.55 * | 6.22 ± 1.23 | 5.14 ± 1.68 | 3.42 ± 0.31 | 4.56 ± 0.25 * | 1.20 ± 0.24 # | 1.89 ± 0.12 |
| IL-2 | 15.3 ± 1.91 | 12.3 ± 1.19 | 13.5 ± 0.78 | 14.9 ± 1.74 | 12.5 ± 1.40 | 12.7 ± 1.06 | 18.2 ± 1.88 | 20.1 ± 1.46 |
| IL-5 | 47.8 ± 3.87 | 29.1 ± 1.96 ** | 42.3 ± 4.71 | 42.2 ± 3.71 | 34.3 ± 1.97 | 58.5 ± 6.12 ** | 51.9 ± 7.47 | 53.9 ± 4.93 |
| KC/GRO | 609 ± 60.2 | 759 ± 77.0 | 536 ± 76.3 | 668 ± 63.3 | 495 ± 58.7 | 521 ± 26.2 | 488 ± 61.6 | 463 ± 48.7 |
| TNFα | 58.6 ± 7.38 | 102 ± 7.62 ** | 70.2 ± 8.64 | 62.4 ± 11.6 | 59.8 ± 11.9 | 99.5 ± 13.9 * | 62.6 ± 13.9 # | 57.9 ± 11.4 |
| Anti-inflammatory | ||||||||
| IL-10 | 106 ± 14.8 | 113 ± 18.4 | 169 ± 12.8 * | 161 ± 10.3 * | 148 ± 17.4 | 126 ± 13.2 | 153 ± 15.9 | 182 ± 15.3 |
| Pleiotropic | ||||||||
| IL-6 | 121 ± 11.9 | 290 ± 31.9 * | 468 ± 93.1 ** | 311 ± 54.8 * | 192 ± 26.8 | 380 ± 51.7 * | 185 ± 51.6 # | 270 ± 19.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merchant, H.J.; Forteath, C.; Gallagher, J.R.; Dinkova-Kostova, A.T.; Ashford, M.L.J.; McCrimmon, R.J.; McNeilly, A.D. Activation of the Nrf2 Pathway by Sulforaphane Improves Hypoglycaemia-Induced Cognitive Impairment in a Rodent Model of Type 1 Diabetes. Antioxidants 2025, 14, 308. https://doi.org/10.3390/antiox14030308
Merchant HJ, Forteath C, Gallagher JR, Dinkova-Kostova AT, Ashford MLJ, McCrimmon RJ, McNeilly AD. Activation of the Nrf2 Pathway by Sulforaphane Improves Hypoglycaemia-Induced Cognitive Impairment in a Rodent Model of Type 1 Diabetes. Antioxidants. 2025; 14(3):308. https://doi.org/10.3390/antiox14030308
Chicago/Turabian StyleMerchant, Heather J., Calum Forteath, Jennifer R. Gallagher, Albena T. Dinkova-Kostova, Michael L. J. Ashford, Rory J. McCrimmon, and Alison D. McNeilly. 2025. "Activation of the Nrf2 Pathway by Sulforaphane Improves Hypoglycaemia-Induced Cognitive Impairment in a Rodent Model of Type 1 Diabetes" Antioxidants 14, no. 3: 308. https://doi.org/10.3390/antiox14030308
APA StyleMerchant, H. J., Forteath, C., Gallagher, J. R., Dinkova-Kostova, A. T., Ashford, M. L. J., McCrimmon, R. J., & McNeilly, A. D. (2025). Activation of the Nrf2 Pathway by Sulforaphane Improves Hypoglycaemia-Induced Cognitive Impairment in a Rodent Model of Type 1 Diabetes. Antioxidants, 14(3), 308. https://doi.org/10.3390/antiox14030308