
Redox Imbalance in Neurological Disorders in Adults and Children

 ,
,  , ,
, ,  , ,
, ,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Cellular Pathways of Redox Imbalance
2.1. Hypoxia
2.2. Hyperoxia and ROS Production
2.3. Role of the Antioxidant System of Cells in Redox Balance
2.4. Heme Proteins and Iron
2.5. Mitochondria and ROS
2.6. Protein Misfolding
2.7. Neuroinflammation
3. Neurodegenerative Diseases
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Amyotrophic Lateral Sclerosis
4. Pediatric Neurological Disorders
4.1. Adrenoleukodystrophy
4.2. Pelizaeus–Merzbacher Disease
4.3. Spinal Muscular Atrophy
4.4. Mucopolysaccharidoses
5. Therapeutic Prospects
5.1. Antioxidants
5.2. Mitochondria-Targeting Drugs
5.3. Metal Protein Attenuating Compounds
5.4. Opioids or Cannabinoids
5.5. Non-Pharmacological Interventions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-Sensing Mechanisms in Cells. FEBS J. 2020, 287, 3888–3906. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Ursini, F.; Maiorino, M. An Overview of Mechanisms of Redox Signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M. Oxygen, Evolution and Redox Signalling in the Human Brain; Quantum in the Quotidian. J. Physiol. 2018, 597, 15–28. [Google Scholar] [CrossRef]
- Leithner, C.; Royl, G. The Oxygen Paradox of Neurovascular Coupling. J. Cereb. Blood Flow Metab. 2014, 34, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Sharp, F.R.; Bernaudin, M. HIF1 and Oxygen Sensing in the Brain. Nat. Rev. Neurosci. 2004, 5, 437–448. [Google Scholar] [CrossRef]
- Terraneo, L.; Paroni, R.; Bianciardi, P.; Giallongo, T.; Carelli, S.; Gorio, A.; Samaja, M. Brain Adaptation to Hypoxia and Hyperoxia in Mice. Redox Biol. 2017, 11, 12–20. [Google Scholar] [CrossRef]
- Rey, F.; Messa, L.; Maghraby, E.; Casili, G.; Ottolenghi, S.; Barzaghini, B.; Raimondi, M.T.; Cereda, C.; Cuzzocrea, S.; Zuccotti, G.; et al. Oxygen Sensing in Neurodegenerative Diseases: Current Mechanisms, Implication of Transcriptional Response and Pharmacological Modulation. Antioxid. Redox Signal. 2022, 38, 160–182. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Stone, T.W.; Smith, R.A. Oxidative Stress in Neurodegeneration and Available Means of Protection. Front. Biosci. 2008, 13, 3288–3311. [Google Scholar] [CrossRef]
- Margaritelis, N.V.; Chatzinikolaou, P.N.; Chatzinikolaou, A.N.; Paschalis, V.; Theodorou, A.A.; Vrabas, I.S.; Kyparos, A.; Nikolaidis, M.G. The Redox Signal: A Physiological Perspective. IUBMB Life 2022, 74, 29–40. [Google Scholar] [CrossRef]
- Murray, A.J.; Horscroft, J.A. Mitochondrial Function at Extreme High Altitude. J. Physiol. 2016, 594, 1137–1149. [Google Scholar] [CrossRef]
- Rey, F.; Balsari, A.; Giallongo, T.; Ottolenghi, S.; Di Giulio, A.M.; Samaja, M.; Carelli, S. Erythropoietin as a Neuroprotective Molecule: An Overview of Its Therapeutic Potential in Neurodegenerative Diseases. ASN Neuro 2019, 11, 1759091419871420. [Google Scholar] [CrossRef] [PubMed]
- Fantacci, M.; Bianciardi, P.; Caretti, A.; Coleman, T.R.; Cerami, A.; Brines, M.; Samaja, M. Carbamylated Erythropoietin Ameliorates the Metabolic Stress Induced in Vivo by Severe Chronic Hypoxia. Proc. Natl. Acad. Sci. USA 2006, 103, 17531–17536. [Google Scholar] [CrossRef] [PubMed]
- Terraneo, L.; Samaja, M. Comparative Response of Brain to Chronic Hypoxia and Hyperoxia. Int. J. Mol. Sci. 2017, 18, 1914. [Google Scholar] [CrossRef]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin Regulates the in Vitro and in Vivo Production of Neuronal Progenitors by Mammalian Forebrain Neural Stem Cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef] [PubMed]
- Lombardero, M.; Kovacs, K.; Scheithauer, B.W. Erythropoietin: A Hormone with Multiple Functions. Pathobiology 2011, 78, 41–53. [Google Scholar] [CrossRef]
- Carelli, S.; Giallongo, T.; Rey, F.; Colli, M.; Tosi, D.; Bulfamante, G.; Di Giulio, A.M.; Gorio, A. Neuroprotection, Recovery of Function and Endogenous Neurogenesis in Traumatic Spinal Cord Injury Following Transplantation of Activated Adipose Tissue. Cells 2019, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wolferts, G.; Veltkamp, R. Oxygen Therapy Does Not Increase Production and Damage Induced by Reactive Oxygen Species in Focal Cerebral Ischemia. Neurosci. Lett. 2014, 577, 1–5. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.-Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.-J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Smuder, A.J.; Turner, S.M.; Schuster, C.M.; Morton, A.B.; Hinkley, J.M.; Fuller, D.D. Hyperbaric Oxygen Treatment Following Mid-Cervical Spinal Cord Injury Preserves Diaphragm Muscle Function. Int. J. Mol. Sci. 2020, 21, 7219. [Google Scholar] [CrossRef] [PubMed]
- Mcdowall, D.G. II: Biochemical Derangements Associated with Hypoxia and Their Measurement. Br. J. Anaesth. 1969, 41, 251–256. [Google Scholar] [CrossRef]
- Chen, P.-S.; Chiu, W.-T.; Hsu, P.-L.; Lin, S.-C.; Peng, I.-C.; Wang, C.-Y.; Tsai, S.-J. Pathophysiological Implications of Hypoxia in Human Diseases. J. Biomed. Sci. 2020, 27, 63. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and Brain Aging: Neurodegeneration or Neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- England, K.; Cotter, T.G. Direct Oxidative Modifications of Signalling Proteins in Mammalian Cells and Their Effects on Apoptosis. Redox. Rep. 2005, 10, 237–245. [Google Scholar] [CrossRef]
- Liochev, S.I. Reactive Oxygen Species and the Free Radical Theory of Aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Rokka, A.; Antonenkov, V.D.; Soininen, R.; Immonen, H.L.; Pirilä, P.L.; Bergmann, U.; Sormunen, R.T.; Weckström, M.; Benz, R.; Hiltunen, J.K. Pxmp2 Is a Channel-Forming Protein in Mammalian Peroxisomal Membrane. PLoS ONE 2009, 4, e5090. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Brown, N.J.; Pockley, A.G.; Wyld, L. The Unfolded Protein Response and Cancer: A Brighter Future Unfolding? J. Mol. Med. 2007, 85, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.; Lee, G.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox Status Expressed as GSH:GSSG Ratio as a Marker for Oxidative Stress in Paediatric Tumour Patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in Brain Disorders and Aging. Molecules 2022, 27, 324. [Google Scholar] [CrossRef] [PubMed]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.S.; Murphy, M.P. The Mitochondrial Dicarboxylate and 2-Oxoglutarate Carriers Do Not Transport Glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Kirchhof, D.; Manning, E.; Joseph, J.W.; Linseman, D.A. Mitochondrial Glutathione Transport Is a Key Determinant of Neuronal Susceptibility to Oxidative and Nitrosative Stress. J. Biol. Chem. 2013, 288, 5091–5101. [Google Scholar] [CrossRef]
- Solmonson, A.; DeBerardinis, R.J. Lipoic Acid Metabolism and Mitochondrial Redox Regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxid. Med. Cell Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef]
- de Araújo, D.P.; De Sousa, C.N.S.; Araújo, P.V.P.; Menezes, C.E.D.S.; Sousa Rodrigues, F.T.; Escudeiro, S.S.; Lima, N.B.C.; Patrocínio, M.C.A.; Aguiar, L.M.V.; Viana, G.S.D.B.; et al. Behavioral and Neurochemical Effects of Alpha-Lipoic Acid in the Model of Parkinson’s Disease Induced by Unilateral Stereotaxic Injection of 6-Ohda in Rat. Evid.-Based Complement. Altern. Med. 2013, 2013, 571378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Baillet, A.; Chanteperdrix, V.; Trocmé, C.; Casez, P.; Garrel, C.; Besson, G. The Role of Oxidative Stress in Amyotrophic Lateral Sclerosis and Parkinson’s Disease. Neurochem. Res. 2010, 35, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, S.; Sau, D.; Guareschi, S.; Bogush, M.; Brown, R.H.; Naniche, N.; Kia, A.; Trotti, D.; Pasinelli, P. ALS-Linked Mutant SOD1 Damages Mitochondria by Promoting Conformational Changes in Bcl-2. Hum. Mol. Genet. 2010, 19, 2974–2986. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M. Quantum Chemical Studies of Manganese Centers in Biology. Curr. Opin. Chem. Biol. 2002, 6, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Nell, H.J.; Au, J.L.; Giordano, C.R.; Terlecky, S.R.; Walton, P.A.; Whitehead, S.N.; Cechetto, D.F. Targeted Antioxidant, Catalase-SKL, Reduces Beta-Amyloid Toxicity in the Rat Brain. Brain Pathol. 2017, 27, 86–94. [Google Scholar] [CrossRef]
- van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Neuroprotective Properties and Mechanisms of Erythropoietin in in Vitro and in Vivo Experimental Models for Hypoxia/Ischemia. Brain Res. Rev. 2008, 59, 22–33. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Guloksuz, S.; Schmidt-Kastner, R.; Kenis, G.; Ince, B.; Rutten, B.P.F. Involvement of Hemoglobins in the Pathophysiology of Alzheimer’s Disease. Exp. Gerontol. 2019, 126, 110680. [Google Scholar] [CrossRef]
- Atamna, H. Heme Binding to Amyloid-Beta Peptide: Mechanistic Role in Alzheimer’s Disease. J. Alzheimers Dis. 2006, 10, 255–266. [Google Scholar] [CrossRef]
- Daglas, M.; Adlard, P.A. The Involvement of Iron in Traumatic Brain Injury and Neurodegenerative Disease. Front. Neurosci. 2018, 12, 981. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.; Ottolenghi, S.; Zuccotti, G.V.; Samaja, M.; Carelli, S. Mitochondrial Dysfunctions in Neurodegenerative Diseases: Role in Disease Pathogenesis, Strategies for Analysis and Therapeutic Prospects. Neural. Regen. Res. 2022, 17, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed Mitochondrial Dynamics and Neurodegenerative Disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2019, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of Mitochondrial Reactive Oxygen Species in Homeostasis Regulation. Redox. Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef]
- Antonucci, S.; Di Lisa, F.; Kaludercic, N. Mitochondrial Reactive Oxygen Species in Physiology and Disease. Cell Calcium. 2021, 94, 102344. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive Oxygen Species Production by Forward and Reverse Electron Fluxes in the Mitochondrial Respiratory Chain. PLoS Comput. Biol. 2011, 7, e1001115. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA Damage and Reactive Oxygen Species in Neurodegenerative Disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Kalra, J. Crosslink between Mutations in Mitochondrial Genes and Brain Disorders: Implications for Mitochondrial-Targeted Therapeutic Interventions. Neural Regen. Res. 2023, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein Folding and Misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. The Formation and Stabilization of Protein Structure. Biochem. J. 1972, 128, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10 (Suppl. S7), S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of Oligomer Toxicity, Fibril Toxicity and Fibril Spreading in Synucleinopathies. Cell. Mol. Life Sci. 2022, 79, 174. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein Misfolding and Aggregation: Implications in Parkinson’s Disease Pathogenesis. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Busquets, M.A.; Espargaró, A.; Estelrich, J.; Sabate, R. Could α -Synuclein Amyloid-Like Aggregates Trigger a Prionic Neuronal Invasion? BioMed Res. Int. 2015, 2015, 172018. [Google Scholar] [CrossRef] [PubMed]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Oh, C.; Zhang, X.; Lipton, S.A. Protein S-Nitrosylation and Oxidation Contribute to Protein Misfolding in Neurodegeneration. Free. Radic. Biol. Med. 2021, 172, 562–577. [Google Scholar] [CrossRef]
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849. [Google Scholar] [CrossRef]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant Protein S-Nitrosylation in Neurodegenerative Diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef]
- Lee, Y.M.; He, W.; Liou, Y.-C. The Redox Language in Neurodegenerative Diseases: Oxidative Post-Translational Modifications by Hydrogen Peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Rodriguez, E.A.; Wang, Y.; Block, M.L. Outdoor Ambient Air Pollution and Neurodegenerative Diseases: The Neuroinflammation Hypothesis. Curr. Environ. Health Rep. 2017, 4, 166–179. [Google Scholar] [CrossRef]
- Carson, M.J.; Thrash, J.C.; Walter, B. The Cellular Response in Neuroinflammation: The Role of Leukocytes, Microglia and Astrocytes in Neuronal Death and Survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Spath, S.; Goverman, J. Cytokine Networks in Neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical Glial Cell Numbers in Human Brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The Multifaceted Role of Astrocytes in Regulating Myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Chen, M.-L.; Yan, B.-S.; Bando, Y.; Kuchroo, V.K.; Weiner, H.L. Latency-Associated Peptide Identifies a Novel CD4+CD25+ Regulatory T Cell Subset with TGFbeta-Mediated Function and Enhanced Suppression of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2008, 180, 7327–7337. [Google Scholar] [CrossRef]
- Weiner, H.L. A Shift from Adaptive to Innate Immunity: A Potential Mechanism of Disease Progression in Multiple Sclerosis. J. Neurol. 2008, 255 (Suppl. S1), 3–11. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Orlacchio, A.; Maccarrone, M. Is Modulation of Oxidative Stress an Answer? The State of the Art of Redox Therapeutic Actions in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7909380. [Google Scholar] [CrossRef]
- Garofalo, M.; Pandini, C.; Bordoni, M.; Pansarasa, O.; Rey, F.; Costa, A.; Minafra, B.; Diamanti, L.; Zucca, S.; Carelli, S.; et al. Alzheimer’s, Parkinson’s Disease and Amyotrophic Lateral Sclerosis Gene Expression Patterns Divergence Reveals Different Grade of RNA Metabolism Involvement. Int. J. Mol. Sci. 2020, 21, 9500. [Google Scholar] [CrossRef]
- Pessoa, J.; Duarte, A. Overcoming Mitochondrial Dysfunction in Neurodegenerative Diseases. Neural Regen. Res. 2023, 18, 1486. [Google Scholar] [CrossRef]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s Disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Arroyo-García, L.E.; Bachiller, S.; Ruiz, R.; Boza-Serrano, A.; Rodríguez-Moreno, A.; Deierborg, T.; Andrade-Talavera, Y.; Fisahn, A. Targeting Galectin-3 to Counteract Spike-Phase Uncoupling of Fast-Spiking Interneurons to Gamma Oscillations in Alzheimer’s Disease. Transl. Neurodegener. 2023, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. The Critical Role of Methionine 35 in Alzheimer’s Amyloid Beta-Peptide (1-42)-Induced Oxidative Stress and Neurotoxicity. Biochim. Biophys. Acta 2005, 1703, 149–156. [Google Scholar] [CrossRef]
- Murray, I.V.; Sindoni, M.E.; Axelsen, P.H. Promotion of Oxidative Lipid Membrane Damage by Amyloid Beta Proteins. Biochemistry 2005, 44, 12606–12613. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Aksenov, M.Y.; Lovell, M.A.; Markesbery, W.R. Decrease in Peptide Methionine Sulfoxide Reductase in Alzheimer’s Disease Brain. J. Neurochem. 1999, 73, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Schweers, O.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Oxidation of Cysteine-322 in the Repeat Domain of Microtubule-Associated Protein Tau Controls the in Vitro Assembly of Paired Helical Filaments. Proc. Natl. Acad. Sci. USA 1995, 92, 8463–8467. [Google Scholar] [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A Gain of a Loss of Function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial Oxidative Stress Causes Hyperphosphorylation of Tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2018, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, H.; Ogawa, M.; Yamauchi, H.; Yamaguchi, S.; Kimura, J.; Yonekura, Y.; Konishi, J. Altered Cerebral Energy Metabolism in Alzheimer’s Disease: A PET Study. J. Nucl. Med. 1994, 35, 1–6. [Google Scholar]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-Amyloid Inhibits Integrated Mitochondrial Respiration and Key Enzyme Activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Santos, S.; Swerdlow, R.H.; Oliveira, C.R. Functional Mitochondria Are Required for Amyloid Beta-Mediated Neurotoxicity. FASEB J. 2001, 15, 1439–1441. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B. Amyloid Beta-Peptide Is Produced by Cultured Cells during Normal Metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Bentahir, M.; Nyabi, O.; Verhamme, J.; Tolia, A.; Horré, K.; Wiltfang, J.; Esselmann, H.; De Strooper, B. Presenilin Clinical Mutations Can Affect Gamma-Secretase Activity by Different Mechanisms. J. Neurochem. 2006, 96, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Almeida, C.G.; Kearney, P.F.; Yu, F.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Oligomerization of Alzheimer’s Beta-Amyloid within Processes and Synapses of Cultured Neurons and Brain. J. Neurosci. 2004, 24, 3592–3599. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A Beta 42(43) and A Beta 40 in Senile Plaques with End-Specific A Beta Monoclonals: Evidence That an Initially Deposited Species Is A Beta 42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of Neurofibrillary Tangles in P301l Tau Transgenic Mice Induced by Abeta 42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Alexander, G.E.; Chen, K.; Pietrini, P.; Rapoport, S.I.; Reiman, E.M. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am. J. Psychiatry 2002, 159, 738–745. [Google Scholar] [CrossRef]
- Park, S.Y.; Ferreira, A. The Generation of a 17 KDa Neurotoxic Fragment: An Alternative Mechanism by Which Tau Mediates Beta-Amyloid-Induced Neurodegeneration. J. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef]
- Shoji, M.; Golde, T.E.; Ghiso, J.; Cheung, T.T.; Estus, S.; Shaffer, L.M.; Cai, X.D.; McKay, D.M.; Tintner, R.; Frangione, B. Production of the Alzheimer Amyloid Beta Protein by Normal Proteolytic Processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; Theuns, J.; Van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; Van Broeckhoven, C. Mean Age-of-Onset of Familial Alzheimer Disease Caused by Presenilin Mutations Correlates with Both Increased Abeta42 and Decreased Abeta40. Hum. Mutat. 2006, 27, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Prasher, V.P.; Farrer, M.J.; Kessling, A.M.; Fisher, E.M.; West, R.J.; Barber, P.C.; Butler, A.C. Molecular Mapping of Alzheimer-Type Dementia in Down’s Syndrome. Ann. Neurol. 1998, 43, 380–383. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing Endogenous Tau Ameliorates Amyloid Beta-Induced Deficits in an Alzheimer’s Disease Mouse Model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Althafar, Z.M. Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules. Molecules 2022, 27, 4124. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy. 1817. J. Neuropsychiatry. Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative Stress and Parkinson’s Disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Rey, F.; Pandini, C.; Messa, L.; Launi, R.; Barzaghini, B.; Zangaglia, R.; Raimondi, M.T.; Gagliardi, S.; Cereda, C.; Zuccotti, G.V.; et al. α-Synuclein Antisense Transcript SNCA-AS1 Regulates Synapses- and Aging-Related Genes Suggesting Its Implication in Parkinson’s Disease. Aging Cell 2021, 20, e13504. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s Disease as a Result of Aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of Parkinson’s Disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and Consequence: Mitochondrial Dysfunction Initiates and Propagates Neuronal Dysfunction, Neuronal Death and Behavioral Abnormalities in Age-Associated Neurodegenerative Diseases. Biochim. Biophys. Acta 2010, 1802, 122–134. [Google Scholar] [CrossRef]
- Sims, N.R.; Finegan, J.M.; Blass, J.P.; Bowen, D.M.; Neary, D. Mitochondrial Function in Brain Tissue in Primary Degenerative Dementia. Brain Res. 1987, 436, 30–38. [Google Scholar] [CrossRef]
- Carelli, S.; Giallongo, T.; Viaggi, C.; Gombalova, Z.; Latorre, E.; Mazza, M.; Vaglini, F.; Di Giulio, A.M.; Gorio, A. Grafted Neural Precursors Integrate Into Mouse Striatum, Differentiate and Promote Recovery of Function Through Release of Erythropoietin in MPTP-Treated Mice. ASN Neuro 2016, 8, 1759091416676147. [Google Scholar] [CrossRef]
- Carelli, S.; Giallongo, T.; Viaggi, C.; Latorre, E.; Gombalova, Z.; Raspa, A.; Mazza, M.; Vaglini, F.; Di Giulio, A.M.; Gorio, A. Recovery from Experimental Parkinsonism by Intrastriatal Application of Erythropoietin or EPO-Releasing Neural Precursors. Neuropharmacology 2017, 119, 76–90. [Google Scholar] [CrossRef]
- Erbaş, O.; Çınar, B.P.; Solmaz, V.; Çavuşoğlu, T.; Ateş, U. The Neuroprotective Effect of Erythropoietin on Experimental Parkinson Model in Rats. Neuropeptides 2015, 49, 1–5. [Google Scholar] [CrossRef]
- Jang, W.; Park, J.; Shin, K.J.; Kim, J.S.; Youn, J.; Cho, J.W.; Oh, E.; Ahn, J.Y.; Oh, K.W.; Kim, H.T. Safety and Efficacy of Recombinant Human Erythropoietin Treatment of Non-Motor Symptoms in Parkinson’s Disease. J. Neurol. Sci. 2014, 337, 47–54. [Google Scholar] [CrossRef]
- Rey, F.; Ottolenghi, S.; Giallongo, T.; Balsari, A.; Martinelli, C.; Rey, R.; Allevi, R.; Giulio, A.M.D.; Zuccotti, G.V.; Mazzucchelli, S.; et al. Mitochondrial Metabolism as Target of the Neuroprotective Role of Erythropoietin in Parkinson’s Disease. Antioxidants 2021, 10, 121. [Google Scholar] [CrossRef]
- Carelli, S.; Giallongo, T.; Gombalova, Z.; Rey, F.; Gorio, M.C.F.; Mazza, M.; Di Giulio, A.M. Counteracting Neuroinflammation in Experimental Parkinson’s Disease Favors Recovery of Function: Effects of Er-NPCs Administration. J. Neuroinflamm. 2018, 15, 333. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative Damage in Nucleic Acids and Parkinson’s Disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Ferrer, I.; Martinez, A.; Blanco, R.; Dalfó, E.; Carmona, M. Neuropathology of Sporadic Parkinson Disease before the Appearance of Parkinsonism: Preclinical Parkinson Disease. J. Neural Transm. 2011, 118, 821–839. [Google Scholar] [CrossRef] [PubMed]
- Hemmati-Dinarvand, M.; Saedi, S.; Valilo, M.; Kalantary-Charvadeh, A.; Alizadeh Sani, M.; Kargar, R.; Safari, H.; Samadi, N. Oxidative Stress and Parkinson’s Disease: Conflict of Oxidant-Antioxidant Systems. Neurosci. Lett. 2019, 709, 134296. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH Oxidase 1-Mediated Oxidative Stress Leads to Dopamine Neuron Death in Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Zawada, W.M.; Banninger, G.P.; Thornton, J.; Marriott, B.; Cantu, D.; Rachubinski, A.L.; Das, M.; Griffin, W.S.; Jones, S.M. Generation of Reactive Oxygen Species in 1-Methyl-4-Phenylpyridinium (MPP+) Treated Dopaminergic Neurons Occurs as an NADPH Oxidase-Dependent Two-Wave Cascade. J. Neuroinflamm. 2011, 8, 129. [Google Scholar] [CrossRef]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s Disease Protein DJ-1 Is Neuroprotective Due to Cysteine-Sulfinic Acid-Driven Mitochondrial Localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological Rescue of Mitochondrial Deficits in IPSC-Derived Neural Cells from Patients with Familial Parkinson’s Disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef]
- Heo, H.Y.; Park, J.M.; Kim, C.H.; Han, B.S.; Kim, K.S.; Seol, W. LRRK2 Enhances Oxidative Stress-Induced Neurotoxicity via Its Kinase Activity. Exp. Cell Res. 2010, 316, 649–656. [Google Scholar] [CrossRef]
- Senger, D.R.; Li, D.; Jaminet, S.C.; Cao, S. Activation of the Nrf2 Cell Defense Pathway by Ancient Foods: Disease Prevention by Important Molecules and Microbes Lost from the Modern Western Diet. PLoS ONE 2016, 11, e0148042. [Google Scholar] [CrossRef]
- Mostafavi-Pour, Z.; Ramezani, F.; Keshavarzi, F.; Samadi, N. The Role of Quercetin and Vitamin C in Nrf2-Dependent Oxidative Stress Production in Breast Cancer Cells. Oncol. Lett. 2017, 13, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging Roles of Nrf2 and Phase II Antioxidant Enzymes in Neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.; Hobson, D.; Ponnampalam, A. High Affinity Hemoglobin and Parkinson’s Disease. Med. Hypotheses 2014, 83, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s Disease: Alterations in Iron and Redox Biology as a Key to Unlock Therapeutic Strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Umehara, T.; Oka, H.; Nakahara, A.; Shiraishi, T.; Sato, T.; Matsuno, H.; Komatsu, T.; Omoto, S.; Murakami, H.; Iguchi, Y. Sympathetic Nervous Activity and Hemoglobin Levels in de Novo Parkinson’s Disease. Clin. Auton. Res. 2020, 30, 273–278. [Google Scholar] [CrossRef]
- Deng, Q.; Zhou, X.; Chen, J.; Pan, M.; Gao, H.; Zhou, J.; Wang, D.; Chen, Q.; Zhang, X.; Wang, Q.; et al. Lower Hemoglobin Levels in Patients with Parkinson’s Disease Are Associated with Disease Severity and Iron Metabolism. Brain Res. 2017, 1655, 145–151. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Carling, P.J.; Mortiboys, H.; Green, C.; Mihaylov, S.; Sandor, C.; Schwartzentruber, A.; Taylor, R.; Wei, W.; Hastings, C.; Wong, S.; et al. Deep Phenotyping of Peripheral Tissue Facilitates Mechanistic Disease Stratification in Sporadic Parkinson’s Disease. Prog. Neurobiol. 2020, 187, 101772. [Google Scholar] [CrossRef]
- Smith, A.M.; Depp, C.; Ryan, B.J.; Johnston, G.I.; Alegre-Abarrategui, J.; Evetts, S.; Rolinski, M.; Baig, F.; Ruffmann, C.; Simon, A.K.; et al. Mitochondrial Dysfunction and Increased Glycolysis in Prodromal and Early Parkinson’s Blood Cells. Mov. Disord. 2018, 33, 1580–1590. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the Edge with Too Many Mouths to Feed: Why Dopamine Neurons Die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef] [PubMed]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a Novel Protein Regulated during the Critical Period for Song Learning in the Zebra Finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative Damage Linked to Neurodegeneration by Selective Alpha-Synuclein Nitration in Synucleinopathy Lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Jakes, R.; Curran, M.D.; Wallace, A. Effects of the Mutations Ala30 to Pro and Ala53 to Thr on the Physical and Morphological Properties of Alpha-Synuclein Protein Implicated in Parkinson’s Disease. FEBS Lett. 1998, 440, 67–70. [Google Scholar] [CrossRef]
- Wang, C.; Tan, J.M.; Ho, M.W.; Zaiden, N.; Wong, S.H.; Chew, C.L.; Eng, P.W.; Lim, T.M.; Dawson, T.M.; Lim, K.L. Alterations in the Solubility and Intracellular Localization of Parkin by Several Familial Parkinson’s Disease-Linked Point Mutations. J. Neurochem. 2005, 93, 422–431. [Google Scholar] [CrossRef]
- Kawahara, K.; Hashimoto, M.; Bar-On, P.; Ho, G.J.; Crews, L.; Mizuno, H.; Rockenstein, E.; Imam, S.Z.; Masliah, E. Alpha-Synuclein Aggregates Interfere with Parkin Solubility and Distribution: Role in the Pathogenesis of Parkinson Disease. J. Biol. Chem. 2008, 283, 6979–6987. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Recabarren-Leiva, D.; Alarcón, M. New Insights into the Gene Expression Associated to Amyotrophic Lateral Sclerosis. Life Sci. 2018, 193, 110–123. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The Genetics and Neuropathology of Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Galán, L.; Gómez-Pinedo, U.; Guerrero, A.; García-Verdugo, J.M.; Matías-Guiu, J. Amyotrophic Lateral Sclerosis Modifies Progenitor Neural Proliferation in Adult Classic Neurogenic Brain Niches. BMC Neurol. 2017, 17, 173. [Google Scholar] [CrossRef]
- Pandini, C.; Garofalo, M.; Rey, F.; Garau, J.; Zucca, S.; Sproviero, D.; Bordoni, M.; Berzero, G.; Davin, A.; Poloni, T.E.; et al. MINCR: A Long Non-Coding RNA Shared between Cancer and Neurodegeneration. Genomics 2021, 113, 4039–4051. [Google Scholar] [CrossRef]
- Rey, F.; Marcuzzo, S.; Bonanno, S.; Bordoni, M.; Giallongo, T.; Malacarne, C.; Cereda, C.; Zuccotti, G.V.; Carelli, S. LncRNAs Associated with Neuronal Development and Oncogenesis Are Deregulated in SOD1-G93A Murine Model of Amyotrophic Lateral Sclerosis. Biomedicines 2021, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein Oxidative Damage in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic Lateral Sclerosis. Orphanet. J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological Evidence for Lipid Peroxidation and Protein Glycoxidation in Spinal Cords from Sporadic Amyotrophic Lateral Sclerosis Patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-Hydroxynonenal in Cerebrospinal Fluid of Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Nobukuni, K.; Takata, H.; Hayabara, T. Oxidative Stress and Metal Content in Blood and Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients with and without a Cu, Zn-Superoxide Dismutase Mutation. Neurol. Res. 2005, 27, 105–108. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P. Amyotrophic Lateral Sclerosis and Structural Defects in Cu,Zn Superoxide Dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 Alters the Motor Neuronal Transcriptome: Implications for Familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial Amyotrophic Lateral Sclerosis-Linked SOD1 Mutants Perturb Fast Axonal Transport to Reduce Axonal Mitochondria Content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to Mitochondrial Dysfunction in ALS Pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic Recruitment and Activation of ALS-Associated TBK1 with Its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and Mitophagy in ALS. Neurobiol. Dis. 2018, 122, 35–40. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of Axonal Transport Defects in Motor Neuron Diseases: Opportunities for Translational Research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 Associated with Motor Neuron Mitochondria Alters Mitochondrial Shape and Distribution Prior to Clinical Onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.L.; Dickson, D.W. Ultrastructural Localization of TDP-43 in Filamentous Neuronal Inclusions in Various Neurodegenerative Diseases. Acta Neuropathol. 2008, 116, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Mizuno, Y.; Fujita, Y. Bunina Bodies in Amyotrophic Lateral Sclerosis. Neuropathology 2008, 28, 109–115. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging Roles in RNA Processing and Neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1 Mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- D’Ambrogio, A.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, M.; Ayala, Y.M.; Baralle, F.E. Functional Mapping of the Interaction between TDP-43 and HnRNP A2 in Vivo. Nucleic Acids Res. 2009, 37, 4116–4126. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP Mutations in Amyotrophic Lateral Sclerosis with TDP-43 Neuropathology: A Genetic and Histopathological Analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R. A New Subtype of Frontotemporal Lobar Degeneration with FUS Pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) Binds RNA in Vivo and Engages in Nucleo-Cytoplasmic Shuttling. J. Cell Sci. 1997, 110 Pt 15, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-Associated Fused in Sarcoma (FUS) Mutations Disrupt Transportin-Mediated Nuclear Import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Cleveland, D.W. The Seeds of Neurodegeneration: Prion-like Spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A Yeast TDP-43 Proteinopathy Model: Exploring the Molecular Determinants of TDP-43 Aggregation and Cellular Toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef] [PubMed]
- Daigle, J.G.; Lanson, N.A.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. RNA-Binding Ability of FUS Regulates Neurodegeneration, Cytoplasmic Mislocalization and Incorporation into Stress Granules Associated with FUS Carrying ALS-Linked Mutations. Hum. Mol. Genet. 2013, 22, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Baarine, M.; Beeson, C.; Singh, A.; Singh, I. ABCD1 Deletion-Induced Mitochondrial Dysfunction Is Corrected by SAHA: Implication for Adrenoleukodystrophy. J. Neurochem. 2015, 133, 380–396. [Google Scholar] [CrossRef]
- James, R.; Chaytow, H.; Ledahawsky, L.M.; Gillingwater, T.H. Revisiting the Role of Mitochondria in Spinal Muscular Atrophy. Cell. Mol. Life Sci. 2021, 78, 4785–4804. [Google Scholar] [CrossRef]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. JCM 2020, 9, 344. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; Poll-The, B.-T. X-Linked Adrenoleukodystrophy: Pathogenesis and Treatment. Curr. Neurol. Neurosci. Rep. 2014, 14, 486. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.; Jones, R.; Hubbard, W.; Tortorelli, S.; Orsini, J.; Caggana, M.; Vogel, B.; Raymond, G. Newborn Screening for X-Linked Adrenoleukodystrophy. Int. J. Neonatal Screen. 2016, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Roerig, P.; Mayerhofer, P.; Holzinger, A.; Gärtner, J. Characterization and Functional Analysis of the Nucleotide Binding Fold in Human Peroxisomal ATP Binding Cassette Transporters. FEBS Lett. 2001, 492, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Roermund, C.W.T.; Visser, W.F.; IJlst, L.; Cruchten, A.; Boek, M.; Kulik, W.; Waterham, H.R.; Wanders, R.J.A. The Human Peroxisomal ABC Half Transporter ALDP Functions as a Homodimer and Accepts Acyl–CoA Esters. FASEB J. 2008, 22, 4201–4208. [Google Scholar] [CrossRef]
- Powers, J.M. Adreno-Leukodystrophy: A Personal Historical Note. Acta Neuropathol. 2005, 109, 124–127. [Google Scholar] [CrossRef]
- Ferrer, I.; Aubourg, P.; Pujol, A. General Aspects and Neuropathology of X-Linked Adrenoleukodystrophy: X-Linked Adrenoleukodystrophy Neuropathology. Brain Pathol. 2009, 20, 817–830. [Google Scholar] [CrossRef]
- Fourcade, S.; Lopez-Erauskin, J.; Galino, J.; Duval, C.; Naudi, A.; Jove, M.; Kemp, S.; Villarroya, F.; Ferrer, I.; Pamplona, R.; et al. Early Oxidative Damage Underlying Neurodegeneration in X-Adrenoleukodystrophy. Hum. Mol. Genet. 2008, 17, 1762–1773. [Google Scholar] [CrossRef]
- van de Beek, M.-C.; Ofman, R.; Dijkstra, I.; Wijburg, F.; Engelen, M.; Wanders, R.; Kemp, S. Lipid-Induced Endoplasmic Reticulum Stress in X-Linked Adrenoleukodystrophy. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2255–2265. [Google Scholar] [CrossRef]
- Launay, N.; Aguado, C.; Fourcade, S.; Ruiz, M.; Grau, L.; Riera, J.; Guilera, C.; Giròs, M.; Ferrer, I.; Knecht, E.; et al. Autophagy Induction Halts Axonal Degeneration in a Mouse Model of X-Adrenoleukodystrophy. Acta Neuropathol. 2015, 129, 399–415. [Google Scholar] [CrossRef]
- Kruska, N.; Schönfeld, P.; Pujol, A.; Reiser, G. Astrocytes and Mitochondria from Adrenoleukodystrophy Protein (ABCD1)-Deficient Mice Reveal That the Adrenoleukodystrophy-Associated Very Long-Chain Fatty Acids Target Several Cellular Energy-Dependent Functions. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 925–936. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Fourcade, S.; Galino, J.; Ruiz, M.; Schlüter, A.; Naudi, A.; Jove, M.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; et al. Antioxidants Halt Axonal Degeneration in a Mouse Model of X-adrenoleukodystrophy. Ann. Neurol. 2011, 70, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Court, F.A.; Coleman, M.P. Mitochondria as a Central Sensor for Axonal Degenerative Stimuli. Trends Neurosci. 2012, 35, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, R.W.; Linehan, W.M.; Knazek, R.A. Effects of Long-Chain, Saturated Fatty Acids on Membrane Microviscosity and Adrenocorticotropin Responsiveness of Human Adrenocortical Cells in Vitro. J. Clin. Investig. 1988, 81, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Osório, M.J.; Goldman, S.A. Neurogenetics of Pelizaeus-Merzbacher Disease. Handb. Clin Neurol. 2018, 148, 701–722. [Google Scholar] [CrossRef]
- Harding, B.N. Pelizaeus-Merzbacher Disease. In Developmental Neuropathology; Adle-Biassette, H., Harding, B.N., Golden, J., Eds.; John Wiley & Sons, Ltd.: Oxford, UK, 2018; pp. 417–425. ISBN 978-1-119-01311-2. [Google Scholar]
- Duan, R.; Li, L.; Yan, H.; He, M.; Gao, K.; Xing, S.; Ji, H.; Wang, J.; Cao, B.; Li, D.; et al. Novel Insight into the Potential Pathogenicity of Mitochondrial Dysfunction Resulting from PLP1 Duplication Mutations in Patients with Pelizaeus-Merzbacher Disease. Neuroscience 2021, 476, 60–71. [Google Scholar] [CrossRef]
- Inoue, K. Pelizaeus-Merzbacher Disease: Molecular and Cellular Pathologies and Associated Phenotypes. Adv. Exp. Med. Biol. 2019, 1190, 201–216. [Google Scholar] [CrossRef]
- Edgar, J.M.; McCulloch, M.C.; Montague, P.; Brown, A.M.; Thilemann, S.; Pratola, L.; Gruenenfelder, F.I.; Griffiths, I.R.; Nave, K.-A. Demyelination and Axonal Preservation in a Transgenic Mouse Model of Pelizaeus-Merzbacher Disease. EMBO Mol. Med. 2010, 2, 42–50. [Google Scholar] [CrossRef]
- Woodward, K.J.; Cundall, M.; Sperle, K.; Sistermans, E.A.; Ross, M.; Howell, G.; Gribble, S.M.; Burford, D.C.; Carter, N.P.; Hobson, D.L.; et al. Heterogeneous Duplications in Patients with Pelizaeus-Merzbacher Disease Suggest a Mechanism of Coupled Homologous and Nonhomologous Recombination. Am. J. Hum. Genet. 2005, 77, 966–987. [Google Scholar] [CrossRef]
- Ruiz, M.; Bégou, M.; Launay, N.; Ranea-Robles, P.; Bianchi, P.; López-Erauskin, J.; Morató, L.; Guilera, C.; Petit, B.; Vaurs-Barriere, C.; et al. Oxidative Stress and Mitochondrial Dynamics Malfunction Are Linked in Pelizaeus-Merzbacher Disease. Brain Pathol. 2018, 28, 611–630. [Google Scholar] [CrossRef]
- Hüttemann, M.; Zhang, Z.; Mullins, C.; Bessert, D.; Lee, I.; Nave, K.-A.; Appikatla, S.; Skoff, R.P. Different Proteolipid Protein Mutants Exhibit Unique Metabolic Defects. ASN Neuro 2009, 1, AN20090028. [Google Scholar] [CrossRef]
- Appikatla, S.; Bessert, D.; Lee, I.; Hüttemann, M.; Mullins, C.; Somayajulu-Nitu, M.; Yao, F.; Skoff, R.P. Insertion of Proteolipid Protein into Oligodendrocyte Mitochondria Regulates Extracellular PH and Adenosine Triphosphate: Functions of PLPs Insertion into Mitochondria. Glia 2014, 62, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Leach, M.E.; Finanger, E. Spinal Muscular Atrophy. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Anhuf, D.; Eggermann, T.; Rudnik-Schöneborn, S.; Zerres, K. Determination of SMN1 and SMN2 Copy Number Using TaqMan Technology. Hum. Mutat. 2003, 22, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Gubitz, A.K.; Feng, W.; Dreyfuss, G. The SMN Complex. Exp. Cell Res. 2004, 296, 51–56. [Google Scholar] [CrossRef]
- Fallini, C.; Zhang, H.; Su, Y.; Silani, V.; Singer, R.H.; Rossoll, W.; Bassell, G.J. The Survival of Motor Neuron (SMN) Protein Interacts with the MRNA-Binding Protein HuD and Regulates Localization of Poly(A) MRNA in Primary Motor Neuron Axons. J. Neurosci. 2011, 31, 3914–3925. [Google Scholar] [CrossRef]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.-C. Motor Neuron Mitochondrial Dysfunction in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef]
- Boyd, P.J.; Tu, W.-Y.; Shorrock, H.K.; Groen, E.J.N.; Carter, R.N.; Powis, R.A.; Thomson, S.R.; Thomson, D.; Graham, L.C.; Motyl, A.A.L.; et al. Bioenergetic Status Modulates Motor Neuron Vulnerability and Pathogenesis in a Zebrafish Model of Spinal Muscular Atrophy. PLoS Genet. 2017, 13, e1006744. [Google Scholar] [CrossRef]
- Acsadi, G.; Lee, I.; Li, X.; Khaidakov, M.; Pecinova, A.; Parker, G.C.; Hüttemann, M. Mitochondrial Dysfunction in a Neural Cell Model of Spinal Muscular Atrophy. J. Neurosci. Res. 2009, 87, 2748–2756. [Google Scholar] [CrossRef]
- Hellbach, N.; Peterson, S.; Haehnke, D.; Shankar, A.; LaBarge, S.; Pivaroff, C.; Saenger, S.; Thomas, C.; McCarthy, K.; Ebeling, M.; et al. Impaired Myogenic Development, Differentiation and Function in HESC-Derived SMA Myoblasts and Myotubes. PLoS ONE 2018, 13, e0205589. [Google Scholar] [CrossRef]
- Thelen, M.P.; Wirth, B.; Kye, M.J. Mitochondrial Defects in the Respiratory Complex I Contribute to Impaired Translational Initiation via ROS and Energy Homeostasis in SMA Motor Neurons. Acta Neuropathol. Commun. 2020, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, C.; Ma, L.; Mou, Y.; Zhang, B.; Zhou, S.; Tian, Y.; Trinh, J.; Zhang, X.; Li, X.-J. Drug Screening with Human SMN2 Reporter Identifies SMN Protein Stabilizers to Correct SMA Pathology. Life Sci. Alliance 2019, 2, e201800268. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-C.; Denton, K.R.; Wang, Z.-B.; Zhang, X.; Li, X.-J. Abnormal Mitochondrial Transport and Morphology as Early Pathological Changes in Human Models of Spinal Muscular Atrophy. Dis. Model. Mech. 2016, 9, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Kariya, S.; Park, G.-H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN Protein Impairs Maturation of the Neuromuscular Junctions in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef]
- Torres-Benito, L.; Neher, M.F.; Cano, R.; Ruiz, R.; Tabares, L. SMN Requirement for Synaptic Vesicle, Active Zone and Microtubule Postnatal Organization in Motor Nerve Terminals. PLoS ONE 2011, 6, e26164. [Google Scholar] [CrossRef]
- Ripolone, M.; Ronchi, D.; Violano, R.; Vallejo, D.; Fagiolari, G.; Barca, E.; Lucchini, V.; Colombo, I.; Villa, L.; Berardinelli, A.; et al. Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy. JAMA Neurol. 2015, 72, 666–675. [Google Scholar] [CrossRef]
- Ando, S.; Funato, M.; Ohuchi, K.; Inagaki, S.; Sato, A.; Seki, J.; Kawase, C.; Saito, T.; Nishio, H.; Nakamura, S.; et al. The Protective Effects of Levetiracetam on a Human IPSCs-Derived Spinal Muscular Atrophy Model. Neurochem. Res. 2019, 44, 1773–1779. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]
- Van Alstyne, M.; Lotti, F.; Dal Mas, A.; Area-Gomez, E.; Pellizzoni, L. Stasimon/Tmem41b Localizes to Mitochondria-Associated ER Membranes and Is Essential for Mouse Embryonic Development. Biochem. Biophys. Res. Commun. 2018, 506, 463–470. [Google Scholar] [CrossRef]
- Anderton, R.S.; Meloni, B.P.; Mastaglia, F.L.; Boulos, S. Spinal Muscular Atrophy and the Antiapoptotic Role of Survival of Motor Neuron (SMN) Protein. Mol. Neurobiol. 2013, 47, 821–832. [Google Scholar] [CrossRef]
- Sato, K.; Eguchi, Y.; Kodama, T.S.; Tsujimoto, Y. Regions Essential for the Interaction between Bcl-2 and SMN, the Spinal Muscular Atrophy Disease Gene Product. Cell Death Differ. 2000, 7, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Garcera, A.; Mincheva, S.; Gou-Fabregas, M.; Caraballo-Miralles, V.; Lladó, J.; Comella, J.X.; Soler, R.M. A New Model to Study Spinal Muscular Atrophy: Neurite Degeneration and Cell Death Is Counteracted by BCL-X(L) Overexpression in Motoneurons. Neurobiol. Dis. 2011, 42, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.; Schiaffino, L.; Boido, M.; Valsecchi, V.; Guglielmotto, M.; De Amicis, E.; Puyal, J.; Garcera, A.; Tamagno, E.; Soler, R.M.; et al. Inhibition of Autophagy Delays Motoneuron Degeneration and Extends Lifespan in a Mouse Model of Spinal Muscular Atrophy. Cell Death Dis. 2017, 8, 3223. [Google Scholar] [CrossRef]
- Simic, G.; Seso-Simic, D.; Lucassen, P.J.; Islam, A.; Krsnik, Z.; Cviko, A.; Jelasic, D.; Barisic, N.; Winblad, B.; Kostovic, I.; et al. Ultrastructural Analysis and TUNEL Demonstrate Motor Neuron Apoptosis in Werdnig-Hoffmann Disease. J. Neuropathol. Exp. Neurol. 2000, 59, 398–407. [Google Scholar] [CrossRef]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of P53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- Young, P.J.; Day, P.M.; Zhou, J.; Androphy, E.J.; Morris, G.E.; Lorson, C.L. A Direct Interaction between the Survival Motor Neuron Protein and P53 and Its Relationship to Spinal Muscular Atrophy. J. Biol. Chem. 2002, 277, 2852–2859. [Google Scholar] [CrossRef]
- Helmken, C.; Hofmann, Y.; Schoenen, F.; Oprea, G.; Raschke, H.; Rudnik-Schöneborn, S.; Zerres, K.; Wirth, B. Evidence for a Modifying Pathway in SMA Discordant Families: Reduced SMN Level Decreases the Amount of Its Interacting Partners and Htra2-Beta1. Hum. Genet. 2003, 114, 11–21. [Google Scholar] [CrossRef]
- Rindt, H.; Feng, Z.; Mazzasette, C.; Glascock, J.J.; Valdivia, D.; Pyles, N.; Crawford, T.O.; Swoboda, K.J.; Patitucci, T.N.; Ebert, A.D.; et al. Astrocytes Influence the Severity of Spinal Muscular Atrophy. Hum. Mol. Genet. 2015, 24, 4094–4102. [Google Scholar] [CrossRef]
- Kuru, S.; Sakai, M.; Konagaya, M.; Yoshida, M.; Hashizume, Y.; Saito, K. An Autopsy Case of Spinal Muscular Atrophy Type III (Kugelberg-Welander Disease). Neuropathology 2009, 29, 63–67. [Google Scholar] [CrossRef]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.-E.A.; Stucky, C.L.; Ebert, A.D. Spinal Muscular Atrophy Astrocytes Exhibit Abnormal Calcium Regulation and Reduced Growth Factor Production. Glia 2013, 61, 1418–1428. [Google Scholar] [CrossRef]
- Deguise, M.-O.; Kothary, R. New Insights into SMA Pathogenesis: Immune Dysfunction and Neuroinflammation. Ann. Clin Transl. Neurol. 2017, 4, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Pierzynowska, K.; Gaffke, L.; Cyske, Z.; Węgrzyn, G.; Buttari, B.; Profumo, E.; Saso, L. Oxidative Stress in Mucopolysaccharidoses: Pharmacological Implications. Molecules 2021, 26, 5616. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment: GAG Targeting in Cancer Cell Biology. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like Receptor 4 Pathway and Use of TNF-α Antagonists for Treatment of the Mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA. 2010, 107, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Mandolfo, O.; Parker, H.; Bigger, B. Innate Immunity in Mucopolysaccharide Diseases. Int. J. Mol. Sci. 2022, 23, 1999. [Google Scholar] [CrossRef]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, Mitochondrial Defects and Neurodegeneration in Mucopolysaccharidosis III Type C Mouse Model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef]
- Villani, G.R.D.; Gargiulo, N.; Faraonio, R.; Castaldo, S.; Gonzalez y Reyero, E.; Di Natale, P. Cytokines, Neurotrophins, and Oxidative Stress in Brain Disease from Mucopolysaccharidosis IIIB. J. Neurosci. Res. 2007, 85, 612–622. [Google Scholar] [CrossRef]
- Polgreen, L.E.; Vehe, R.K.; Rudser, K.; Kunin-Batson, A.; Utz, J.J.; Dickson, P.; Shapiro, E.; Whitley, C.B. Elevated TNF-α Is Associated with Pain and Physical Disability in Mucopolysaccharidosis Types I, II, and VI. Mol. Genet. Metab. 2016, 117, 427–430. [Google Scholar] [CrossRef]
- Pereira, V.G.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and Oxidative Stress Analysis in Patients with Mucopolysaccharidosis Type I Undergoing Enzyme Replacement Therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef]
- Filippon, L.; Vanzin, C.S.; Biancini, G.B.; Pereira, I.N.; Manfredini, V.; Sitta, A.; Peralba, M.d.C.R.; Schwartz, I.V.D.; Giugliani, R.; Vargas, C.R. Oxidative Stress in Patients with Mucopolysaccharidosis Type II before and during Enzyme Replacement Therapy. Mol. Genet. Metab. 2011, 103, 121–127. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential Subcellular Localization of Cholesterol, Gangliosides, and Glycosaminoglycans in Murine Models of Mucopolysaccharide Storage Disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef]
- Jacques, C.E.D.; Donida, B.; Mescka, C.P.; Rodrigues, D.G.B.; Marchetti, D.P.; Bitencourt, F.H.; Burin, M.G.; de Souza, C.F.M.; Giugliani, R.; Vargas, C.R. Oxidative and Nitrative Stress and Pro-Inflammatory Cytokines in Mucopolysaccharidosis Type II Patients: Effect of Long-Term Enzyme Replacement Therapy and Relation with Glycosaminoglycan Accumulation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef] [PubMed]
- Constantopoulos, G.; Dekaban, A.S. Neurochemistry of The Mucopolysaccharidoses: Brain Lipids and Lysosomal Enzymes in Patients with Four Types of Mucopolysaccharidosis and in Normal Controls. J. Neurochem. 1978, 30, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cheng, Y.-S.; Yang, S.; Swaroop, M.; Xu, M.; Huang, W.; Zheng, W. Disease Modeling for Mucopolysaccharidosis Type IIIB Using Patient Derived Induced Pluripotent Stem Cells. Exp. Cell Res. 2021, 407, 112785. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, L.; Futerman, A.H. Defective Calcium Homeostasis in the Cerebellum in a Mouse Model of Niemann-Pick A Disease. J. Neurochem. 2005, 95, 1619–1628. [Google Scholar] [CrossRef]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.-J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal Fusion and SNARE Function Are Impaired by Cholesterol Accumulation in Lysosomal Storage Disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef]
- Finkbeiner, S. The Autophagy Lysosomal Pathway and Neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a033993. [Google Scholar] [CrossRef]
- Chai, P.; Ni, H.; Zhang, H.; Fan, X. The Evolving Functions of Autophagy in Ocular Health: A Double-Edged Sword. Int. J. Biol. Sci. 2016, 12, 1332–1340. [Google Scholar] [CrossRef]
- Sambri, I.; D’Alessio, R.; Ezhova, Y.; Giuliano, T.; Sorrentino, N.C.; Cacace, V.; De Risi, M.; Cataldi, M.; Annunziato, L.; De Leonibus, E.; et al. Lysosomal Dysfunction Disrupts Presynaptic Maintenance and Restoration of Presynaptic Function Prevents Neurodegeneration in Lysosomal Storage Diseases. EMBO Mol. Med. 2017, 9, 112–132. [Google Scholar] [CrossRef]
- Monaco, A.; Maffia, V.; Sorrentino, N.C.; Sambri, I.; Ezhova, Y.; Giuliano, T.; Cacace, V.; Nusco, E.; De Risi, M.; De Leonibus, E.; et al. The Amyloid Inhibitor CLR01 Relieves Autophagy and Ameliorates Neuropathology in a Severe Lysosomal Storage Disease. Mol. Ther. 2020, 28, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of Neurodegeneration in Mucopolysaccharidoses II and IIIB: Analysis of Human Brain Tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Winder-Rhodes, S.E.; Garcia-Reitböck, P.; Ban, M.; Evans, J.R.; Jacques, T.S.; Kemppinen, A.; Foltynie, T.; Williams-Gray, C.H.; Chinnery, P.F.; Hudson, G.; et al. Genetic and Pathological Links between Parkinson’s Disease and the Lysosomal Disorder Sanfilippo Syndrome: Parkinson’s Disease and Sanfilippo Syndrome. Mov. Disord. 2012, 27, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Bhattacharya, N.; Mishra, S.; Bhattacharya, A.; Banerjee, P.; Senapati, S.; Mishra, R. Microglia Specific Drug Targeting Using Natural Products for the Regulation of Redox Imbalance in Neurodegeneration. Front. Pharmacol. 2021, 12, 654489. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural. Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Rajasekar, N.; Dwivedi, S.; Tota, S.K.; Kamat, P.K.; Hanif, K.; Nath, C.; Shukla, R. Neuroprotective Effect of Curcumin on Okadaic Acid Induced Memory Impairment in Mice. Eur. J. Pharmacol. 2013, 715, 381–394. [Google Scholar] [CrossRef]
- Sancheti, H.; Kanamori, K.; Patil, I.; Díaz Brinton, R.; Ross, B.D.; Cadenas, E. Reversal of Metabolic Deficits by Lipoic Acid in a Triple Transgenic Mouse Model of Alzheimer’s Disease: A 13C NMR Study. J. Cereb Blood Flow Metab. 2014, 34, 288–296. [Google Scholar] [CrossRef]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A Controlled Trial of Selegiline, Alpha-Tocopherol, or Both as Treatment for Alzheimer’s Disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of Vitamin E and Memantine on Functional Decline in Alzheimer Disease: The TEAM-AD VA Cooperative Randomized Trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral Curcumin for Alzheimer’s Disease: Tolerability and Efficacy in a 24-Week Randomized, Double Blind, Placebo-Controlled Study. Alzheimers Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A Review on Mitochondrial Restorative Mechanism of Antioxidants in Alzheimer’s Disease and Other Neurological Conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Köstel, A.S.; Bora-Tatar, G.; Erdem-Yurter, H. Spinal Muscular Atrophy: An Oxidative Stress Response Counteracted with curcumin. Biomed. Aging Pathol. 2012, 2, 61–66. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B.; Puszczewicz, M. Oxidative Damage and Antioxidative Therapy in Systemic Sclerosis. Mediat. Inflamm. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Casasnovas, C.; Ruiz, M.; Schlüter, A.; Naudí, A.; Fourcade, S.; Veciana, M.; Castañer, S.; Albertí, A.; Bargalló, N.; Johnson, M.; et al. Biomarker Identification, Safety, and Efficacy of High-Dose Antioxidants for Adrenomyeloneuropathy: A Phase II Pilot Study. Neurotherapeutics 2019, 16, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M.; Hervé, V.; Ben Khedher, M.R.; Rabanel, J.-M.; Ramassamy, C. Glutathione: An Old and Small Molecule with Great Functions and New Applications in the Brain and in Alzheimer’s Disease. Antioxid. Redox Signal. 2021, 35, 270–292. [Google Scholar] [CrossRef]
- Taylor, S.; Wheeler, L.C.; Taylor, J.R.; Griffin, H.C. Nutrition: An Issue of Concern for Children with Disabilities. Nurse Pract. 1996, 21, 17–18, 20. [Google Scholar]
- Pocernich, C.B.; Butterfield, D.A. Elevation of Glutathione as a Therapeutic Strategy in Alzheimer Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 625–630. [Google Scholar] [CrossRef]
- Zarka, M.H.; Bridge, W.J. Oral Administration of γ-Glutamylcysteine Increases Intracellular Glutathione Levels above Homeostasis in a Randomised Human Trial Pilot Study. Redox Biol. 2017, 11, 631–636. [Google Scholar] [CrossRef]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-Acetylcysteine, Oral Glutathione (GSH) and a Novel Sublingual Form of GSH on Oxidative Stress Markers: A Comparative Crossover Study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-Glutamylcysteine (γ-GC) Lessens Oxidative Stress, Brain Inflammation and Amyloid Pathology and Improves Spatial Memory in a Murine Model of AD. Neurochem. Int. 2021, 144, 104931. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 1 January 2021).
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive Oxygen Species: From Health to Disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, I.; Bringas, M.L.; Aguiar, A.; Morales, L.; Alvarez, M.; Valdés, P.A.; Alvarez, L. Use of Cuban Recombinant Human Erythropoietin in Parkinson’s Disease Treatment. MEDICC Rev. 2012, 14, 11–17. [Google Scholar] [PubMed]
- Langan, A.R.; Khan, M.A.; Yeung, I.W.T.; Van Dyk, J.; Hill, R.P. Partial Volume Rat Lung Irradiation: The Protective/Mitigating Effects of Eukarion-189, a Superoxide Dismutase-Catalase Mimetic. Radiother. Oncol. 2006, 79, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Stevenson, F.F.; Doctrow, S.R.; Andersen, J.K. Superoxide Dismutase/Catalase Mimetics Are Neuroprotective against Selective Paraquat-Mediated Dopaminergic Neuron Death in the Substantial Nigra: Implications for Parkinson Disease. J. Biol. Chem. 2005, 280, 29194–29198. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Rong, Y.; Doctrow, S.; Baudry, M.; Malfroy, B.; Xu, Z. Synthetic Superoxide Dismutase/Catalase Mimetics Reduce Oxidative Stress and Prolong Survival in a Mouse Amyotrophic Lateral Sclerosis Model. Neurosci. Lett. 2001, 304, 157–160. [Google Scholar] [CrossRef]
- Melov, S.; Wolf, N.; Strozyk, D.; Doctrow, S.R.; Bush, A.I. Mice Transgenic for Alzheimer Disease Beta-Amyloid Develop Lens Cataracts That Are Rescued by Antioxidant Treatment. Free Radic. Biol. Med. 2005, 38, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Marcus, C.B.; Huffman, K.; Kruk, H.; Malfroy, B.; Doctrow, S.R. Synthetic Combined Superoxide Dismutase/Catalase Mimetics Are Protective as a Delayed Treatment in a Rat Stroke Model: A Key Role for Reactive Oxygen Species in Ischemic Brain Injury. J. Pharmacol. Exp. Ther. 1998, 284, 215–221. [Google Scholar]
- Hallows, W.C.; Albaugh, B.N.; Denu, J.M. Where in the Cell Is SIRT3?--Functional Localization of an NAD+-Dependent Protein Deacetylase. Biochem. J. 2008, 411, e11–e13. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, Q.; Shi, J.; Zhou, S. Regulation of SIRT3 on Mitochondrial Functions and Oxidative Stress in Parkinson’s Disease. Biomed. Pharm. 2020, 132, 110928. [Google Scholar] [CrossRef]
- Anamika; Khanna, A.; Acharjee, P.; Acharjee, A.; Trigun, S.K. Mitochondrial SIRT3 and Neurodegenerative Brain Disorders. J. Chem. Neuroanat. 2019, 95, 43–53. [Google Scholar] [CrossRef]
- Zeng, R.; Wang, X.; Zhou, Q.; Fu, X.; Wu, Q.; Lu, Y.; Shi, J.; Klaunig, J.E.; Zhou, S. Icariin Protects Rotenone-Induced Neurotoxicity through Induction of SIRT3. Toxicol. Appl. Pharmacol. 2019, 379, 114639. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wang, H.; Hua, X.; Chen, D.; Huang, C.; Chen, Z. An Outline for the Pharmacological Effect of Icariin in the Nervous System. Eur. J. Pharmacol. 2019, 842, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Metsämuuronen, S.; Sirén, H. Bioactive Phenolic Compounds, Metabolism and Properties: A Review on Valuable Chemical Compounds in Scots Pine and Norway Spruce. Phytochem. Rev. 2019, 18, 623–664. [Google Scholar] [CrossRef]
- Mathieu, L.; Lopes Costa, A.; Le Bachelier, C.; Slama, A.; Lebre, A.S.; Taylor, R.W.; Bastin, J.; Djouadi, F. Resveratrol Attenuates Oxidative Stress in Mitochondrial Complex I Deficiency: Involvement of SIRT3. Free Radic. Biol. Med. 2016, 96, 190–198. [Google Scholar] [CrossRef]
- Chen, H.H.; Chang, P.C.; Wey, S.P.; Chen, P.M.; Chen, C.; Chan, M.H. Therapeutic Effects of Honokiol on Motor Impairment in Hemiparkinsonian Mice Are Associated with Reversing Neurodegeneration and Targeting PPARγ Regulation. Biomed. Pharm. 2018, 108, 254–262. [Google Scholar] [CrossRef]
- Li, S.; Pu, X.P. Neuroprotective Effect of Kaempferol against a 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Mouse Model of Parkinson’s Disease. Biol. Pharm. Bull. 2011, 34, 1291–1296. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Sastre, J.; Vendemiale, G.; Altomare, E. Targeting Mitochondria: A New Promising Approach for the Treatment of Liver Diseases. Curr. Med. Chem. 2010, 17, 2325–2337. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Drug Delivery to Mitochondria: The Key to Mitochondrial Medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef]
- Szeto, H.H. Cell-Permeable, Mitochondrial-Targeted, Peptide Antioxidants. AAPS J. 2006, 8, E277–E283. [Google Scholar] [CrossRef]
- Bordet, T.; Berna, P.; Abitbol, J.-L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef]
- Singh, J.; Olle, B.; Suhail, H.; Felicella, M.M.; Giri, S. Metformin-Induced Mitochondrial Function and ABCD2 up-Regulation in X-Linked Adrenoleukodystrophy Involves AMP-Activated Protein Kinase. J. Neurochem. 2016, 138, 86–100. [Google Scholar] [CrossRef]
- Kenche, V.B.; Barnham, K.J. Alzheimer’s Disease & Metals: Therapeutic Opportunities. Br. J. Pharmacol. 2011, 163, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits Beta-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.-X.; Tammer, A.; et al. Metal-Protein Attenuation with Iodochlorhydroxyquin (Clioquinol) Targeting Abeta Amyloid Deposition and Toxicity in Alzheimer Disease: A Pilot Phase 2 Clinical Trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 Rapidly Improves Cognition in Alzheimer’s Disease: Additional Phase II Analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef]
- Petri, S.; Calingasan, N.Y.; Alsaied, O.A.; Wille, E.; Kiaei, M.; Friedman, J.E.; Baranova, O.; Chavez, J.C.; Beal, M.F. The Lipophilic Metal Chelators DP-109 and DP-460 Are Neuroprotective in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2007, 102, 991–1000. [Google Scholar] [CrossRef]
- Watanabe, S.; Nagano, S.; Duce, J.; Kiaei, M.; Li, Q.-X.; Tucker, S.M.; Tiwari, A.; Brown, R.H.; Beal, M.F.; Hayward, L.J.; et al. Increased Affinity for Copper Mediated by Cysteine 111 in Forms of Mutant Superoxide Dismutase 1 Linked to Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2007, 42, 1534–1542. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 2019, 16, 9–29. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The Marijuana Component Cannabidiol Inhibits Beta-Amyloid-Induced Tau Protein Hyperphosphorylation through Wnt/Beta-Catenin Pathway Rescue in PC12 Cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.; Pertwee, R.; Fernández-Ruiz, J. Symptom-Relieving and Neuroprotective Effects of the Phytocannabinoid Δ9-THCV in Animal Models of Parkinson’s Disease. Br. J. Pharm. 2011, 163, 1495–1506. [Google Scholar] [CrossRef]
- Sieradzan, K.A.; Fox, S.H.; Hill, M.; Dick, J.P.; Crossman, A.R.; Brotchie, J.M. Cannabinoids Reduce Levodopa-Induced Dyskinesia in Parkinson’s Disease: A Pilot Study. Neurology 2001, 57, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, J.L.; Seely, K.A.; Reed, R.L.; Crow, J.P.; Prather, P.L. The CB2 Cannabinoid Agonist AM-1241 Prolongs Survival in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis When Initiated at Symptom Onset. J. Neurochem. 2007, 101, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Mendiola-Precoma, J.; Berumen, L.C.; Padilla, K.; Garcia-Alcocer, G. Therapies for Prevention and Treatment of Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 2589276. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Snyder, H.M.; Carrillo, M.C.; Fazio, S.; Kim, H.; Johns, H. Summary of the Evidence on Modifiable Risk Factors for Cognitive Decline and Dementia: A Population-Based Perspective. Alzheimers Dement. 2015, 11, 718–726. [Google Scholar] [CrossRef]
- Navarro-Yepes, J.; Zavala-Flores, L.; Anandhan, A.; Wang, F.; Skotak, M.; Chandra, N.; Li, M.; Pappa, A.; Martinez-Fong, D.; Del Razo, L.M.; et al. Antioxidant Gene Therapy against Neuronal Cell Death. Pharm. Ther. 2014, 142, 206–230. [Google Scholar] [CrossRef]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and Neuroprotective Functions of Cathepsin B: Implications for Alzheimer’s Disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef]
- Singer, O.; Marr, R.A.; Rockenstein, E.; Crews, L.; Coufal, N.G.; Gage, F.H.; Verma, I.M.; Masliah, E. Targeting BACE1 with SiRNAs Ameliorates Alzheimer Disease Neuropathology in a Transgenic Model. Nat. Neurosci. 2005, 8, 1343–1349. [Google Scholar] [CrossRef]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene Abeparvovec Gene Therapy for Symptomatic Infantile-Onset Spinal Muscular Atrophy in Patients with Two Copies of SMN2 (STR1VE): An Open-Label, Single-Arm, Multicentre, Phase 3 Trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef]
- Mirea, A.; Shelby, E.-S.; Axente, M.; Badina, M.; Padure, L.; Leanca, M.; Dima, V.; Sporea, C. Combination Therapy with Nusinersen and Onasemnogene Abeparvovec-Xioi in Spinal Muscular Atrophy Type I. J. Clin. Med. 2021, 10, 5540. [Google Scholar] [CrossRef]
- Gidaro, T.; Servais, L. Nusinersen Treatment of Spinal Muscular Atrophy: Current Knowledge and Existing Gaps. Dev. Med. Child. Neurol. 2019, 61, 19–24. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, A.; Dunbar, C.E. Hematopoietic Stem Cell Gene Therapy: Assessing the Relevance of Preclinical Models. Semin. Hematol. 2013, 50, 101–130. [Google Scholar] [CrossRef] [PubMed]



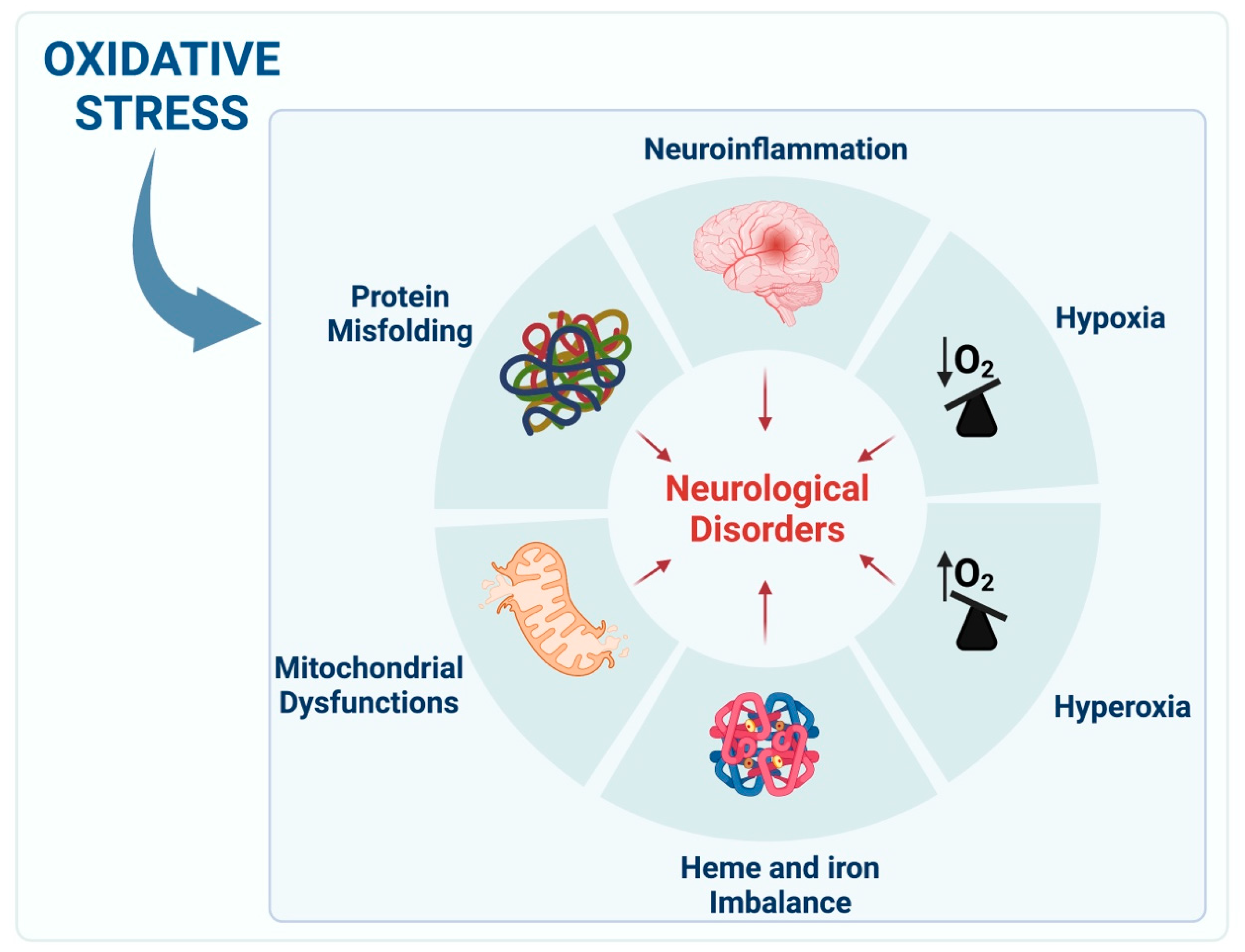{kind=link}
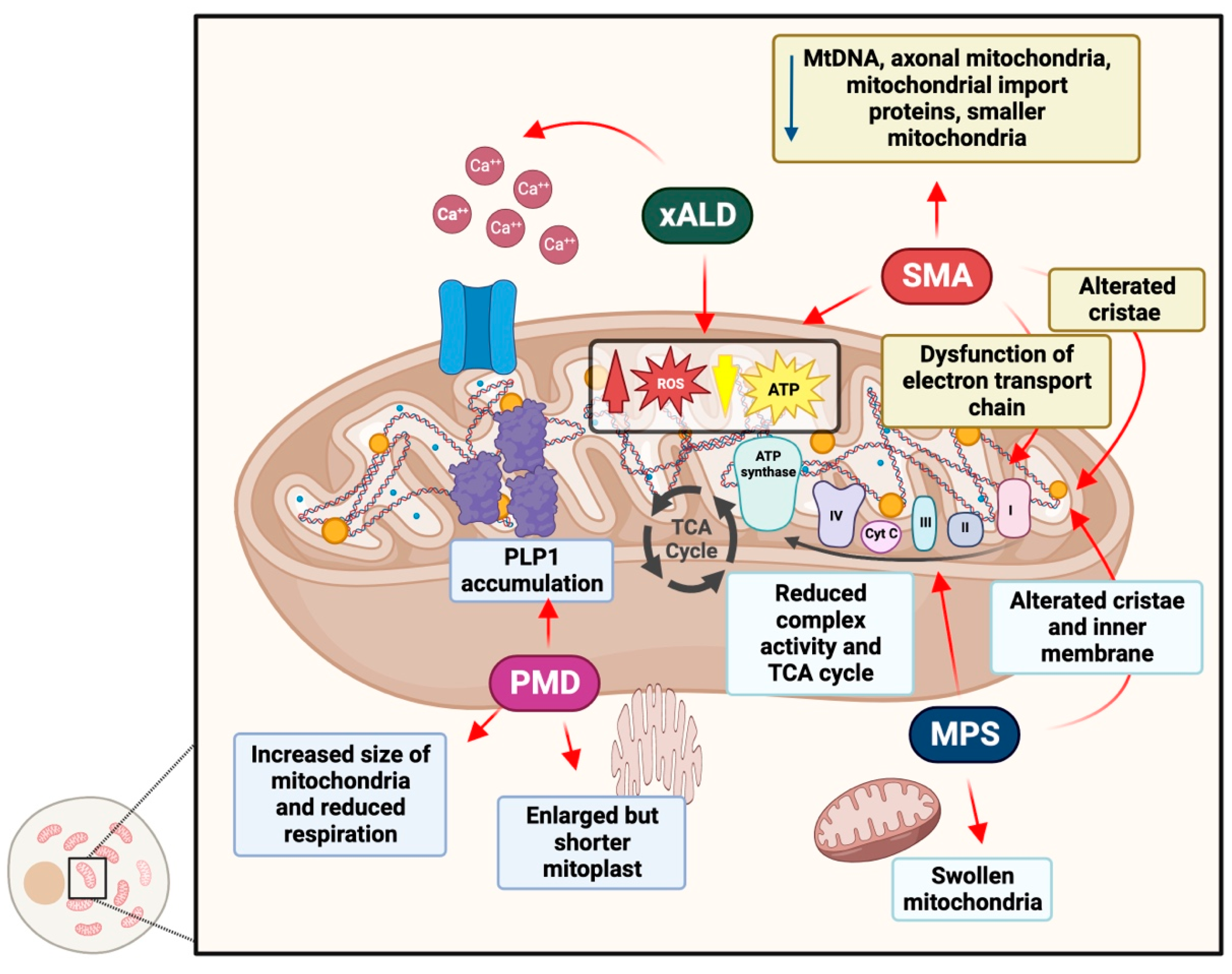{kind=link}
| Pathological Feature | Alzheimer’sh Disease | Parkinson’sh Disease | Amyotrophich Lateral Sclerosis |
|---|---|---|---|
| Hypoxia | Aberrant amyloid β metabolism [97] | β-synuclein accumulationh [98] | / |
| Hyperoxia | Tau polymerizationh [99,100] | NADPH oxidase activation [101] | Increased markers for lipid and DNA oxidationh [102] |
| Heme proteinsh and iron | Decrease in hemoglobin transcription [50] | Altered iron metabolism, low SN hemoglobin [103] | / |
| Mithocondriah dysfunctions | Krebs cycle enzymes deficiencies [104] | Increased mitophagyh [105,106] | Impaired mitophagy, h altered mitochondria morphology [107,108] |
| Neuroinflammation | Microglia activationh [109] | Microglia activation, lymphocytes infiltrationh [110,111,112] | Inflammatory cytokine production [113] |
| Protein misfolding | Amyloid plaques formationh [114,115] | α-syn aggregationh [116] | Cytoplasmic inclusionsh [117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey, F.; Berardo, C.; Maghraby, E.; Mauri, A.; Messa, L.; Esposito, L.; Casili, G.; Ottolenghi, S.; Bonaventura, E.; Cuzzocrea, S.; et al. Redox Imbalance in Neurological Disorders in Adults and Children. Antioxidants 2023, 12, 965. https://doi.org/10.3390/antiox12040965
Rey F, Berardo C, Maghraby E, Mauri A, Messa L, Esposito L, Casili G, Ottolenghi S, Bonaventura E, Cuzzocrea S, et al. Redox Imbalance in Neurological Disorders in Adults and Children. Antioxidants. 2023; 12(4):965. https://doi.org/10.3390/antiox12040965
Chicago/Turabian StyleRey, Federica, Clarissa Berardo, Erika Maghraby, Alessia Mauri, Letizia Messa, Letizia Esposito, Giovanna Casili, Sara Ottolenghi, Eleonora Bonaventura, Salvatore Cuzzocrea, and et al. 2023. "Redox Imbalance in Neurological Disorders in Adults and Children" Antioxidants 12, no. 4: 965. https://doi.org/10.3390/antiox12040965
APA StyleRey, F., Berardo, C., Maghraby, E., Mauri, A., Messa, L., Esposito, L., Casili, G., Ottolenghi, S., Bonaventura, E., Cuzzocrea, S., Zuccotti, G., Tonduti, D., Esposito, E., Paterniti, I., Cereda, C., & Carelli, S. (2023). Redox Imbalance in Neurological Disorders in Adults and Children. Antioxidants, 12(4), 965. https://doi.org/10.3390/antiox12040965