


Exercise-Intervened Circulating Extracellular Vesicles Alleviate Oxidative Stress in Cerebral Microvascular Endothelial Cells Under Hypertensive Plus Hypoxic Conditions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Treadmill Exercise Protocol
2.3. Cell Culture
2.4. cEV Isolation and Characterization
2.5. Internalization Assay of cEVs into mBMECs
2.6. cEV Uptake Pathway Determination
2.7. Co-Incubation of cEVs with mBMECs Challenged by Ang II Plus Hypoxia
2.8. Migration Assay of mBMECs After cEV Treatment
2.9. Reactive Oxygen Species Quantification in mBMECs After cEV Treatment
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
3.1. Treadmill Exercise Intervention Stimulates cEV Release in Hypertensive Transgenic Mice and Increases the Internalization of cEVs by mBMECs
3.2. ET-cEVs Incorporate into mBMECs Through Endocytosis and Macropinocytosis
3.3. Both nET-cEVs and ET-cEVs Improve the Migration Capability of mBMECs Compromised by Ang II Plus Hypoxia Injury
3.4. ET-cEVs Exhibit Anti-Oxidative Effects in mBMECs Challenged by Ang II Plus Hypoxia
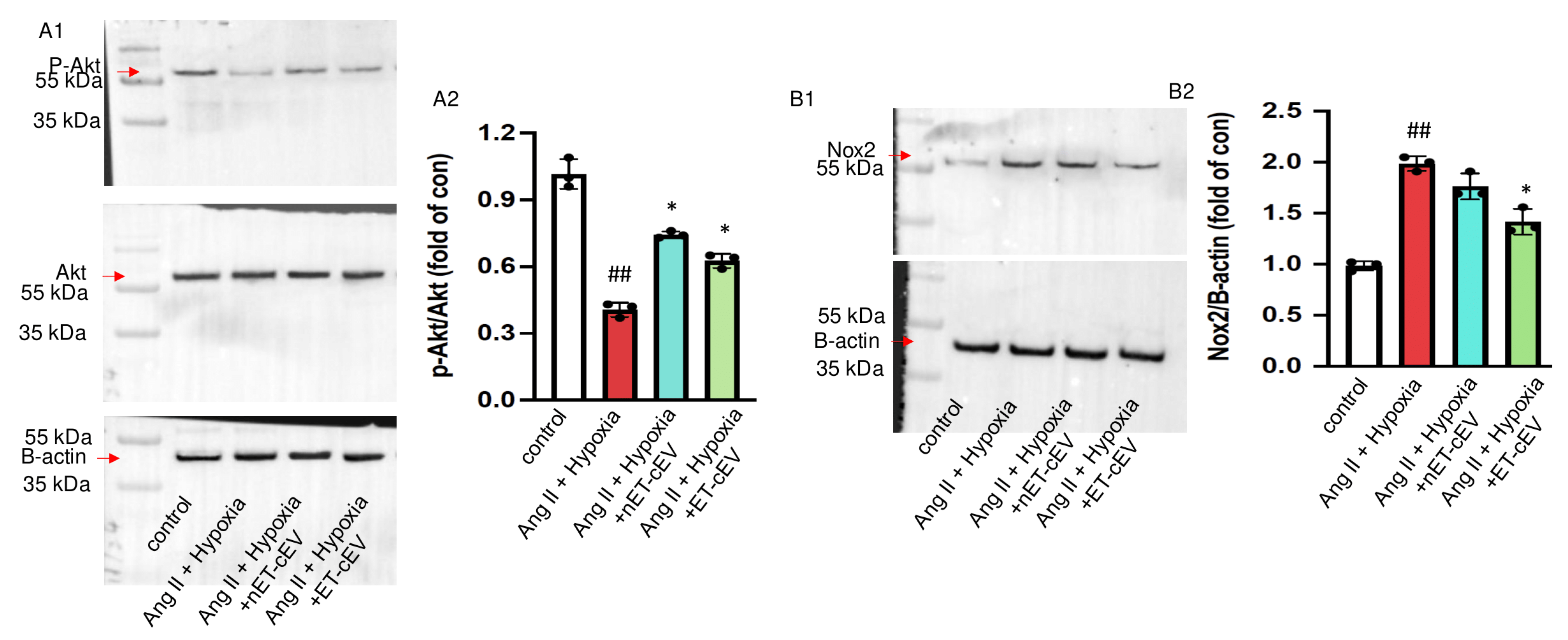
3.5. ET-cEVs Increase p-Akt/Akt and Decrease Nox2 Expressions in mBMECs Post-Ang II Plus Hypoxia Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Ye, T.; Ye, P.; Borghi, C.; Cro, S.; Damasceno, A.; Khan, N.; Nilsson, P.M.; Prabhakaran, D.; Ramirez, A.; et al. Hypertension in stroke survivors and associations with national premature stroke mortality: Data for 2.5 million participants from multinational screening campaigns. Lancet Glob. Health 2022, 10, e1141–e1149. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [PubMed]
- Dorans, K.S.; Mills, K.T.; Liu, Y.; He, J. Trends in Prevalence and Control of Hypertension According to the 2017 American College of Cardiology/American Heart Association (ACC/AHA) Guideline. J. Am. Heart Assoc. 2018, 7, e008888. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Iadecola, C. Hypertension, dietary salt and cognitive impairment. J. Cereb. Blood Flow Metab. 2018, 38, 2112–2128. [Google Scholar] [CrossRef]
- Pires, P.W.; Dams Ramos, C.M.; Matin, N.; Dorrance, A.M. The effects of hypertension on the cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1598–H1614. [Google Scholar] [CrossRef]
- Di Chiara, T.; Del Cuore, A.; Daidone, M.; Scaglione, S.; Norrito, R.L.; Puleo, M.G.; Scaglione, R.; Pinto, A.; Tuttolomondo, A. Pathogenetic Mechanisms of Hypertension-Brain-Induced Complications: Focus on Molecular Mediators. Int. J. Mol. Sci. 2022, 23, 2445. [Google Scholar] [CrossRef]
- Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Hua, Y.; Xi, G.; Wang, M.M.; Keep, R.F. Endothelial Targets in Stroke: Translating Animal Models to Human. Arter. Thromb. Vasc. Biol. 2019, 39, 2240–2247. [Google Scholar] [CrossRef]
- Hou, L.; Li, M.; Wang, J.; Li, Y.; Zheng, Q.; Zhang, L.; Yao, Q.; Zhang, J.; Dong, S.; Zhou, M.; et al. Association between physical exercise and stroke recurrence among first-ever ischemic stroke survivors. Sci. Rep. 2021, 11, 13372. [Google Scholar] [CrossRef]
- Howard, V.J.; McDonnell, M.N. Physical activity in primary stroke prevention: Just do it! Stroke 2015, 46, 1735–1739. [Google Scholar] [CrossRef]
- Edward, J.A.; Cornwell III, W.K. Impact of Exercise on Cerebrovascular Physiology and Risk of Stroke. Stroke 2022, 53, 2404–2410. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Exercise is a double-edged sword for endothelial function. Hypertens. Res. 2016, 39, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, J.; Liu, H.; Chen, Y.; Ma, X.; Chen, S.; Chen, Y.; Bihl, J.; Yang, Y. Moderate Exercise Enhances Endothelial Progenitor Cell Exosomes Release and Function. Med. Sci. Sports Exerc. 2018, 50, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, H.; Chen, S.; Zhang, W.; Chen, Y.; Yang, Y. Moderate exercise has beneficial effects on mouse ischemic stroke by enhancing the functions of circulating endothelial progenitor cell-derived exosomes. Exp. Neurol. 2020, 330, 113325. [Google Scholar] [CrossRef]
- Yang, J.S.; Lee, J.C.; Byeon, S.K.; Rha, K.H.; Moon, M.H. Size Dependent Lipidomic Analysis of Urinary Exosomes from Patients with Prostate Cancer by Flow Field-Flow Fractionation and Nanoflow Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chem. 2017, 89, 2488–2496. [Google Scholar] [CrossRef]
- Sigdel, S.; Swenson, S.; Wang, J. Extracellular Vesicles in Neurodegenerative Diseases: An Update. Int. J. Mol. Sci. 2023, 24, 13161. [Google Scholar] [CrossRef]
- Nopp, S.; van der Bent, M.L.; Kraemmer, D.; Königsbrügge, O.; Wojta, J.; Pabinger, I.; Ay, C.; Nossent, A.Y. Circulatory miR-411-5p as a Novel Prognostic Biomarker for Major Adverse Cardiovascular Events in Patients with Atrial Fibrillation. Int. J. Mol. Sci. 2023, 24, 3861. [Google Scholar] [CrossRef]
- Bei, Y.; Xu, T.; Lv, D.; Yu, P.; Xu, J.; Che, L.; Das, A.; Tigges, J.; Toxavidis, V.; Ghiran, I.; et al. Exercise-induced circulating extracellular vesicles protect against cardiac ischemia-reperfusion injury. Basic Res. Cardiol. 2017, 112, 38. [Google Scholar] [CrossRef]
- Hou, Z.; Qin, X.; Hu, Y.; Zhang, X.; Li, G.; Wu, J.; Li, J.; Sha, J.; Chen, J.; Xia, J.; et al. Longterm Exercise-Derived Exosomal miR-342-5p: A Novel Exerkine for Cardioprotection. Circ. Res. 2019, 124, 1386–1400. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Kalani, A.; Medina, I.; Familtseva, A.; Tyagi, S.C. Cardiosome mediated regulation of MMP9 in diabetic heart: Role of mir29b and mir455 in exercise. J. Cell Mol. Med. 2015, 19, 2153–2161. [Google Scholar] [CrossRef]
- Conkright, W.R.; Beckner, M.E.; Sterczala, A.J.; Mi, Q.; Lovalekar, M.; Sahu, A.; Krajewski, K.T.; Martin, B.J.; Flanagan, S.D.; Greeves, J.P.; et al. Resistance exercise differentially alters extracellular vesicle size and subpopulation characteristics in healthy men and women: An observational cohort study. Physiol. Genom. 2022, 54, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Eguchi, A.; Tamai, Y.; Fukuda, S.; Tempaku, M.; Izuoka, K.; Iwasa, M.; Takei, Y.; Togashi, K. Protein Composition of Circulating Extracellular Vesicles Immediately Changed by Particular Short Time of High-Intensity Interval Training Exercise. Front. Physiol. 2021, 12, 693007. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, R.; Yang, Y.; Jacobs, B.; Chen, S.; Iwuchukwu, I.; Gaines, K.J.; Chen, Y.; Simman, R.; Lv, G.; et al. The Novel Methods for Analysis of Exosomes Released from Endothelial Cells and Endothelial Progenitor Cells. Stem. Cells Int. 2016, 2016, 2639728. [Google Scholar] [CrossRef] [PubMed]
- Kwok, Z.H.; Wang, C.; Jin, Y. Extracellular Vesicle Transportation and Uptake by Recipient Cells: A Critical Process to Regulate Human Diseases. Processes 2021, 9, 273. [Google Scholar] [CrossRef]
- Swanson, J.A.; Watts, C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. [Google Scholar] [CrossRef]
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef]
- Morales-Ruiz, M.; Fulton, D.; Sowa, G.; Languino, L.R.; Fujio, Y.; Walsh, K.; Sessa, W.C. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ. Res. 2000, 86, 892–896. [Google Scholar] [CrossRef]
- Satoh, M.; Ohkubo, T.; Asayama, K.; Murakami, Y.; Sugiyama, D.; Yamada, M.; Saitoh, S.; Sakata, K.; Irie, F.; Sairenchi, T.; et al. Lifetime Risk of Stroke and Coronary Heart Disease Deaths According to Blood Pressure Level: EPOCH-JAPAN (Evidence for Cardiovascular Prevention From Observational Cohorts in Japan). Hypertension 2019, 73, 52–59. [Google Scholar] [CrossRef]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.-B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar]
- Fu, S.; Zhang, Y.; Li, Y.; Luo, L.; Zhao, Y.; Yao, Y. Extracellular vesicles in cardiovascular diseases. Cell Death Discov. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Buffolo, F.; Monticone, S.; Camussi, G.; Aikawa, E. Role of Extracellular Vesicles in the Pathogenesis of Vascular Damage. Hypertension 2022, 79, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeis, C.; Helmig, S.; Tug, S.; Simon, P.; Kramer-Albers, E.M. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J. Extracell. Vesicles 2015, 4, 28239. [Google Scholar] [CrossRef]
- Whitham, M.; Parker, B.L.; Friedrichsen, M.; Hingst, J.R.; Hjorth, M.; Hughes, W.E.; Egan, C.L.; Cron, L.; Watt, K.I.; Kuchel, R.P.; et al. Extracellular Vesicles Provide a Means for Tissue Crosstalk during Exercise. Cell Metab. 2018, 27, 237–251e4. [Google Scholar] [CrossRef]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108. [Google Scholar] [CrossRef]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef]
- Ajikumar, A.; Long, M.B.; Heath, P.R.; Wharton, S.B.; Ince, P.G.; Ridger, V.C.; Simpson, J.E. Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. Int. J. Mol. Sci. 2019, 20, 5227. [Google Scholar] [CrossRef]
- Evans, C.E.; Iruela-Arispe, M.L.; Zhao, Y.Y. Mechanisms of Endothelial Regeneration and Vascular Repair and Their Application to Regenerative Medicine. Am. J. Pathol. 2021, 191, 52–65. [Google Scholar] [CrossRef]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006, 34, 249–270. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Yang, Y.; Xiao, X.; Chen, S.; Zhang, C.; Jacobs, B.; Bin Zhao, B.; Bihl, J.; Chen, Y. Endothelial progenitor cells and neural progenitor cells synergistically protect cerebral endothelial cells from Hypoxia/reoxygenation-induced injury via activating the PI3K/Akt pathway. Mol. Brain 2016, 9, 12. [Google Scholar] [CrossRef]
- Tian, Y.; Zhao, L.; Gui, Z.; Liu, S.; Liu, C.; Yu, T.; Zhang, L. PI3K/AKT signaling activates HIF1alpha to modulate the biological effects of invasive breast cancer with microcalcification. NPJ Breast Cancer 2023, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Lee, M.Y.; Ryu, J.M.; Song, C.H.; Han, H.J. Role of HIF-1alpha and VEGF in human mesenchymal stem cell proliferation by 17beta-estradiol: Involvement of PKC, PI3K/Akt, and MAPKs. Am. J. Physiol. Cell Physiol. 2009, 296, C317–C326. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular Noxs, and hypertension: Focus on translational and clinical research. Antioxid. Redox Signal. 2014, 20, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L.; Touyz, R.M. From bedside to bench to bedside: Role of renin-angiotensin-aldosterone system in remodeling of resistance arteries in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H435–H446. [Google Scholar] [CrossRef]
- Yao, J.; Zheng, J.; Cai, J.; Zeng, K.; Zhou, C.; Zhang, J.; Li, S.; Li, H.; Chen, L.; He, L.; et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate rat hepatic ischemia-reperfusion injury by suppressing oxidative stress and neutrophil inflammatory response. FASEB J. 2019, 33, 1695–1710. [Google Scholar] [CrossRef]
- Nie, Y.; Sato, Y.; Garner, R.T.; Kargl, C.; Wang, C.; Kuang, S.; Gilpin, C.J.; Gavin, T.P. Skeletal muscle-derived exosomes regulate endothelial cell functions via reactive oxygen species-activated nuclear factor-kappaB signalling. Exp. Physiol. 2019, 104, 1262–1273. [Google Scholar] [CrossRef]
- Kisielewska, M.; Rakoczy, K.; Skowron, I.; Górczyńska, J.; Kacer, J.; Bocheńska, A.; Choromańska, A. Utilizing Extracellular Vesicles for Eliminating ’Unwanted Molecules’: Harnessing Nature’s Structures in Modern Therapeutic Strategies. Molecules 2024, 29, 948. [Google Scholar] [CrossRef]
- Chen, S.; Sigdel, S.; Sawant, H.; Bihl, J.; Wang, J. Exercise-Intervened Endothelial Progenitor Cell Exosomes Protect N2a Cells by Improving Mitochondrial Function. Int. J. Mol. Sci. 2024, 25, 1148. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigdel, S.; Chen, S.; Udoh, G.; Wang, J. Exercise-Intervened Circulating Extracellular Vesicles Alleviate Oxidative Stress in Cerebral Microvascular Endothelial Cells Under Hypertensive Plus Hypoxic Conditions. Antioxidants 2025, 14, 77. https://doi.org/10.3390/antiox14010077
Sigdel S, Chen S, Udoh G, Wang J. Exercise-Intervened Circulating Extracellular Vesicles Alleviate Oxidative Stress in Cerebral Microvascular Endothelial Cells Under Hypertensive Plus Hypoxic Conditions. Antioxidants. 2025; 14(1):77. https://doi.org/10.3390/antiox14010077
Chicago/Turabian StyleSigdel, Smara, Shuzhen Chen, Gideon Udoh, and Jinju Wang. 2025. "Exercise-Intervened Circulating Extracellular Vesicles Alleviate Oxidative Stress in Cerebral Microvascular Endothelial Cells Under Hypertensive Plus Hypoxic Conditions" Antioxidants 14, no. 1: 77. https://doi.org/10.3390/antiox14010077
APA StyleSigdel, S., Chen, S., Udoh, G., & Wang, J. (2025). Exercise-Intervened Circulating Extracellular Vesicles Alleviate Oxidative Stress in Cerebral Microvascular Endothelial Cells Under Hypertensive Plus Hypoxic Conditions. Antioxidants, 14(1), 77. https://doi.org/10.3390/antiox14010077