1. Introduction
Vascular dementia (VaD), traditionally recognized as the second most common type of dementia after Alzheimer’s disease (AD), has recently been suggested to be the most prevalent form of dementia among the elderly [
1,
2,
3]. As the global population ages rapidly, the number of VaD cases is expected to rise, placing additional pressure on healthcare systems. Studies estimate that VaD accounts for approximately 15–20% of all dementia cases, and this percentage may be higher in regions with elevated rates of hypertension, diabetes, and stroke [
4]. Furthermore, nearly 50% of stroke survivors may develop VaD-related symptoms within 25 years [
5]. Additionally, cerebrovascular disease (CVD), the underlying condition of VaD, is the second leading cause of death worldwide and significantly contributes to cognitive decline and disability across populations [
6].
VaD significantly impairs cognitive functions, including memory, executive function, language, and attention, thereby severely diminishing patients’ quality of life. Furthermore, VaD is associated with high mortality rates. Compared to AD, VaD is linked to a shorter life expectancy, with an average of 3.9 years from dementia onset to death, versus 7.1 years for AD [
7]. Additionally, the chronic nature of VaD exacerbates mortality rates, especially when compounded by other CVDs, such as stroke [
8].
From a societal perspective, the rise in VaD cases imposes substantial economic burdens on society. Health statistics indicate that VaD patients incur the highest annual costs among all dementia patients, including those with CVDs, as observed in New York, USA [
9]. Direct costs include expenses for long-term care facilities and nursing homes, while indirect costs arise from reduced productivity, as patients and their caregivers may leave the workforce. As VaD progresses, individuals often require long-term care, increasing their dependence and placing considerable stress on caregivers. These financial pressures impact individuals, families, and national healthcare systems, with dementia-related expenses expected to escalate in the coming decades [
7,
10,
11].
Despite its significant impact, treatment options for VaD are limited. Managing vascular risk factors like hypertension, dyslipidemia, and diabetes can help slow the disease’s progression, but there is no established cure or long-term therapy that can fully stop or reverse cognitive decline [
12,
13]. Current medications, such as acetylcholinesterase inhibitors, provide only temporary symptom relief. Antiplatelet agents may help to prevent further cerebrovascular events, but no drug has been proven to significantly improve symptoms once the disease has advanced [
14,
15,
16,
17]. Therefore, treatments for VaD have been mainly focused on controlling stroke risk factors.
Recently, molecular hydrogen (H
2) has emerged as a promising therapeutic agent [
18,
19]. Studies have shown that H
2 has anti-oxidative, anti-apoptotic, and anti-inflammatory effects in various conditions, including cancer, lung damage, and neurodegenerative diseases, by neutralizing ROS, which cause damage to DNA, proteins, and cells [
20,
21,
22]. Given the significant role of ROS in exacerbating neurological damage, H
2 presents a potential strategy for mitigating oxidative stress in dementia-related conditions. Regarding oxidative stress in disease, a VaD differs in its etiology from AD, as it occurs due to neurovascular dysfunction that induces ischemia or reperfusion, which can greatly increase the ROS that harm tissue. In contrast, AD primarily occurs through the accumulation of amyloid beta plaques. Therefore, managing the excessive accumulation of ROS in the brains affected by VaD constitutes a more critical challenge compared to AD [
1,
2,
3].
In detail, H
2 can exert its therapeutic effects on VaD through multiple cellular interactions at the molecular level. Primarily, H
2 acts as a selective antioxidant, neutralizing harmful reactive oxygen species such as hydrogen peroxide (H
2O
2) and peroxynitrite (ONOO
−), thereby reducing oxidative stress and preventing neuronal damage [
23]. It also modulates inflammatory responses by suppressing pro-inflammatory cytokines and inhibiting transcription factors like NF-κB, which helps mitigate neural inflammation associated with VaD [
20,
22]. H
2 further enhances the cell’s antioxidant defenses by activating the Nrf2 pathway, leading to increased expression of antioxidant enzymes [
24]
Although hydrogen shows great potential, its effects on mammals, especially regarding immune-related mechanisms in neurodegenerative diseases, are not fully understood. Since neurodegenerative diseases often involve inflammation, understanding hydrogen’s role in immune modulation is crucial. Regarding this, we investigated the anti-oxidative effects of H
2 in a mouse model of cerebral ischemia and reperfusion injury, demonstrating its ability to remove free radicals in our previous study published in 2021. We observed changes in cytokine levels, such as reduced IL-6 and increased IL-10 and IL-2, suggesting that T cells play a role in this process [
18,
22].
Building on our previous findings, the present study aims to elucidate the therapeutic potential and underlying mechanisms of H
2 in treating VaD, with a specific focus on inflammation-related pathways. Given that VaD is characterized by chronic cerebrovascular insufficiency leading to neuronal damage, cognitive decline, and persistent neuroinflammation, effective treatment strategies must address both oxidative stress and immune dysregulation [
25,
26]. This study evaluates the impact of H
2 on key inflammation-related factors involved in neuronal regeneration and regulatory T cell (Tregs) pathways using reliable techniques such as flow cytometry and polymerase chain reaction (PCR) analyses. We specifically measured alterations in cytokine levels, Treg populations, and the expression of genes associated with neuroprotection and antioxidant responses. Additionally, we assessed H
2’s ability to reduce ROS accumulation. By providing comprehensive evidence of H
2’s multifaceted role in mitigating oxidative stress and modulating immune responses, this research underscores the potential of H
2 as a novel therapeutic agent for slowing the progression of vascular dementia and improving cognitive and neuronal health in affected individuals.
2. Materials and Methods
2.1. Animals
This study was approved by the Institutional Animal Care and Use Committee (IACUC) at the Korea University Medical Center (approval no. KOREA-2021-0176). We conducted a study using a male 30 g 12-week-old C57BL/6 mouse (DBL, Eumseong, Republic of Korea). Animals were group-housed with no more than 4 animals per cage and acclimatized to standard laboratory conditions on a 12 h light/dark cycle. Food and water were provided ad libitum.
2.2. Experimental Design
We conducted a study designed to establish the effect of hydrogen-rich water (HRW) on a mouse model of VaD and to analyze the mechanism of action of H2. Surgery to make a VaD model was performed on 33 mice and half of them were randomly selected and treated with hydrogen. Thus, the group was divided into an untreated VaD group (VaD; n = 15) and a hydrogen-treated VaD group (VaD + H2; n = 18), and this was compared with the Sham control group (n = 15). HRW was generated by the HRW machine (H2B-H20, Hommage, Yongchang, China). Hydrogen concentration in HRW was maintained above 1.000 ppm by refreshing it every morning until the end of the experiment. HRW was provided only to the mice of the H2 group instead of regular water and they were allowed to drink it ad libitum. In contrast, the Sham and VaD group mice were provided with regular water.
2.3. Bilateral Common Carotid Artery Stenosis (BCCAS) Operation
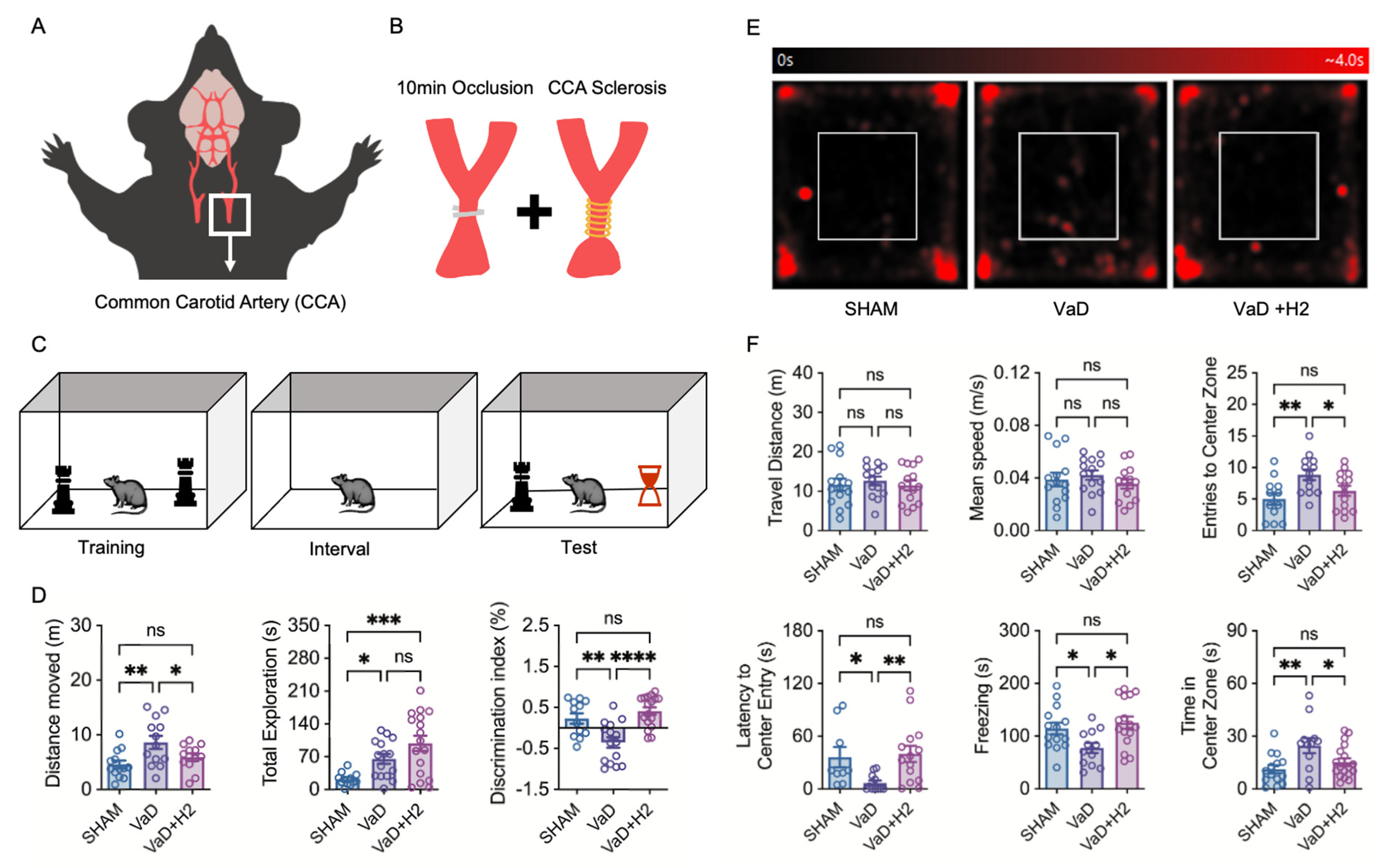
Anesthesia was induced in mice through inhalation of 5% isoflurane in 1 L of oxygen using an animal anesthesia machine (L-PAS-01D, LMS Korea, Pyeongtaek, Republic of Korea). Full anesthesia was achieved within 3 min. During the surgical procedure, anesthesia was maintained with 3% isoflurane in 600 mL of oxygen. A vertical midline incision was made along the ventral neck of each mouse, and the soft tissues and muscles were carefully dissected to sequentially isolate both common carotid arteries (CCAs). Both CCAs were occluded for 10 min using bulldog clips. To mitigate the risk of mortality due to reperfusion injury, the clips were removed at 3 min intervals. Subsequently, a 0.16 mm coil (SWPA 0.16, Sawane SPRING Co., Ltd., Hamamatsu, Japan) was wrapped around each common carotid artery to induce stenosis. After suturing the incision, the mice were housed individually until consciousness was restored, after which they were returned to their original cages. All procedures were completed within 30 min. This model was designed to mimic the conditions of VaD, characterized by reperfusion-induced damage and continuous cerebral hypoperfusion, by employing an occlusion method aligned with Bilateral CCA stenosis (BCCAS) operational protocols.
2.4. Open Field Test (OFT)
For cognitive function tests, mice were translocated to a dimly lit test room 30 min before testing and the testing chamber was sanitized with 70% ethanol. In the open field test (OFT), we placed the mouse in the center of the chamber (acryl, 50 × 50 × 30 cm3) and allowed it to move freely for 5 min. Recording was started 2 min after leaving the mouse in the chamber and the movement pattern was analyzed using ANY-maze software Version 7.48 (Stoelting, Wood Dale, IL, USA).
2.5. Novel Object Recognition Test (NORT)
In the novel object recognition test (NORT), mice were individually placed in an empty acrylic chamber (33 × 20 × 30 cm3) for habituation over 3 consecutive days, with each habituation session lasting 5 min. On the test day, each mouse was placed in the chamber containing two identical objects positioned in opposite corners and allowed to explore for 5 min. After a 2 h interval, one of the two objects was replaced with a novel object, and the mouse was returned to the chamber for a second 5 min exploration period. Using this approach, we assessed the mice’s ability to recognize familiar objects and their propensity to explore novel objects compared to familiar ones. The mice’s movements were recorded and analyzed using ANY-maze software.
2.6. H&E
Mouse brain tissue was sectioned to a thickness of 6 μm. Brain tissue slices were stained with hematoxylin and eosin (H&E). The slices were mounted on cover glasses using a quick hardening mounting medium (03989, Sigma-Aldrich, St. Louis, MO, USA). The slides were digitized using a Pannoramic Digital Slide Scanner (3DHISTECH Ltd., Budapest, Hungary).
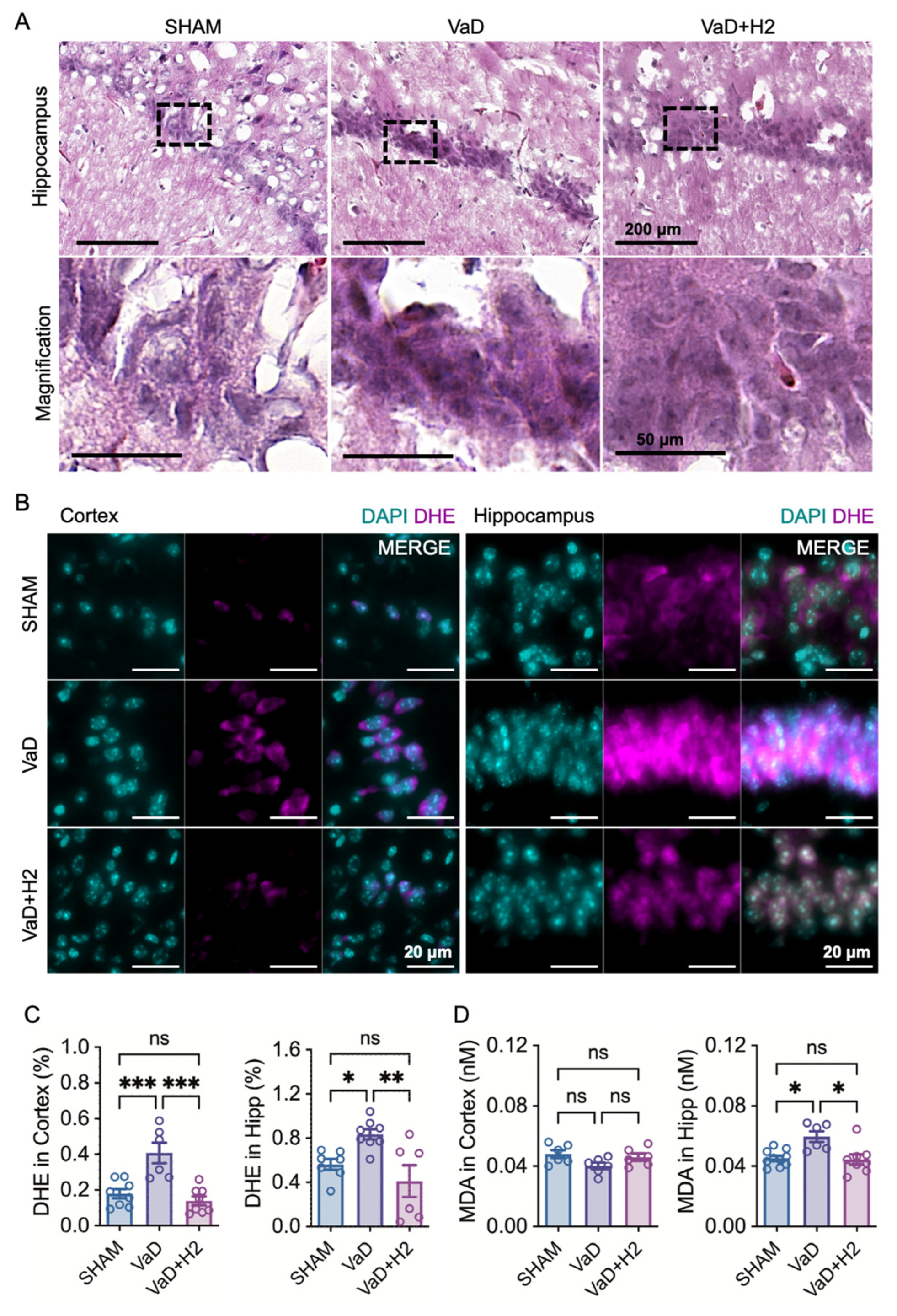
2.7. DHE Assay
Brain tissue slices (6 μm) were incubated with 10 μM dihydroethidium (D7008, Sigma-Aldrich, St. Louis, MO, USA) for 1 h in the dark at room temperature. The slices were washed three times with 1X PBS, with each wash lasting 5 min. Cover glasses were mounted onto slides containing the brain tissue slices using an antifade mounting medium containing DAPI (H-1200-10, Vector Laboratories, Burlingame, CA, USA). The tissue slices were observed under a fluorescence microscope (Axiovert 200M, Carl Zeiss Meditec, Dublin, CA, USA). The DHE intensity relative to DAPI intensity was calculated for each region of interest (ROI) using ImageJ software 1.53a (National Institutes of Health, Bethesda, MD, USA).
2.8. Malondialdehyde (MDA) Assay
Mouse hippocampal brain tissues were harvested and homogenized in 1 mL of cold assay buffer from the TBARS Assay Kit (STA-330, Cell Biolabs, Inc., San Diego, CA, USA). The homogenized samples were then centrifuged at 10,000× g for 10 min at 4 °C. The resulting supernatant was transferred to fresh 1.5 mL microcentrifuge tubes and butylated hydroxytoluene (BHT) was added from a 100X stock to achieve a final concentration of 1X in each sample. Malondialdehyde (MDA) standards were prepared by serially diluting MDA in double-distilled water to concentrations ranging from 0 μM to 120 μM. Next, 100 µL of either the MDA standards or the samples were placed into separate microcentrifuge tubes, followed by the addition of 100 µL of SDS lysis solution to each tube. After thorough mixing, the tubes were incubated at room temperature for 5 min. Subsequently, 250 µL of TBA reagent was added to each tube and mixed well. The tubes were then incubated at 95 °C for 60 min before being rapidly cooled on ice for 5 min. Following incubation, the samples were centrifuged at 3000 rpm for 15 min at room temperature, and the clear supernatants were carefully transferred to new microcentrifuge tubes. For spectrophotometric measurements, 200 µL of each standard and sample was aliquoted into the wells of a 96-well plate and the absorbance was read at 532 nm.
2.9. Immunofluorescence Microscopy
Immediately after sacrifice, mouse brains were fixed by immersion in 4% paraformaldehyde (PFA) for 48 h at 4 °C. The fixed brain specimens were then thoroughly washed multiple times with 1X PBS and cryoprotected by soaking in a 50% sucrose buffer for 48 h. Frozen brains were subsequently sectioned into 6 μm thick slices and blocked using a 5% bovine serum albumin (BSA) solution diluted in PBS. Each section was incubated with a primary antibody against BCL2 (ab59348, Abcam, Cambridge, UK) or IL-4 (SC-53984, Santa Cruz Biotechnology Inc., Paso Robles, CA, USA) diluted in the 2.5% BSA solution for 1 h at room temperature. Following primary antibody incubation, sections were treated with a secondary antibody conjugated to Alexa Fluor 568 goat anti-rabbit IgG (F0257, Sigma-Aldrich, St. Louis, MO, USA) for another 1 h at room temperature. After antibody labeling, a mounting medium containing DAPI was applied to each section, which was then covered with a cover glass. Immunolabeled proteins were visualized and analyzed using a fluorescence microscope.
2.10. Flow Cytometry
For flow cytometry analysis of the Treg population, a Foxp3 staining kit (560133, BD Biosciences, San Jose, CA, USA) was utilized. Mouse brain cells were isolated and resuspended in FACS buffer. Cells were stained for surface markers CD4 and CD25 using fluorescently labeled antibodies. Following surface staining, cells were fixed and permeabilized with the BD Transcription Factor Staining Buffer Set for intracellular Foxp3 detection. Intracellular staining was then performed using an anti-Foxp3 antibody. Stained cells were analyzed on a BD FACSCalibur flow cytometer. CD4+Foxp3+ Tregs were quantified within the CD4+ population.
2.11. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Mouse hippocampal brain tissue cells were lysed and homogenized in 1 mL Trizol reagent (T9424, Sigma-Aldrich) in Eppendorf microcentrifuge tubes for 5 min. Subsequently, 0.2 mL chloroform (366927, Sigma-Aldrich) was added to the sample, thoroughly mixed, and incubated for 2 min at room temperature. The sample was centrifuged for 15 min at 12,000× g and 4 °C. The colorless upper aqueous phase was transferred to a new tube and 0.5 mL isopropanol (278475, Sigma-Aldrich) was added to the sample and incubated for 10 min at 4 °C. The sample was centrifuged for 10 min at 12,000× g at 4 °C. The supernatant was discarded and the pellet was resuspended with 1 mL 75% ethanol. The mixture was vortexed and centrifuged for 5 min at 7500× g at 4 °C. The supernatant was discarded. The RNA pellet was air-dried and resuspended in 50 μL RNase-free water (W2004, Biosesang, Seoul, Republic of Korea). The isolated RNA was reverse-transcribed to cDNA using the High-Capacity RNA-to-cDNA Kit (4387406, Thermo Fisher Scientific, Waltham, MA, USA). cDNA was mixed with Accutarget qPCR Screening Kit primers (SM-0005, SM-0087, SM-0130, Bioneer, Seoul, Republic of Korea) and SYBR™ Green Universal Master Mix (4309155, Thermo Fisher Scientific) and amplified in a two-step process with a 55 °C annealing temperature using the QuantStudio 5 Real-Time PCR System (A34322, Applied Biosystems, Foster City, CA, USA).
2.12. Statistical Analysis
All results are expressed as mean ± standard error of the mean (SEM). Outliers were excluded by conducting an outlier assessment per every analysis. To compare differences among the three groups, a one-way analysis of variance (ANOVA) was performed, followed by post hoc analysis. Statistical analyses were conducted using Prism 9 software (GraphPad Software, San Diego, CA, USA). A p-value of <0.05 was considered statistically significant.
4. Discussion
The present study demonstrates that H
2 treatment effectively restores cognitive and emotional functions in a VaD mouse model induced by BCCAS. Various methods exist for creating VaD models, each with unique strengths and limitations. For instance, the multiple infarct and thromboembolism model, involving the injection of micro-emboli into the internal carotid artery, is considered clinically relevant but fails to sustain long-term deficits, making it unsuitable for this study, which aimed to evaluate treatment responses over an extended period [
27].
Another model, unilateral CCA occlusion, results in only mild cerebral blood flow reduction, producing weak symptoms and limited pathology, and was therefore deemed inappropriate for assessing the efficacy of our treatment [
28,
29]. In contrast, the BCCAS model predominantly induces white matter lesions in the corpus callosum and enhances the inflammatory response [
30]. Although this model can sometimes lead to only mild cerebral blood flow reduction, we addressed this limitation by using a smaller diameter coil (0.16 mm) instead of the standard 0.18 mm coil. This modification enabled the establishment of a VaD model that exhibited measurable anxiety-impaired behavior and memory deficits, as demonstrated through cognitive behavioral tests (
Figure 1F). These findings parallel clinical observations in human VaD patients, who experience a gradual reduction in anxiety during the advanced stages of the disease and exhibit significant dementia-related impairments in learning and memory following a stroke [
31,
32].
Our current model also represents an improvement over our previous approach. Previously, we induced ischemia–reperfusion injury by temporarily clamping both CCAs and then releasing them after a set duration [
22]. While this method generated degenerative changes in the brains of mice, it produced a singular, transient injury event. In the current study, we established permanent stenosis of the CCAs, thus creating a sustained hypoperfusion environment. This model more closely mimics the chronic hypoperfusion observed in patients with atherosclerotic internal carotid artery stenosis. Although the outcomes appear to be like those of the previous model, the underlying principle differs significantly. Here, we demonstrated the effectiveness of H
2 in delaying cognitive decline in a persistent, rather than transient, environment.
To assess the effect of H
2 treatment, we compared mice that drank H
2-rich water (HRW) with those that did not. Compared to the VaD group, the H
2-treated mice displayed anxiety and memory performances similar to those of the Sham group. This clinical improvement aligns with previous studies, suggesting that H
2 provides therapeutic benefits in VaD models [
22]. Results from the NORT revealed that VaD mice exhibited increased movement distance, potentially reflecting characteristic symptoms associated with dementia such as irritability [
33]. In contrast, H
2-treated mice demonstrated improved exploratory behavior and a stronger preference for the novel object, indicating a notable enhancement in learning and memory abilities due to H
2 treatment. Interestingly, this behavioral pattern differed from that of the Sham group, suggesting unique cognitive recovery dynamics in the H
2-treated mice (
Figure 1D).
Histological and biochemical analyses further revealed that H
2 reduces oxidative stress in the VaD brain. H&E staining showed reversal of pyknotic changes in hippocampal neurons, while DHE staining confirmed reduced ROS levels in both the cortex and hippocampus of H
2-treated mice (
Figure 2). Additionally, MDA assays demonstrated attenuated lipid peroxidation in the H
2-treated group. At the molecular level, H
2 treatment restored Gpx5 mRNA expression, a critical ROS scavenger, to near-Sham levels, thereby augmenting antioxidant defenses and minimizing oxidative stress-induced neuronal damage (
Figure 4A). These results are in line with previous work showing that H
2 can serve as a therapeutic agent by reducing ROS [
20,
21,
22,
34].
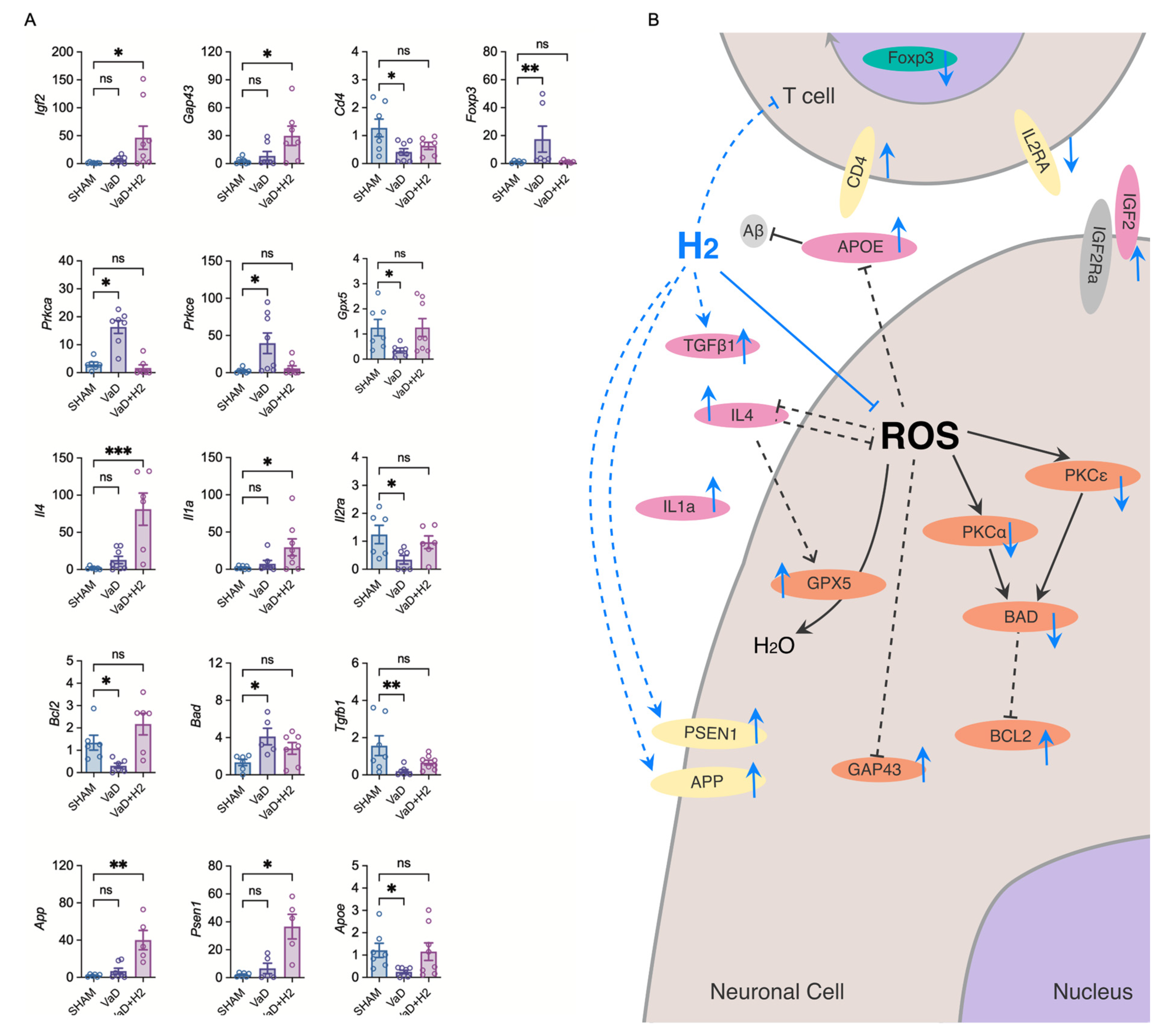
H
2 treatment also influenced apoptotic and survival signaling pathways (
Figure 4A). In VaD mice, the pro-apoptotic gene
Bad was upregulated and the anti-apoptotic gene
Bcl2 was downregulated, indicating heightened neuronal vulnerability. H
2 treatment restored
Bcl2 and suppressed
Bad, shifting the balance toward cell survival [
35].
Tgfb1, a cytokine that promotes neuronal survival, was also normalized by H
2 treatment, suggesting improved neuronal integrity and survival through the precise regulation of microglial activation [
36]. Additionally, H
2 enhanced the expression of neurogenesis and synaptic remodeling markers such as Igf2 and Gap43, suggesting improved synaptic plasticity and regeneration [
37]. It also upregulated App and
Psen1, associated with amyloid regulation, and restored
Apoe expression, a key factor in neuronal repair [
38,
39,
40,
41,
42]. Altogether, these changes suggest that H
2 supports structural and functional recovery and may impact pathways relevant to Alzheimer’s disease.
Immunologically, H
2 promoted an anti-inflammatory environment. Anti-inflammatory cytokines
IL-4 and
IL-1a were significantly elevated and
Il2ra (encoding CD25) expression was restored, indicating an improved immunological balance. Flow cytometry revealed that H
2 normalized CD4
+ T cell populations and reduced the proportion of Tregs (CD4
+Foxp3
+) elevated in VaD mice. Thus, H
2 modulates immune responses, maintaining homeostasis and limiting excessive inflammation. The reduction in activated T cells is indicative of a non-toxic brain environment marked by cellular equilibrium [
43]. Accordingly, the flow cytometry results likely represent a low inflammatory state in the brain, eliminating the need for toxic mechanisms to remove cells (
Figure 3E,F).
A schematic diagram (
Figure 4B) illustrates the hypothesized mechanisms of H
2 action in VaD cases. Under ROS-excessive conditions, GPX5 scavenges ROS, and apoptotic markers like BAD and BCL2 mediate neuronal fate. PKCα and PKCε regulate these apoptotic processes, while IL-4, GAP43, and APOE expression is diminished under oxidative stress. H
2 treatment is thought to counter these processes, restoring GPX5, BCL2, GAP43, PSEN1, and APP, while inhibiting BAD, PKCα, and PKCε. H
2 also potentially promotes IGF2 for neurogenesis and synaptic plasticity and elevates anti-inflammatory cytokines such as TGFβ1, IL-4, and IL-1a. Together, these adjustments improve cognitive and emotional functions by restoring both neuronal and immunological homeostasis.
Nonetheless, our approach presents several limitations. The administration of H
2 poses significant challenges. Although intraperitoneal injection allows for precise dosage control, it can induce stress in animals, potentially confounding behavioral outcomes. While H
2 gas inhalation serves as an alternative, its technical complexity led us to opt for oral administration via HRW. However, despite consistently observing a significant reduction in hippocampal ROS levels, the standard error of the mean in the DHE assays indicates variability among individual mice, likely attributable to irregular oral intake (
Figure 2C,D). While this method simplifies treatment delivery, it complicates the accurate quantification of individual intake. Future studies could benefit from indirect analytical techniques, such as measuring MDA concentrations in blood, to assess H
2 consumption per animal and determine its influence on treatment efficacy.
Furthermore, our VaD model exhibits certain limitations. Although we have advanced the design to overcome previous challenges in mouse model development, this model results in high inter-animal variance due to inherent vascular conditions, rendering some animals unsuitable for use as VaD models and introducing outliers. Additionally, the coil insertion procedure is technically demanding, requiring highly skilled personnel to perform the surgery effectively. Moreover, the gold micro-coil material is not fully biodegradable, raising concerns about potential long-term effects on the mice’s physiology while the coil remains in their bodies. Further considerations of the methodology for establishing the model are required for future advanced studies.
Additionally, as MDA and DHE levels measured in the cortex vary depending on the assay method used, careful selection of ROS measurement techniques is crucial to ensure accurate and reliable results. To address this issue, we plan to implement multiple ROS measurement methods to achieve more accurate and reliable quantification of ROS levels in animals.
In conclusion, this study presents a refined VaD mouse model that more closely mimics chronic hypoperfusion and demonstrates the protective role of H2 treatment in cognitive and emotional functions. H2 attenuates oxidative stress, reduces apoptosis, enhances neurogenesis and synaptic plasticity, and maintains immunological balance. Although further research is needed to optimize H2 dosing and elucidate its precise molecular mechanisms, our findings suggest that H2 may serve as a promising therapeutic approach for VaD.



{kind=link}
{kind=link}
{kind=link}
{kind=link}