
The Multifaceted Roles of NRF2 in Cancer: Friend or Foe?

 ,
,  , and
, and
Abstract
1. Introduction
2. The Transcription Factor NRF2
2.1. NRF Family Members
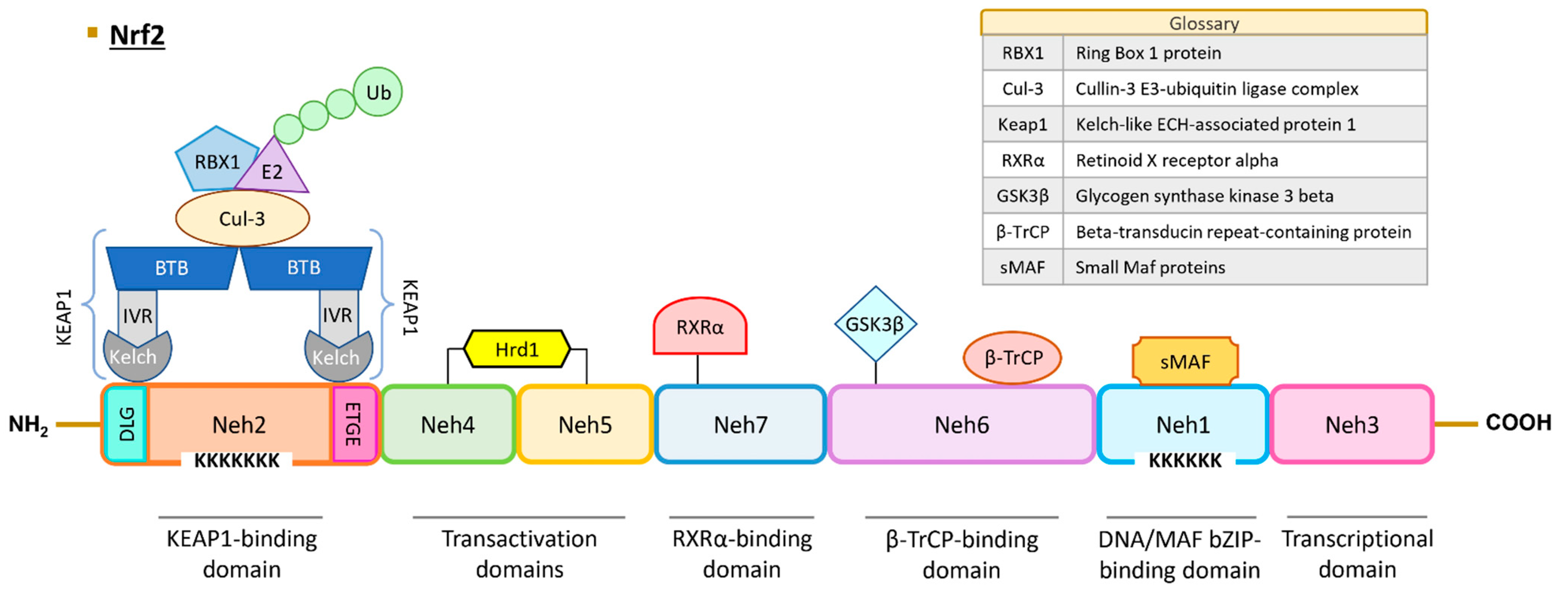
2.2. Domains and Interactions
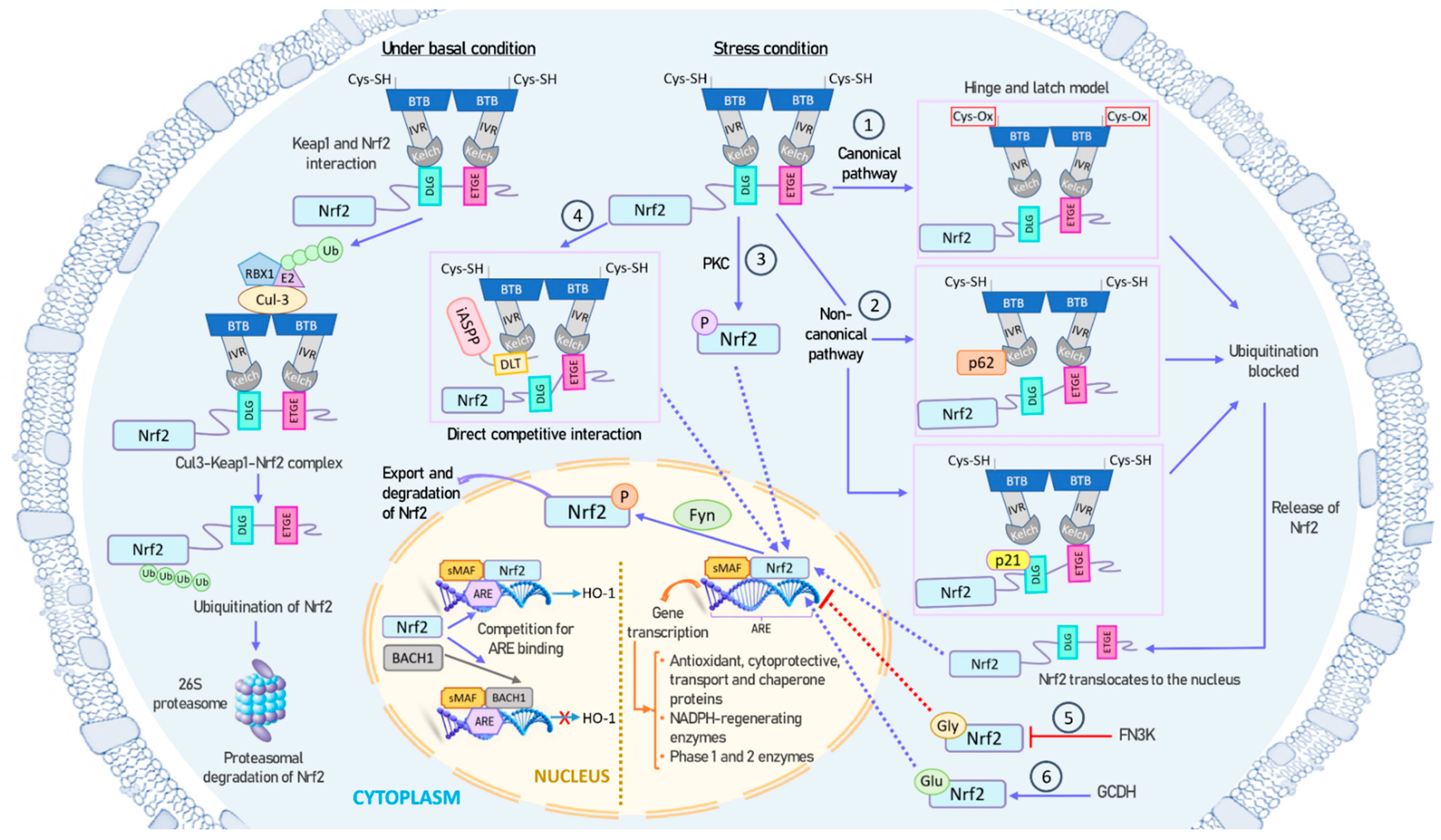
2.3. Mechanisms of NRF2 Activation and Inhibition
- (1)
- KEAP1 is the main repressor of NRF2 activation and is rich in redox-sensitive cysteine residues. Notably, Cys151, Cys273, and Cys288 in the KEAP1 protein are involved in the interaction with NRF2 [67]. During oxidative stress, these cysteine residues, functioning as ROS sensors, become oxidized, causing a structural alteration in KEAP1. In the canonical pathway, this oxidative change disrupts the hinge-and-latch complex and severs the connection with the NRF2 DLG pattern. Consequently, the NRF2–KEAP1 complex undergoes a conformational shift that prevents NRF2 ubiquitination [47,55].
- (2)
- Other proteins such as p21 or p62 can compete with KEAP1 for binding to the NRF2 DLG motif through the non-canonical pathway. This competitive binding results in a conformational change that hinders NRF2 degradation [68].
- (3)
- Different phosphorylation events involving NRF2 can lead to its dissociation from KEAP1. For instance, protein kinase C (PKC) induces phosphorylation of the Ser40 residue [69], which is situated in the Neh2 domain that interacts with KEAP1 [56]. This phosphorylation hinders the binding of NRF2 to KEAP1, preventing its sequestration by KEAP1.
- (4)
- The antioxidant iASPP competes with NRF2 for KEAP1 binding via a DLT motif and induces NRF2 activation [70].
- (5)
- NRF2 can undergo glycation, rendering it unstable and impairing its binding to small MAF proteins and transcriptional activation. Fructosamine-3-kinase (FN3K) can promote deglycation of the NRF2 protein [71].
- (6)
- NRF2 glutarylation, regulated by the mitochondrial protein glutaryl-CoA dehydrogenase (GCDH), enhances protein stability and transcriptional activity [72].
2.4. Genes Regulated by NRF2
- (1)
- Antioxidant proteins: NRF2 finely regulates redox homeostasis by controlling the expression of antioxidant enzymes and facilitating the production of glutathione (GSH). Some of these antioxidant proteins include biliverdin reductase B, ferritin, glutamate-cysteine ligase (GCL), superoxide dismutases (SODs), glutathione peroxidases (GPXs), peroxiredoxins (PRXs), and glutathione reductase (GR). Notably, GCL is a critical enzyme involved in the synthesis of the potent antioxidant GSH.
- (2)
- NADPH-regenerating enzymes: NRF2 plays a crucial role in governing metabolic reprogramming and the generation of NADPH, which is pivotal in cellular antioxidant systems. Key enzymes in this category include glucose-6-phosphate dehydrogenase (G6PD), malic enzyme 1 (ME1), and 6-phosphogluconate dehydrogenase (6PGD).
- (3)
- Cytoprotective proteins: NRF2 regulates important proteins like HO-1 and metallothioneins. HO-1 catalyzes the breakdown of heme, resulting in the production of various compounds, including biliverdin, carbon monoxide, and iron. The cytoprotective effect of HO-1 is mediated indirectly through the generation of biliverdin and the potent antioxidant bilirubin. Notably, HO-1 expression has been observed to be higher in NRF2 knockout K-rasG12V 293T cells compared to wild-type NRF2 cells, suggesting that these enzymes can also be regulated by other transcription factors and signaling pathways [80].
- (4)
- Phase 1 enzymes: Phase I metabolism involves the reduction, oxidation, or hydrolysis of molecules such as drugs or toxic compounds. NRF2 regulates a range of enzymes in this category, including alcohol dehydrogenases (ADHs), aldehyde dehydrogenases (ALDHs), cytochromes P450 (CYPs), NQO1, and carboxyl esterase (CES).
- (5)
- Phase 2 enzymes: NRF2-dependent conjugation reactions are crucial for the detoxification of various xenobiotics. These reactions are carried out by glutathione S-transferases (GSTs), sulfotransferases (SULTs), and UDP-glucuronosyl transferases (UGTs).
- (6)
- Transport proteins: NRF2 also regulates transport proteins such as multi-drug resistance-associated proteins (MRPs) and neutral amino acid transporters through ARE sequences in their promoters. MRPs play a role in drug resistance.
- (7)
- Chaperone proteins: Chaperone proteins are responsible for ensuring the proper three-dimensional folding of other proteins, thus facilitating their maturation. NRF2 regulates various chaperone proteins, including heat-shock proteins (HSPs).
- (8)
- Transcription factors: NRF2 regulates the expression of MAF proteins, as well as BACH1 and NRF2 itself, through the ARE sequences in their promoters.
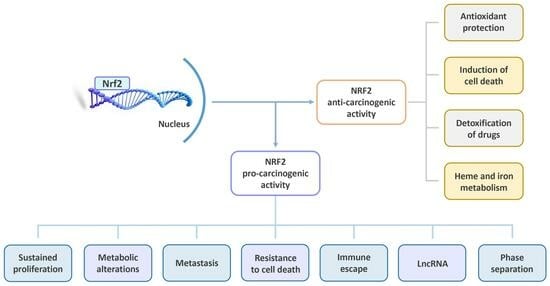
2.5. NRF2: A Double-Edged Sword
2.5.1. NRF2: The Bright Side
2.5.2. NRF2: The Dark Side
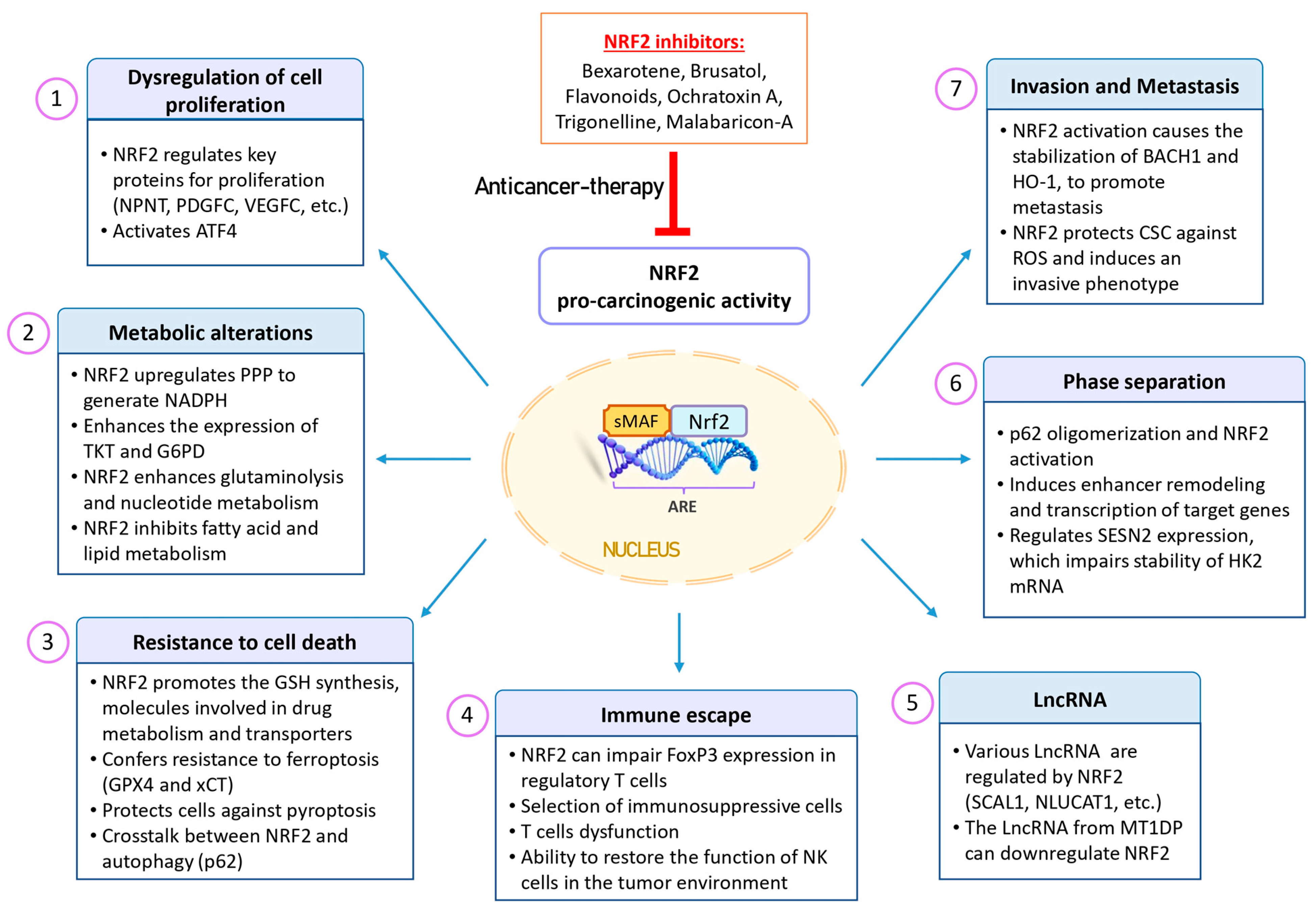
3. NRF2 in the Context of Cancer Promotion
3.1. Pro-Oncogenic Functions of the KEAP1–NRF2 Pathway
3.2. Role of NRF2 in the Dysregulation of Cell Proliferation
3.3. Role of NRF2 in Tumor Metabolism
3.4. Role of NRF2 in Cell Death
4. Role of NRF2 and ROS on Some Critical Cellular Processes
4.1. NRF2 and ROS in Tumor Immunology
4.2. Interplays between NRF2, ROS and LncRNAs in Cancer
4.3. Crosstalk between NRF2 Activation and Phase Separation
4.4. Role of NRF2 and BACH1 in Cancer Stem Cells and Metastasis
5. NRF2 in Cancer Prevention and Its Therapeutic Implications
5.1. NRF2 Activators
5.2. NRF2 Inhibitors
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- MacRosty, C.R.; Rivera, M.P. Lung Cancer in Women: A Modern Epidemic. Clin. Chest Med. 2020, 41, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Alexander, E.E.; Singh, R.; Shan, A.; Qian, J.; Santella, R.M.; Oberley, L.W.; Yan, T.; Zhong, W.; Jiang, X.; et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer 2000, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Sandoval, J.M.; Fattaccioli, A.; Dejeans, N.; Garbe, J.C.; Dieu, M.; Verrax, J.; Renard, P.; Huang, P.; Calderon, P.B. Chromatin remodeling regulates catalase expression during cancer cells adaptation to chronic oxidative stress. Free Radic. Biol. Med. 2016, 99, 436–450. [Google Scholar] [CrossRef]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metsabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agents Med. Chem. 2013, 13, 502–514. [Google Scholar] [PubMed]
- Dejeans, N.; Glorieux, C.; Guenin, S.; Beck, R.; Sid, B.; Rousseau, R.; Bisig, B.; Delvenne, P.; Buc Calderon, P.; Verrax, J. Overexpression of GRP94 in breast cancer cells resistant to oxidative stress promotes high levels of cancer cell proliferation and migration: Implications for tumor recurrence. Free Radic. Biol. Med. 2012, 52, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase down-regulation in cancer cells exposed to arsenic trioxide is involved in their increased sensitivity to a pro-oxidant treatment. Cancer Cell Int. 2018, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Pedrosa, R.C.; Beck, R.; Dejeans, N.; Taper, H.; Calderon, P.B. In situ modulation of oxidative stress: A novel and efficient strategy to kill cancer cells. Curr. Med. Chem. 2009, 16, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Stockis, J.; Tison, A.; Taper, H.S.; Calderon, P.B. Oxidative stress by ascorbate/menadione association kills K562 human chronic myelogenous leukaemia cells and inhibits its tumour growth in nude mice. Biochem. Pharmacol. 2006, 72, 671–680. [Google Scholar] [CrossRef]
- Glorieux, C.; Xia, X.; He, Y.Q.; Hu, Y.; Cremer, K.; Robert, A.; Liu, J.; Wang, F.; Ling, J.; Chiao, P.J.; et al. Regulation of PD-L1 expression in K-ras-driven cancers through ROS-mediated FGFR1 signaling. Redox Biol. 2021, 38, 101780. [Google Scholar] [CrossRef]
- He, M.; Wang, M.; Xu, T.; Zhang, M.; Dai, H.; Wang, C.; Ding, D.; Zhong, Z. Reactive oxygen species-powered cancer immunotherapy: Current status and challenges. J. Control. Release 2023, 356, 623–648. [Google Scholar] [CrossRef]
- Ralph, S.J.; Reynolds, M.J. Intratumoral pro-oxidants promote cancer immunotherapy by recruiting and reprogramming neutrophils to eliminate tumors. Cancer Immunol. Immunother. 2023, 72, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, N.; Jiang, H.; Li, Q.; Xing, D. Reactive Oxygen Species in Anticancer Immunity: A Double-Edged Sword. Front. Bioeng. Biotechnol. 2021, 9, 784612. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.S.; Kan, Y.W. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Kobayashi, A. Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers 2020, 12, 2681. [Google Scholar] [CrossRef]
- Chevillard, G.; Blank, V. NFE2L3 (NRF3): The Cinderella of the Cap’n’Collar transcription factors. Cell Mol. Life Sci. 2011, 68, 3337–3348. [Google Scholar] [CrossRef]
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.C.; Blank, V. Complexity of CNC transcription factors as revealed by gene targeting of the Nrf3 locus. Mol. Cell Biol. 2004, 24, 3286–3294. [Google Scholar] [CrossRef]
- Immonen, A.; Haapasaari, K.M.; Skarp, S.; Karihtala, P.; Teppo, H.R. NRF3 Decreases during Melanoma Carcinogenesis and Is an Independent Prognostic Marker in Melanoma. Oxid. Med. Cell Longev. 2022, 2022, 2240223. [Google Scholar] [CrossRef]
- Siegenthaler, B.; Defila, C.; Muzumdar, S.; Beer, H.D.; Meyer, M.; Tanner, S.; Bloch, W.; Blank, V.; Schafer, M.; Werner, S. Nrf3 promotes UV-induced keratinocyte apoptosis through suppression of cell adhesion. Cell Death Differ. 2018, 25, 1749–1765. [Google Scholar] [CrossRef] [PubMed]
- Waku, T.; Kobayashi, A. Pathophysiological Potentials of NRF3-Regulated Transcriptional Axes in Protein and Lipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 12686. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.; Vani, K.; Leung, S.; Epstein, A. Segmentally restricted, cephalic expression of a leucine zipper gene during Drosophila embryogenesis. Mech. Dev. 1991, 34, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Han, X.L.; Kan, Y.W. Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc. Natl. Acad. Sci. USA 1993, 90, 11371–11375. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tian, W.; Whitman, S.A.; Zhang, D.D. The predicted molecular weight of Nrf2: It is what it is not. Antioxid. Redox Signal. 2013, 18, 91–93. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [PubMed]
- Velichkova, M.; Hasson, T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol. Cell Biol. 2005, 25, 4501–4513. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.S.; McIntosh, D.J.; Ingram, S.M.; Tillery, L.; Motley, E.D.; Arinze, I.J.; Misra, S. SUMO-Modification of Human Nrf2 at K(110) and K(533) Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell Physiol. Biochem. 2021, 55, 141–159. [Google Scholar] [PubMed]
- Kang, H.J.; Hong, Y.B.; Kim, H.J.; Bae, I. CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J. Biol. Chem. 2010, 285, 21258–21268. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.Y.; Spatola, B.N.; Curran, S.P. WDR23 regulates NRF2 independently of KEAP1. PLoS Genet. 2017, 13, e1006762. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzym. Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef]
- Cores, A.; Piquero, M.; Villacampa, M.; Leon, R.; Menendez, J.C. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef]
- Li, W.; Yu, S.W.; Kong, A.N. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J. Biol. Chem. 2006, 281, 27251–27263. [Google Scholar] [CrossRef] [PubMed]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12475–12480. [Google Scholar] [CrossRef]
- Ge, W.; Zhao, K.; Wang, X.; Li, H.; Yu, M.; He, M.; Xue, X.; Zhu, Y.; Zhang, C.; Cheng, Y.; et al. iASPP Is an Antioxidative Factor and Drives Cancer Growth and Drug Resistance by Competing with Nrf2 for Keap1 Binding. Cancer Cell 2017, 32, 561–573.e6. [Google Scholar] [CrossRef]
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; de Stanchina, E.; et al. The Oncogenic Action of NRF2 Depends on De-glycation by Fructosamine-3-Kinase. Cell 2019, 178, 807–819.e21. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Crawford, D.; Khateb, A.; Feng, Y.; Sergienko, E.; Pathria, G.; Ma, C.T.; Olson, S.H.; Scott, D.; Murad, R.; et al. NRF2 mediates melanoma addiction to GCDH by modulating apoptotic signalling. Nat. Cell Biol. 2022, 24, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Hoshino, H.; Muto, A.; Suwabe, N.; Nishikawa, S.; Nakauchi, H.; Yamamoto, M. Multivalent DNA binding complex generated by small Maf and Bach1 as a possible biochemical basis for beta-globin locus control region complex. J. Biol. Chem. 1998, 273, 11783–11790. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Jaiswal, A.K. Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J. Biol. Chem. 2010, 285, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Jyrkkanen, H.K.; Kuosmanen, S.; Heinaniemi, M.; Laitinen, H.; Kansanen, E.; Mella-Aho, E.; Leinonen, H.; Yla-Herttuala, S.; Levonen, A.L. Novel insights into the regulation of antioxidant-response-element-mediated gene expression by electrophiles: Induction of the transcriptional repressor BACH1 by Nrf2. Biochem. J. 2011, 440, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Shao, J.; Glorieux, C.; Liao, J.; Chen, P.; Lu, W.; Liang, Z.; Wen, S.; Hu, Y.; Huang, P. Impact of Nrf2 on tumour growth and drug sensitivity in oncogenic K-ras-transformed cells in vitro and in vivo. Free Radic. Res. 2018, 52, 661–671. [Google Scholar] [CrossRef]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Imbeault, S.; Barakauskas, V.; Erb, H.; Jiang, L.; Li, P.; Murphy, T.H. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem. 2005, 280, 22925–22936. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Kan, Y.W. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12731–12736. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Kozumbo, W.J. The hormetic dose-response mechanism: Nrf2 activation. Pharmacol. Res. 2021, 167, 105526. [Google Scholar] [CrossRef]
- Li, J.; Xu, C.; Liu, Q. Roles of NRF2 in DNA damage repair. Cell Oncol. 2023, 46, 1577–1593. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef]
- Homma, S.; Ishii, Y.; Morishima, Y.; Yamadori, T.; Matsuno, Y.; Haraguchi, N.; Kikuchi, N.; Satoh, H.; Sakamoto, T.; Hizawa, N.; et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 2009, 15, 3423–3432. [Google Scholar] [CrossRef]
- Pillai, R.; Hayashi, M.; Zavitsanou, A.M.; Papagiannakopoulos, T. NRF2: KEAPing Tumors Protected. Cancer Discov. 2022, 12, 625–643. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [PubMed]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Mori, E.; Mori, K.; Goto, S.; Urata, Y.; Oka, M.; Kohno, S.; Kondo, T. Co-expression of gamma-glutamylcysteine synthetase sub-units in response to cisplatin and doxorubicin in human cancer cells. Int. J. Cancer 1999, 82, 405–411. [Google Scholar] [CrossRef]
- Kaur, T.; Khanduja, K.L.; Gupta, R.; Gupta, N.M.; Vaiphei, K. Changes in antioxidant defense status in response to cisplatin and 5-FU in esophageal carcinoma. Dis. Esophagus 2008, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef]
- Cho, J.M.; Manandhar, S.; Lee, H.R.; Park, H.M.; Kwak, M.K. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: Implication to cancer cell resistance. Cancer Lett. 2008, 260, 96–108. [Google Scholar] [CrossRef]
- Tao, S.; Rojo de la Vega, M.; Chapman, E.; Ooi, A.; Zhang, D.D. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 2018, 57, 182–192. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, S.; Kunwar, A.; Sandur, S.K.; Pandey, B.N.; Chaubey, R.C. Differential response of DU145 and PC3 prostate cancer cells to ionizing radiation: Role of reactive oxygen species, GSH and Nrf2 in radiosensitivity. Biochim. Biophys. Acta 2014, 1840, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Kansanen, E.; Polonen, P.; Heinaniemi, M.; Levonen, A.L. Role of the Keap1-Nrf2 pathway in cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar]
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318. [Google Scholar] [CrossRef]
- Bartman, C.R.; Weilandt, D.R.; Shen, Y.; Lee, W.D.; Han, Y.; TeSlaa, T.; Jankowski, C.S.R.; Samarah, L.; Park, N.R.; da Silva-Diz, V.; et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 2023, 614, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Seagroves, T.N.; Ryan, H.E.; Lu, H.; Wouters, B.G.; Knapp, M.; Thibault, P.; Laderoute, K.; Johnson, R.S. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell Biol. 2001, 21, 3436–3444. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Shah, N.M.; Rushworth, S.A.; Murray, M.Y.; Bowles, K.M.; MacEwan, D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget 2013, 4, 1130–1142. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Kitteringham, N.R.; Abdullah, A.; Walsh, J.; Randle, L.; Jenkins, R.E.; Sison, R.; Goldring, C.E.; Powell, H.; Sanderson, C.; Williams, S.; et al. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J. Proteom. 2010, 73, 1612–1631. [Google Scholar] [CrossRef]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Cahill, J.M.; Liu, J.; Kuang, X.; Liu, N.; Scofield, V.L.; Voorhees, J.R.; Reid, A.J.; Yan, M.; Lynn, W.S.; et al. Activation of transcription factor Nrf-2 and its downstream targets in response to moloney murine leukemia virus ts1-induced thiol depletion and oxidative stress in astrocytes. J. Virol. 2004, 78, 11926–11938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxid. Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Kudo, Y.; Sugimoto, M.; Arias, E.; Kasashima, H.; Cordes, T.; Linares, J.F.; Duran, A.; Nakanishi, Y.; Nakanishi, N.; L’Hermitte, A.; et al. PKClambda/iota Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 2020, 38, 247–262.e11. [Google Scholar] [CrossRef]
- Weiss-Sadan, T.; Ge, M.; Hayashi, M.; Gohar, M.; Yao, C.H.; de Groot, A.; Harry, S.; Carlin, A.; Fischer, H.; Shi, L.; et al. NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer. Cell Metab. 2023, 35, 487–503.e7. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, X.; Qiu, J. Emerging role of NRF2 in ROS-mediated tumor chemoresistance. Biomed. Pharmacother. 2020, 131, 110676. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Dargelos, E.; Brule, C.; Stuelsatz, P.; Mouly, V.; Veschambre, P.; Cottin, P.; Poussard, S. Up-regulation of calcium-dependent proteolysis in human myoblasts under acute oxidative stress. Exp. Cell Res. 2010, 316, 115–125. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.M.; Judge, A.R.; Talbert, E.E.; Powers, S.K. Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. Am. J. Physiol. Cell Physiol. 2009, 296, C363–C371. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhao, Q.; Zhang, Y.; Shi, W.; Wang, H.; Zheng, Z.; Meng, L.; Xin, Y.; Jiang, X. Sulforaphane-Mediated Nrf2 Activation Prevents Radiation-Induced Skin Injury through Inhibiting the Oxidative-Stress-Activated DNA Damage and NLRP3 Inflammasome. Antioxidants 2021, 10, 1850. [Google Scholar] [CrossRef] [PubMed]
- Garstkiewicz, M.; Strittmatter, G.E.; Grossi, S.; Sand, J.; Fenini, G.; Werner, S.; French, L.E.; Beer, H.D. Opposing effects of Nrf2 and Nrf2-activating compounds on the NLRP3 inflammasome independent of Nrf2-mediated gene expression. Eur. J. Immunol. 2017, 47, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.; Ma, J.; Chen, X.; Chen, K.; Jiang, Z.; Zong, L.; Yu, S.; Li, X.; Xu, Q.; et al. The Relevance of Nrf2 Pathway and Autophagy in Pancreatic Cancer Cells upon Stimulation of Reactive Oxygen Species. Oxid. Med. Cell Longev. 2016, 2016, 3897250. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Vendramini-Costa, D.B.; Carvalho, J.E. Molecular link mechanisms between inflammation and cancer. Curr. Pharm. Des. 2012, 18, 3831–3852. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Cosio, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Dumenil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef]
- Duwe, A.K.; Werkmeister, J.; Roder, J.C.; Lauzon, R.; Payne, U. Natural killer cell-mediated lysis involves an hydroxyl radical-dependent step. J. Immunol. 1985, 134, 2637–2644. [Google Scholar] [CrossRef]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: Selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef]
- Wheeler, M.L.; Defranco, A.L. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J. Immunol. 2012, 189, 4405–4416. [Google Scholar] [CrossRef]
- Hildeman, D.A.; Mitchell, T.; Teague, T.K.; Henson, P.; Day, B.J.; Kappler, J.; Marrack, P.C. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 1999, 10, 735–744. [Google Scholar] [CrossRef]
- Aksoylar, H.I.; Patsoukis, N. Treatment with Exogenously Added Catalase Alters CD8 T Cell Memory Differentiation and Function. Adv. Biol. 2022, 7, 2101320. [Google Scholar] [CrossRef]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef]
- Griess, B.; Mir, S.; Datta, K.; Teoh-Fitzgerald, M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic. Biol. Med. 2020, 147, 48–60. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef]
- Kim, J.; Surh, Y.J. The Role of Nrf2 in Cellular Innate Immune Response to Inflammatory Injury. Toxicol. Res. 2009, 25, 159–173. [Google Scholar] [CrossRef]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar]
- Morzadec, C.; Macoch, M.; Sparfel, L.; Kerdine-Romer, S.; Fardel, O.; Vernhet, L. Nrf2 expression and activity in human T lymphocytes: Stimulation by T cell receptor activation and priming by inorganic arsenic and tert-butylhydroquinone. Free Radic. Biol. Med. 2014, 71, 133–145. [Google Scholar] [CrossRef]
- Pyaram, K.; Kumar, A.; Kim, Y.H.; Noel, S.; Reddy, S.P.; Rabb, H.; Chang, C.H. Keap1-Nrf2 System Plays an Important Role in Invariant Natural Killer T Cell Development and Homeostasis. Cell Rep. 2019, 27, 699–707.e4. [Google Scholar] [CrossRef]
- Klemm, P.; Rajendiran, A.; Fragoulis, A.; Wruck, C.; Schippers, A.; Wagner, N.; Bopp, T.; Tenbrock, K.; Ohl, K. Nrf2 expression driven by Foxp3 specific deletion of Keap1 results in loss of immune tolerance in mice. Eur. J. Immunol. 2020, 50, 515–524. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar]
- Zhang, D.; Rennhack, J.; Andrechek, E.R.; Rockwell, C.E.; Liby, K.T. Identification of an Unfavorable Immune Signature in Advanced Lung Tumors from Nrf2-Deficient Mice. Antioxid. Redox Signal. 2018, 29, 1535–1552. [Google Scholar] [CrossRef]
- Singh, A.; Daemen, A.; Nickles, D.; Jeon, S.M.; Foreman, O.; Sudini, K.; Gnad, F.; Lajoie, S.; Gour, N.; Mitzner, W.; et al. NRF2 Activation Promotes Aggressive Lung Cancer and Associates with Poor Clinical Outcomes. Clin. Cancer Res. 2021, 27, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Yamashita, N.; Ogawa, T.; Koseki, K.; Warabi, E.; Ohue, T.; Komatsu, M.; Matsushita, H.; Kakimi, K.; Kawakami, E.; et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes. J. Immunother. Cancer 2021, 9, e002954. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021, 33, 1205–1220.e5. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Jahraus, B.; Balta, E.; Ziegler, J.D.; Hubner, K.; Blank, N.; Niesler, B.; Wabnitz, G.H.; Samstag, Y. Sulforaphane Inhibits Inflammatory Responses of Primary Human T-Cells by Increasing ROS and Depleting Glutathione. Front. Immunol. 2018, 9, 2584. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Hansch, G.M.; Hubner, K.; Samstag, Y. Sulforaphane as anticancer agent: A double-edged sword? Tricky balance between effects on tumor cells and immune cells. Adv. Biol. Regul. 2019, 71, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Renken, S.; Nakajima, T.; Magalhaes, I.; Mattsson, J.; Lundqvist, A.; Arner, E.S.J.; Kiessling, R.; Wickstrom, S.L. Targeting of Nrf2 improves antitumoral responses by human NK cells, TIL and CAR T cells during oxidative stress. J. Immunother. Cancer 2022, 10, e004458. [Google Scholar] [CrossRef] [PubMed]
- Kesner, J.S.; Chen, Z.; Shi, P.; Aparicio, A.O.; Murphy, M.R.; Guo, Y.; Trehan, A.; Lipponen, J.E.; Recinos, Y.; Myeku, N.; et al. Noncoding translation mitigation. Nature 2023, 617, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qiu, Y.; Xiang, R.; Huang, P. Characterization of H(2)O(2)-Induced Alterations in Global Transcription of mRNA and lncRNA. Antioxidants 2022, 11, 495. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Sparaneo, A.; Muscarella, L.A. NRF2 Regulation by Noncoding RNAs in Cancers: The Present Knowledge and the Way Forward. Cancers 2020, 12, 3621. [Google Scholar] [CrossRef]
- Moreno Leon, L.; Gautier, M.; Allan, R.; Ilie, M.; Nottet, N.; Pons, N.; Paquet, A.; Lebrigand, K.; Truchi, M.; Fassy, J.; et al. The nuclear hypoxia-regulated NLUCAT1 long non-coding RNA contributes to an aggressive phenotype in lung adenocarcinoma through regulation of oxidative stress. Oncogene 2019, 38, 7146–7165. [Google Scholar] [CrossRef]
- Gai, C.; Liu, C.; Wu, X.; Yu, M.; Zheng, J.; Zhang, W.; Lv, S.; Li, W. MT1DP loaded by folate-modified liposomes sensitizes erastin-induced ferroptosis via regulating miR-365a-3p/NRF2 axis in non-small cell lung cancer cells. Cell Death Dis. 2020, 11, 751. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; McAtee, C.K.; Su, X. Phase separation in immune signalling. Nat. Rev. Immunol. 2022, 22, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Gudmundsson, S.R.; Sou, Y.S.; Ichimura, Y.; Tamura, N.; Kazuno, S.; Ueno, T.; Miura, Y.; Noshiro, D.; Abe, M.; et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 2021, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, P.; Sou, Y.S.; Kageyama, S.; Koike, M.; Waguri, S.; Komatsu, M. NBR1-mediated p62-liquid droplets enhance the Keap1-Nrf2 system. EMBO Rep. 2020, 21, e48902. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Valionyte, E.; Kelly, J.; Luo, S. Histone H3F3/H3.3 chaperone DAXX converts to modulate SQSTM1 phase condensation for NFE2L2 activation. Autophagy 2020, 16, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Willis, T.L.; Button, R.W.; Strang, C.J.; Fu, Y.; Wen, X.; Grayson, P.R.C.; Evans, T.; Sipthorpe, R.J.; Roberts, S.L.; et al. Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response. Nat. Commun. 2019, 10, 3759. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.; Noshiro, D.; Morishita, H.; Takada, S.; Kageyama, S.; Fujioka, Y.; Funakoshi, T.; Komatsu-Hirota, S.; Arai, R.; Ryzhii, E.; et al. Phosphorylation of phase-separated p62 bodies by ULK1 activates a redox-independent stress response. EMBO J. 2023, 42, e113349. [Google Scholar] [CrossRef]
- Gao, K.; Shi, Q.; Liu, Y.; Wang, C. Enhanced autophagy and NFE2L2/NRF2 pathway activation in SPOP mutation-driven prostate cancer. Autophagy 2022, 18, 2013–2015. [Google Scholar] [CrossRef]
- Tan, C.T.; Chang, H.C.; Zhou, Q.; Yu, C.; Fu, N.Y.; Sabapathy, K.; Yu, V.C. MOAP-1-mediated dissociation of p62/SQSTM1 bodies releases Keap1 and suppresses Nrf2 signaling. EMBO Rep. 2021, 22, e50854. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, Y.; Liu, Z.; Lu, Y.; Zhu, X.; Lan, B.; Mi, Z.; Dang, L.; Li, N.; Zhan, W.; et al. Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci. Transl. Med. 2020, 12, eaba3613. [Google Scholar] [CrossRef]
- Li, M.; Thorne, R.F.; Wang, R.; Cao, L.; Cheng, F.; Sun, X.; Wu, M.; Ma, J.; Liu, L. Sestrin2-mediated disassembly of stress granules dampens aerobic glycolysis to overcome glucose starvation. Cell Death Discov. 2023, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86.e6. [Google Scholar] [CrossRef] [PubMed]
- Hallis, S.P.; Kim, J.M.; Kwak, M.K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 2023, 46, 153–164. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Zhou, F.; Su, A.; Zhang, Y.; Xing, Z.; Mi, L.; Li, Z.; Wu, W. Brusatol: A potential sensitizing agent for cancer therapy from Brucea javanica. Biomed. Pharmacother. 2023, 158, 114134. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e18. [Google Scholar] [CrossRef]
- Consonni, F.M.; Bleve, A.; Totaro, M.G.; Storto, M.; Kunderfranco, P.; Termanini, A.; Pasqualini, F.; Ali, C.; Pandolfo, C.; Sgambelluri, F.; et al. Heme catabolism by tumor-associated macrophages controls metastasis formation. Nat. Immunol. 2021, 22, 595–606. [Google Scholar] [CrossRef]
- Sato, M.; Matsumoto, M.; Saiki, Y.; Alam, M.; Nishizawa, H.; Rokugo, M.; Brydun, A.; Yamada, S.; Kaneko, M.K.; Funayama, R.; et al. BACH1 Promotes Pancreatic Cancer Metastasis by Repressing Epithelial Genes and Enhancing Epithelial-Mesenchymal Transition. Cancer Res. 2020, 80, 1279–1292. [Google Scholar] [CrossRef]
- Panieri, E.; Buha, A.; Telkoparan-Akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential Applications of NRF2 Modulators in Cancer Therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Copple, I.M. Advances and challenges in therapeutic targeting of NRF2. Trends Pharmacol. Sci. 2023, 44, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, H.X.; Zhu, J.Q.; Dou, Y.X.; Xian, Y.F.; Lin, Z.X. Natural Nrf2 Inhibitors: A Review of Their Potential for Cancer Treatment. Int. J. Biol. Sci. 2023, 19, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Jin, X.; Zhang, J.; Li, L.; Zhao, J. Metformin suppresses Nrf2-mediated chemoresistance in hepatocellular carcinoma cells by increasing glycolysis. Aging 2020, 12, 17582–17600. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Kim, K.; Norris, A.J.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; Della Donna, L.; Ratikan, J.; Szelag, H.; Hlatky, L.; et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhe, H.; Zhao, R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef]
- Winkel, A.F.; Engel, C.K.; Margerie, D.; Kannt, A.; Szillat, H.; Glombik, H.; Kallus, C.; Ruf, S.; Gussregen, S.; Riedel, J.; et al. Characterization of RA839, a Noncovalent Small Molecule Binder to Keap1 and Selective Activator of Nrf2 Signaling. J. Biol. Chem. 2015, 290, 28446–28455. [Google Scholar] [CrossRef]
- Brennan, M.S.; Matos, M.F.; Richter, K.E.; Li, B.; Scannevin, R.H. The NRF2 transcriptional target, OSGIN1, contributes to monomethyl fumarate-mediated cytoprotection in human astrocytes. Sci. Rep. 2017, 7, 42054. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Hartsock, M.J.; Xu, Z.; He, M.; Duh, E.J. Monomethyl fumarate promotes Nrf2-dependent neuroprotection in retinal ischemia-reperfusion. J. Neuroinflamm. 2015, 12, 239. [Google Scholar] [CrossRef] [PubMed]
- Serrya, M.S.; El-Sheakh, A.R.; Makled, M.N. Evaluation of the therapeutic effects of mycophenolate mofetil targeting Nrf-2 and NLRP3 inflammasome in acetic acid induced ulcerative colitis in rats. Life Sci. 2021, 271, 119154. [Google Scholar] [CrossRef] [PubMed]
- Issac, J.; Raveendran, P.S.; Kunnummal, M.; Angelin, M.; Ravindran, S.; Basu, B.; Das, A.V. RXR agonist, Bexarotene, effectively reduces drug resistance via regulation of RFX1 in embryonic carcinoma cells. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119510. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Tian, W.; La Clair, J.J.; Tan, A.C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef]
- Liu, X.; Peng, X.; Cen, S.; Yang, C.; Ma, Z.; Shi, X. Wogonin induces ferroptosis in pancreatic cancer cells by inhibiting the Nrf2/GPX4 axis. Front. Pharmacol. 2023, 14, 1129662. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Ananda Sadagopan, S.K.; Dharmalingam, P.; Ganapasam, S. Luteolin, a bioflavonoid inhibits Azoxymethane-induced colorectal cancer through activation of Nrf2 signaling. Toxicol. Mech. Methods 2014, 24, 13–20. [Google Scholar] [CrossRef]
- Limonciel, A.; Jennings, P. A review of the evidence that ochratoxin A is an Nrf2 inhibitor: Implications for nephrotoxicity and renal carcinogenicity. Toxins 2014, 6, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.L.; Schreiber, S.; Schafer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Manna, A.; De Sarkar, S.; De, S.; Bauri, A.K.; Chattopadhyay, S.; Chatterjee, M. The variable chemotherapeutic response of Malabaricone-A in leukemic and solid tumor cell lines depends on the degree of redox imbalance. Phytomedicine 2015, 22, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Gazaryan, I.G.; Thomas, B. The status of Nrf2-based therapeutics: Current perspectives and future prospects. Neural Regen. Res. 2016, 11, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Singh, A.K.; Singh, M.; Tewari, M.; Shukla, H.S.; Gambhir, I.S. The see-saw of Keap1-Nrf2 pathway in cancer. Crit. Rev. Oncol. Hematol. 2017, 116, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Jyrkkanen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Devel Ther. 2018, 12, 3181–3197. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Chu, X.Y.; Jiang, L.H.; Liu, M.Y.; Mei, Z.L.; Zhang, H.Y. Identification of Non-Electrophilic Nrf2 Activators from Approved Drugs. Molecules 2017, 22, 28587109. [Google Scholar] [CrossRef] [PubMed]
- Tarumoto, T.; Nagai, T.; Ohmine, K.; Miyoshi, T.; Nakamura, M.; Kondo, T.; Mitsugi, K.; Nakano, S.; Muroi, K.; Komatsu, N.; et al. Ascorbic acid restores sensitivity to imatinib via suppression of Nrf2-dependent gene expression in the imatinib-resistant cell line. Exp. Hematol. 2004, 32, 375–381. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NRF2 Activators | Modes of Action | References |
|---|---|---|
| Sulforaphane | Covalent binding to KEAP1 cysteine residues | [196,197] |
| Triterpenoids (CDDO) | Target KEAP1 and activation of NRF2 response | [198,199] |
| RA839 | Selective inhibitor of the KEAP1/NRF2 interaction | [200] |
| MMF/DMF | Activation of NRF2 and upregulation of its target genes | [201,202,203] |
| NRF2 Inhibitors | Modes of Action | References |
| Bexarotene | Interaction with the Neh7 domain of NRF2 | [204,205,206] |
| Brusatol | Global protein synthesis inhibitor | [207,208] |
| Flavonoids | Increase in NRF2 instability | [209,210] |
| Ochratoxin A | Interference with NRF2 translocation and its DNA binding | [211] |
| Trigonelline | Reduced nuclear accumulation of the NRF2 protein | [212,213] |
| Malabaricon-A | Inhibition of NRF2 transcriptional activity | [214] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glorieux, C.; Enríquez, C.; González, C.; Aguirre-Martínez, G.; Buc Calderon, P. The Multifaceted Roles of NRF2 in Cancer: Friend or Foe? Antioxidants 2024, 13, 70. https://doi.org/10.3390/antiox13010070
Glorieux C, Enríquez C, González C, Aguirre-Martínez G, Buc Calderon P. The Multifaceted Roles of NRF2 in Cancer: Friend or Foe? Antioxidants. 2024; 13(1):70. https://doi.org/10.3390/antiox13010070
Chicago/Turabian StyleGlorieux, Christophe, Cinthya Enríquez, Constanza González, Gabriela Aguirre-Martínez, and Pedro Buc Calderon. 2024. "The Multifaceted Roles of NRF2 in Cancer: Friend or Foe?" Antioxidants 13, no. 1: 70. https://doi.org/10.3390/antiox13010070
APA StyleGlorieux, C., Enríquez, C., González, C., Aguirre-Martínez, G., & Buc Calderon, P. (2024). The Multifaceted Roles of NRF2 in Cancer: Friend or Foe? Antioxidants, 13(1), 70. https://doi.org/10.3390/antiox13010070