
The Contribution of Oxidative Stress to NF1-Altered Tumors

 , ,
, ,
Abstract
1. Introduction
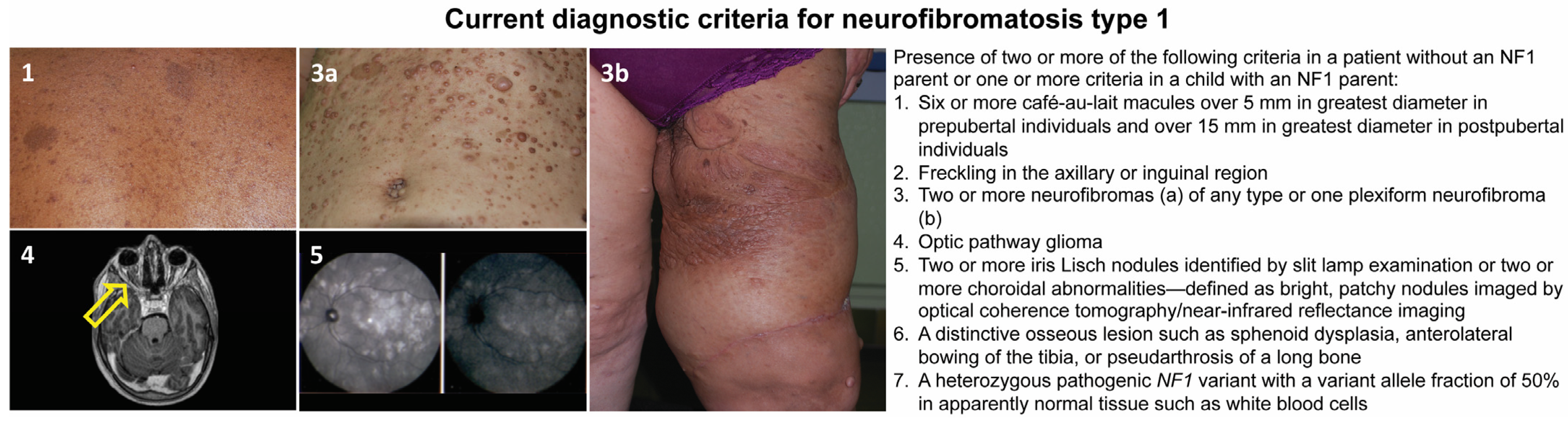
2. Neurofibromatosis Type 1
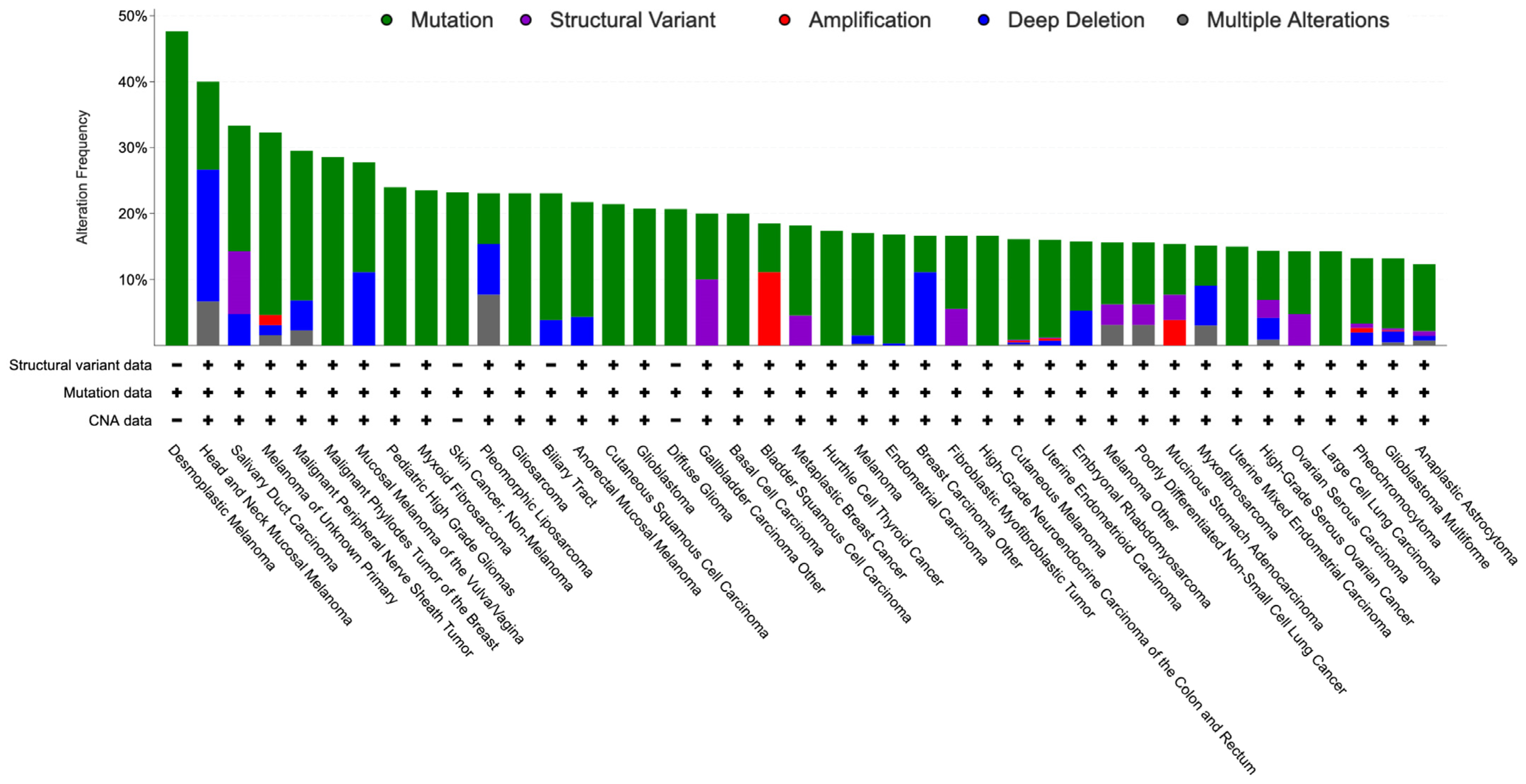
3. NF1-Altered Sporadic Tumors
4. Molecular Genetics of NF1 Inactivation
5. NF1-Related Tumorigenic Mechanisms
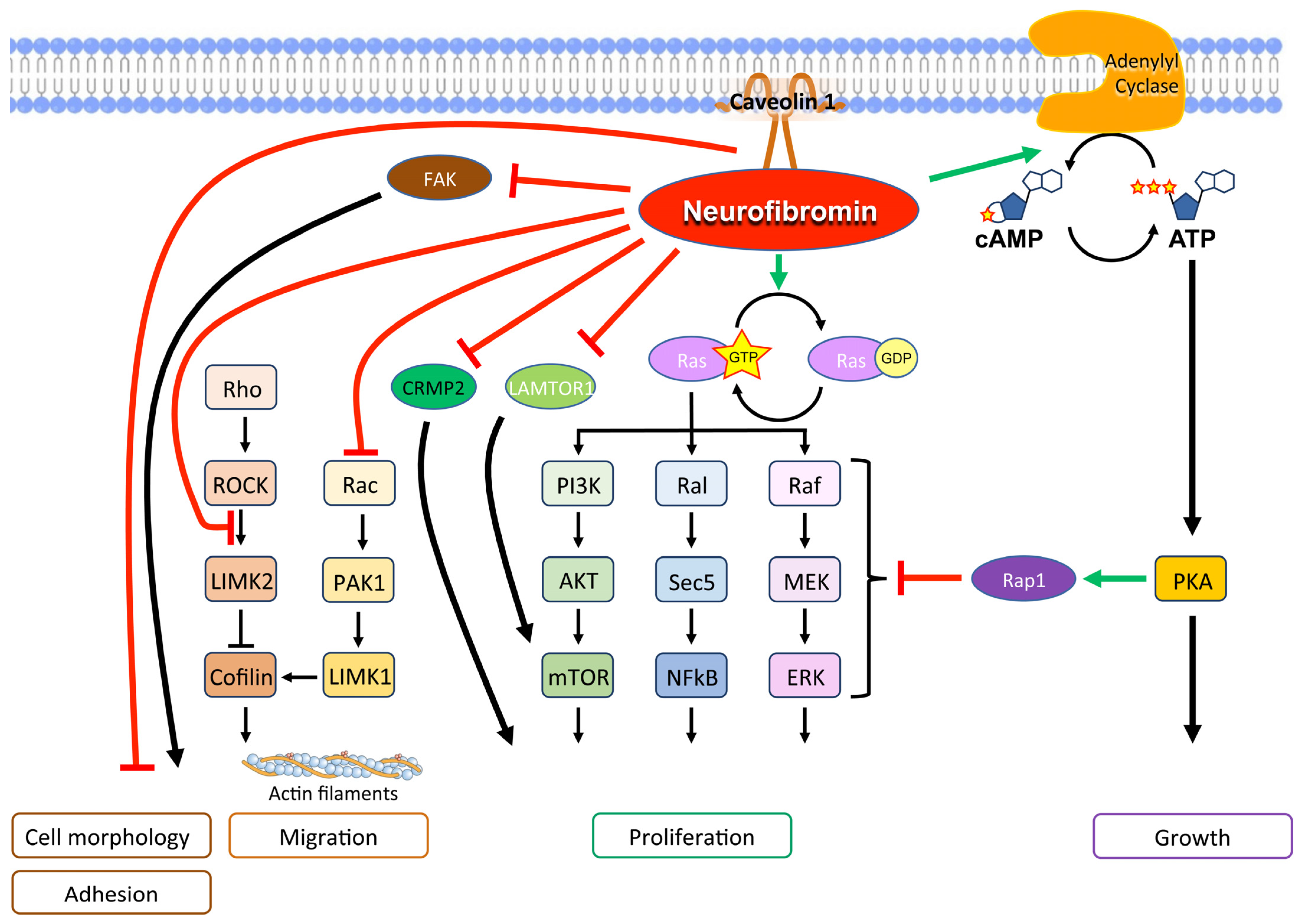
Neurofibromin Cellular Functions
6. Oxidative Stress and Cancer
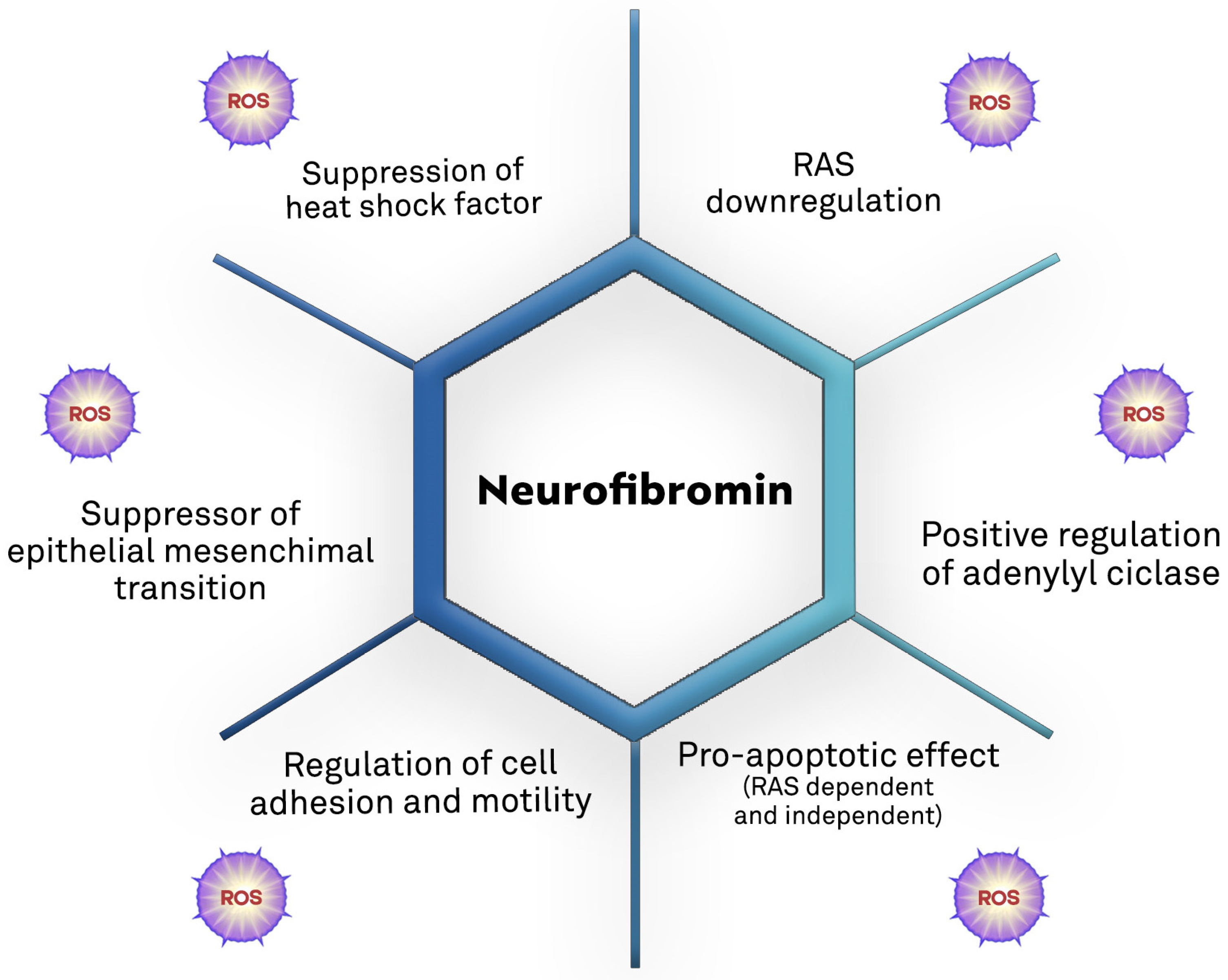
7. Neurofibromin and Oxidative Stress
8. Therapeutic Implications of NF1-Related Oxidative Damage
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox. Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Ohba, Y.; Mochizuki, N.; Yamashita, S.; Chan, A.M.; Schrader, J.W.; Hattori, S.; Nagashima, K.; Matsuda, M. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J. Biol. Chem. 2000, 275, 20020–20026. [Google Scholar] [CrossRef]
- Li, X.; Gao, M.; Choi, J.M.; Kim, B.J.; Zhou, M.T.; Chen, Z.; Jain, A.N.; Jung, S.Y.; Yuan, J.; Wang, W.; et al. Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-coupled Affinity Purification/Mass Spectrometry Analysis Revealed a Novel Role of Neurofibromin in mTOR Signaling. Mol. Cell. Proteom. 2017, 16, 594–607. [Google Scholar] [CrossRef]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Brosius, S. A history of von Recklinghausen’s NF1. J. Hist. Neurosci. 2010, 19, 333–348. [Google Scholar] [CrossRef]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Arbiser, J.; Epstein, J.A.; Gutmann, D.H.; Huot, S.J.; Lin, A.E.; McManus, B.; Korf, B.R. Cardiovascular disease in neurofibromatosis 1: Report of the NF1 Cardiovascular Task Force. Genet. Med. 2002, 4, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.L.; Arthur Shores, E.; North, K.N. Learning disabilities in children with neurofibromatosis type 1: Subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev. Med. Child Neurol. 2006, 48, 973–977. [Google Scholar] [CrossRef]
- Seminog, O.O.; Goldacre, M.J. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: Population-based record-linkage study. Br. J. Cancer 2013, 108, 193–198. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Yang, Q.; Friedman, J.M. Mortality in neurofibromatosis 1: An analysis using U.S. death certificates. Am. J. Hum. Genet. 2001, 68, 1110–1118. [Google Scholar] [CrossRef]
- Landry, J.P.; Schertz, K.L.; Chiang, Y.J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients with Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population from 1985 to 2020. JAMA Netw. Open 2021, 4, e210945. [Google Scholar] [CrossRef]
- Stiller, C.A.; Chessells, J.M.; Fitchett, M. Neurofibromatosis and childhood leukaemia/lymphoma: A population-based UKCCSG study. Br. J. Cancer 1994, 70, 969–972. [Google Scholar] [CrossRef]
- Carton, C.; Evans, D.G.; Blanco, I.; Friedrich, R.E.; Ferner, R.E.; Farschtschi, S.; Salvador, H.; Azizi, A.A.; Mautner, V.; Röhl, C.; et al. ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. EClinicalMedicine 2023, 56, 101818. [Google Scholar] [CrossRef]
- Pasmant, E.; Vidaud, M.; Vidaud, D.; Wolkenstein, P. Neurofibromatosis type 1: From genotype to phenotype. J. Med. Genet. 2012, 49, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Ruggieri, M.; Maynard, J.; Osborn, M.; Hartog, C.; Mudd, S.; Penttinen, M.; Cordeiro, I.; Ponder, M.; Ponder, B.A.; et al. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum. Genet. 1998, 102, 591–597. [Google Scholar] [CrossRef]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p. Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef]
- Side, L.; Taylor, B.; Cayouette, M.; Conner, E.; Thompson, P.; Luce, M.; Shannon, K. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N. Engl. J. Med. 1997, 336, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Sawada, S.; Florell, S.; Purandare, S.M.; Ota, M.; Stephens, K.; Viskochil, D. Identification of NF1 mutations in both alleles of a dermal neurofibroma. Nat. Genet. 1996, 14, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Martelotto, L.G.; De Filippo, M.R.; Ng, C.K.; Natrajan, R.; Fuhrmann, L.; Cyrta, J.; Piscuoglio, S.; Wen, H.C.; Lim, R.S.; Shen, R.; et al. Genomic landscape of adenoid cystic carcinoma of the breast. J. Pathol. 2015, 237, 179–189. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Shain, A.H. The landscape of driver mutations in cutaneous squamous cell carcinoma. NPJ Genom. Med. 2021, 6, 61. [Google Scholar] [CrossRef]
- Gambini, D.; Passoni, E.; Nazzaro, G.; Beltramini, G.; Tomasello, G.; Ghidini, M.; Kuhn, E.; Garrone, O. Basal Cell Carcinoma and Hedgehog Pathway Inhibitors: Focus on Immune Response. Front. Med. 2022, 9, 893063. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih Ie, M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Trovó-Marqui, A.B.; Tajara, E.H. Neurofibromin: A general outlook. Clin. Genet. 2006, 70, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Yao, S.; Claes, K.; Kehrer-Sawatzki, H.; Tinschert, S.; De Raedt, T.; Legius, E.; Callens, T.; Beiglböck, H.; Maertens, O.; et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer 2006, 45, 265–276. [Google Scholar] [CrossRef]
- Martín, Y.; Dopazo, A.; Hernández-Chico, C. Progress and challenges in developing a molecular diagnostic test for neurofibromatosis type 1. Expert. Rev. Mol. Diagn. 2011, 11, 671–673. [Google Scholar] [CrossRef]
- Luijten, M.; Wang, Y.; Smith, B.T.; Westerveld, A.; Smink, L.J.; Dunham, I.; Roe, B.A.; Hulsebos, T.J. Mechanism of spreading of the highly related neurofibromatosis type 1 (NF1) pseudogenes on chromosomes 2, 14 and 22. Eur. J. Hum. Genet. 2000, 8, 209–214. [Google Scholar] [CrossRef][Green Version]
- Ars, E.; Serra, E.; García, J.; Kruyer, H.; Gaona, A.; Lázaro, C.; Estivill, X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum. Mol. Genet. 2000, 9, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Peltonen, J.; Jaakkola, S.; Lebwohl, M.; Renvall, S.; Risteli, L.; Virtanen, I.; Uitto, J. Cellular differentiation and expression of matrix genes in type 1 neurofibromatosis. Lab. Investig. 1988, 59, 760–771. [Google Scholar]
- Koczkowska, M.; Chen, Y.; Xie, J.; Callens, T.; Gomes, A.; Wimmer, K.; Messiaen, L.M. Analysis of 200 unrelated individuals with a constitutional NF1 deep intronic pathogenic variant reveals that variants flanking the alternatively spliced NF1 exon 31 [23a] cause a classical neurofibromatosis type 1 phenotype while altering predominantly NF1 isoform type II. Hum. Genet. 2023, 142, 849–886. [Google Scholar] [CrossRef]
- Kluwe, L.; Siebert, R.; Gesk, S.; Friedrich, R.E.; Tinschert, S.; Kehrer-Sawatzki, H.; Mautner, V.F. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum. Mutat. 2004, 23, 111–116. [Google Scholar] [CrossRef]
- Jacks, T.; Shih, T.S.; Schmitt, E.M.; Bronson, R.T.; Bernards, A.; Weinberg, R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994, 7, 353–361. [Google Scholar] [CrossRef]
- Brannan, C.I.; Perkins, A.S.; Vogel, K.S.; Ratner, N.; Nordlund, M.L.; Reid, S.W.; Buchberg, A.M.; Jenkins, N.A.; Parada, L.F.; Copeland, N.G. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994, 8, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Devine, W.P.; Shieh, J.T. Tumor and Constitutional Sequencing for Neurofibromatosis Type 1. JCO Precis. Oncol. 2022, 6, e2100540. [Google Scholar] [CrossRef] [PubMed]
- Serra, E.; Rosenbaum, T.; Nadal, M.; Winner, U.; Ars, E.; Estivill, X.; Lázaro, C. Mitotic recombination effects homozygosity for NF1 germline mutations in neurofibromas. Nat. Genet. 2001, 28, 294–296. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, L.T.; Fromm, J.A.; Hollstein, P.E.; Kubek, S.; Beroukhim, R.; De Raedt, T.; Johnson, B.W.; Williams, S.M.; Nghiemphu, P.; Liau, L.M.; et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 2009, 16, 44–54. [Google Scholar] [CrossRef]
- Khosravi, T.; Oladnabi, M. The role of miRNAs and lncRNAs in neurofibromatosis type 1. J. Cell Biochem. 2023, 124, 17–30. [Google Scholar] [CrossRef]
- Fishbein, L.; Eady, B.; Sanek, N.; Muir, D.; Wallace, M.R. Analysis of somatic NF1 promoter methylation in plexiform neurofibromas and Schwann cells. Cancer Genet. Cytogenet. 2005, 157, 181–186. [Google Scholar] [CrossRef]
- Peta, C.; Tsirimonaki, E.; Samouil, D.; Georgiadou, K.; Mangoura, D. Nuclear Isoforms of Neurofibromin Are Required for Proper Spindle Organization and Chromosome Segregation. Cells 2020, 9, 2348. [Google Scholar] [CrossRef]
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Tong, J.; Hannan, F.; Zhu, Y.; Bernards, A.; Zhong, Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat. Neurosci. 2002, 5, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Jang, H.; Zhang, M.; Tsai, C.J.; Sablina, A.A. The Mystery of Rap1 Suppression of Oncogenic Ras. Trends Cancer 2020, 6, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Yi, Y.; Chen, D.Y.; Weber, J.D.; Gutmann, D.H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005, 65, 2755–2760. [Google Scholar] [CrossRef]
- Boyanapalli, M.; Lahoud, O.B.; Messiaen, L.; Kim, B.; Anderle de Sylor, M.S.; Duckett, S.J.; Somara, S.; Mikol, D.D. Neurofibromin binds to caveolin-1 and regulates ras, FAK, and Akt. Biochem. Biophys. Res. Commun. 2006, 340, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Starinsky-Elbaz, S.; Faigenbloom, L.; Friedman, E.; Stein, R.; Kloog, Y. The pre-GAP-related domain of neurofibromin regulates cell migration through the LIM kinase/cofilin pathway. Mol. Cell. Neurosci. 2009, 42, 278–287. [Google Scholar] [CrossRef]
- Ozawa, T.; Araki, N.; Yunoue, S.; Tokuo, H.; Feng, L.; Patrakitkomjorn, S.; Hara, T.; Ichikawa, Y.; Matsumoto, K.; Fujii, K.; et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J. Biol. Chem. 2005, 280, 39524–39533. [Google Scholar] [CrossRef] [PubMed]
- Kweh, F.; Zheng, M.; Kurenova, E.; Wallace, M.; Golubovskaya, V.; Cance, W.G. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 2009, 48, 1005–1017. [Google Scholar] [CrossRef]
- Errico, A.; Stocco, A.; Riccardi, V.M.; Gambalunga, A.; Bassetto, F.; Grigatti, M.; Ferlosio, A.; Tadini, G.; Garozzo, D.; Ferraresi, S.; et al. Neurofibromin Deficiency and Extracellular Matrix Cooperate to Increase Transforming Potential through FAK-Dependent Signaling. Cancers 2021, 13, 2329. [Google Scholar] [CrossRef]
- Arima, Y.; Hayashi, H.; Kamata, K.; Goto, T.M.; Sasaki, M.; Kuramochi, A.; Saya, H. Decreased expression of neurofibromin contributes to epithelial-mesenchymal transition in neurofibromatosis type 1. Exp. Dermatol. 2010, 19, e136–e141. [Google Scholar] [CrossRef]
- Luo, G.; Kim, J.; Song, K. The C-terminal domains of human neurofibromin and its budding yeast homologs Ira1 and Ira2 regulate the metaphase to anaphase transition. Cell Cycle 2014, 13, 2780–2789. [Google Scholar] [CrossRef]
- Koliou, X.; Fedonidis, C.; Kalpachidou, T.; Mangoura, D. Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J. Neurochem. 2016, 136, 78–91. [Google Scholar] [CrossRef]
- Patrakitkomjorn, S.; Kobayashi, D.; Morikawa, T.; Wilson, M.M.; Tsubota, N.; Irie, A.; Ozawa, T.; Aoki, M.; Arimura, N.; Kaibuchi, K.; et al. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J. Biol. Chem. 2008, 283, 9399–9413. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; Villa, L.S.; Yeon, S.K.; Householder, K.T.; Park, K.D.; Sirianni, R.W.; Khanna, R. CRMP2 Phosphorylation Drives Glioblastoma Cell Proliferation. Mol. Neurobiol. 2018, 55, 4403–4416. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox. Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Rose Li, Y.; Halliwill, K.D.; Adams, C.J.; Iyer, V.; Riva, L.; Mamunur, R.; Jen, K.Y.; Del Rosario, R.; Fredlund, E.; Hirst, G.; et al. Mutational signatures in tumours induced by high and low energy radiation in Trp53 deficient mice. Nat. Commun. 2020, 11, 394. [Google Scholar] [CrossRef]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Song, N.Y.; Zhu, F.; Wang, Z.; Willette-Brown, J.; Xi, S.; Sun, Z.; Su, L.; Wu, X.; Ma, B.; Nussinov, R.; et al. IKKα inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E812–E821. [Google Scholar] [CrossRef]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Giorgini, S.; Morichetti, D.; Goteri, G.; Sartini, D.; Serritelli, E.N.; Emanuelli, M. Paraoxonase-2 is upregulated in triple negative breast cancer and contributes to tumor progression and chemoresistance. Hum. Cell. 2023, 36, 1108–1119. [Google Scholar] [CrossRef]
- Bacchetti, T.; Campagna, R.; Sartini, D.; Cecati, M.; Morresi, C.; Bellachioma, L.; Martinelli, E.; Rocchetti, G.; Lucini, L.; Ferretti, G.; et al. C. spinosa L. subsp. rupestris Phytochemical Profile and Effect on Oxidative Stress in Normal and Cancer Cells. Molecules 2022, 27, 6488. [Google Scholar] [CrossRef]
- Campagna, R.; Belloni, A.; Pozzi, V.; Salvucci, A.; Notarstefano, V.; Togni, L.; Mascitti, M.; Sartini, D.; Giorgini, E.; Salvolini, E.; et al. Role Played by Paraoxonase-2 Enzyme in Cell Viability, Proliferation and Sensitivity to Chemotherapy of Oral Squamous Cell Carcinoma Cell Lines. Int. J. Mol. Sci. 2022, 24, 338. [Google Scholar] [CrossRef] [PubMed]
- Hamza, A.A.; Heeba, G.H.; Hassanin, S.O.; Elwy, H.M.; Bekhit, A.A.; Amin, A. Hibiscus-cisplatin combination treatment decreases liver toxicity in rats while increasing toxicity in lung cancer cells via oxidative stress- apoptosis pathway. Biomed. Pharmacother. 2023, 165, 115148. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; Zirpoli, G.R.; Hutson, A.D.; McCann, W.E.; McCann, S.E.; Barlow, W.E.; Kelly, K.M.; Cannioto, R.; Sucheston-Campbell, L.E.; Hershman, D.L.; et al. Dietary Supplement Use During Chemotherapy and Survival Outcomes of Patients with Breast Cancer Enrolled in a Cooperative Group Clinical Trial (SWOG S0221). J. Clin. Oncol. 2020, 38, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E.; Mucha, J.; Hagenlocher, C.; Ehemann, V.; Kluwe, L.; Mautner, V.; von Deimling, A. Sensitivity of malignant peripheral nerve sheath tumor cells to TRAIL is augmented by loss of NF1 through modulation of MYC/MAD and is potentiated by curcumin through induction of ROS. PLoS ONE 2013, 8, e57152. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.Y.; Kopnin, P.B.; Vasiliev, J.M.; Kopnin, B.P. ROS up-regulation mediates Ras-induced changes of cell morphology and motility. Exp. Cell Res. 2006, 312, 2066–2073. [Google Scholar] [CrossRef]
- Dai, C.; Santagata, S.; Tang, Z.; Shi, J.; Cao, J.; Kwon, H.; Bronson, R.T.; Whitesell, L.; Lindquist, S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Investig. 2012, 122, 3742–3754. [Google Scholar] [CrossRef]
- Szyller, J.; Bil-Lula, I. Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits from Physical Exercises: A Review to the Current Knowledge. Oxid. Med. Cell. Longev. 2021, 2021, 6678457. [Google Scholar] [CrossRef]
- Yap, Y.S.; McPherson, J.R.; Ong, C.K.; Rozen, S.G.; Teh, B.T.; Lee, A.S.; Callen, D.F. The NF1 gene revisited-from bench to bedside. Oncotarget 2014, 5, 5873–5892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, R.; Qu, Y.; Shu, M.; Guo, S.; Bai, Z. Lipoamide Inhibits NF1 Deficiency-induced Epithelial-Mesenchymal Transition in Murine Schwann Cells. Arch. Med. Res. 2017, 48, 498–505. [Google Scholar] [CrossRef]
- Tong, J.J.; Schriner, S.E.; McCleary, D.; Day, B.J.; Wallace, D.C. Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nat. Genet. 2007, 39, 476–485. [Google Scholar] [CrossRef]
- Mayes, D.A.; Rizvi, T.A.; Titus-Mitchell, H.; Oberst, R.; Ciraolo, G.M.; Vorhees, C.V.; Robinson, A.P.; Miller, S.D.; Cancelas, J.A.; Stemmer-Rachamimov, A.O.; et al. Nf1 loss and Ras hyperactivation in oligodendrocytes induce NOS-driven defects in myelin and vasculature. Cell Rep. 2013, 4, 1197–1212. [Google Scholar] [CrossRef]
- Bessler, W.K.; Hudson, F.Z.; Zhang, H.; Harris, V.; Wang, Y.; Mund, J.A.; Downing, B.; Ingram, D.A.; Case, J.; Fulton, D.J.; et al. Neurofibromin is a novel regulator of Ras-induced reactive oxygen species production in mice and humans. Free Radic. Biol. Med. 2016, 97, 212–222. [Google Scholar] [CrossRef]
- Masgras, I.; Ciscato, F.; Brunati, A.M.; Tibaldi, E.; Indraccolo, S.; Curtarello, M.; Chiara, F.; Cannino, G.; Papaleo, E.; Lambrughi, M.; et al. Absence of Neurofibromin Induces an Oncogenic Metabolic Switch via Mitochondrial ERK-Mediated Phosphorylation of the Chaperone TRAP1. Cell Rep. 2017, 18, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Masgras, I.; Cannino, G.; Ciscato, F.; Sanchez-Martin, C.; Darvishi, F.B.; Scantamburlo, F.; Pizzi, M.; Menga, A.; Fregona, D.; Castegna, A.; et al. Tumor growth of neurofibromin-deficient cells is driven by decreased respiration and hampered by NAD. Cell Death Differ. 2022, 29, 1996–2008. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef]
- Ferro, E.; Goitre, L.; Baldini, E.; Retta, S.F.; Trabalzini, L. Ras GTPases are both regulators and effectors of redox agents. Methods Mol. Biol. 2014, 1120, 55–74. [Google Scholar] [CrossRef]
- Adachi, Y.; Shibai, Y.; Mitsushita, J.; Shang, W.H.; Hirose, K.; Kamata, T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene 2008, 27, 4921–4932. [Google Scholar] [CrossRef]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Goodwin, C.B.; Nabinger, S.C.; Richine, B.M.; Yang, Z.; Hanenberg, H.; Ohnishi, H.; Matozaki, T.; Feng, G.S.; Chan, R.J. Protein-tyrosine phosphatase Shp2 positively regulates macrophage oxidative burst. J. Biol. Chem. 2015, 290, 3894–3909. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal. Transduct. Target Ther. 2020, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Mellier, G.; Pervaiz, S. The three Rs along the TRAIL: Resistance, re-sensitization and reactive oxygen species (ROS). Free Radic Res. 2012, 46, 996–1003. [Google Scholar] [CrossRef]
- Allaway, R.J.; Wood, M.D.; Downey, S.L.; Bouley, S.J.; Traphagen, N.A.; Wells, J.D.; Batra, J.; Melancon, S.N.; Ringelberg, C.; Seibel, W.; et al. Exploiting mitochondrial and metabolic homeostasis as a vulnerability in NF1 deficient cells. Oncotarget 2018, 9, 15860–15875. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Bekaii-Saab, T. Selumetinib for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 24, 111–123. [Google Scholar] [CrossRef]
- Casey, D.; Demko, S.; Sinha, A.; Mishra-Kalyani, P.S.; Shen, Y.L.; Khasar, S.; Goheer, M.A.; Helms, W.S.; Pan, L.; Xu, Y.; et al. FDA Approval Summary: Selumetinib for Plexiform Neurofibroma. Clin. Cancer Res. 2021, 27, 4142–4146. [Google Scholar] [CrossRef] [PubMed]
- Decroocq, J.; Birsen, R.; Montersino, C.; Chaskar, P.; Mano, J.; Poulain, L.; Friedrich, C.; Alary, A.S.; Guermouche, H.; Sahal, A.; et al. RAS activation induces synthetic lethality of MEK inhibition with mitochondrial oxidative metabolism in acute myeloid leukemia. Leukemia 2022, 36, 1237–1252. [Google Scholar] [CrossRef]
- Feng, J.; Lian, Z.; Xia, X.; Lu, Y.; Hu, K.; Zhang, Y.; Liu, Y.; Hu, L.; Yuan, K.; Sun, Z.; et al. Targeting metabolic vulnerability in mitochondria conquers MEK inhibitor resistance in KRAS-mutant lung cancer. Acta Pharm. Sin. B 2023, 13, 1145–1163. [Google Scholar] [CrossRef]
- Esposito, T.; Schettino, C.; Polverino, P.; Allocca, S.; Adelfi, L.; D’Amico, A.; Capaldo, G.; Varriale, B.; Di Salle, A.; Peluso, G.; et al. Synergistic Interplay between Curcumin and Polyphenol-Rich Foods in the Mediterranean Diet: Therapeutic Prospects for Neurofibromatosis 1 Patients. Nutrients 2017, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef]
- Asleh, J.; Shofty, B.; Cohen, N.; Kavushansky, A.; López-Juárez, A.; Constantini, S.; Ratner, N.; Kahn, I. Brain-wide structural and functional disruption in mice with oligodendrocyte-specific. Proc. Natl. Acad. Sci. USA 2020, 117, 22506–22513. [Google Scholar] [CrossRef]
- Lee, M.J.; Hung, S.H.; Huang, M.C.; Tsai, T.; Chen, C.T. Doxycycline potentiates antitumor effect of 5-aminolevulinic acid-mediated photodynamic therapy in malignant peripheral nerve sheath tumor cells. PLoS ONE 2017, 12, e0178493. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Peng, P.C.; Tsai, T.; Chien, H.F.; Lee, M.J. A Novel Treatment Modality for Malignant Peripheral Nerve Sheath Tumor Using a Dual-Effect Liposome to Combine Photodynamic Therapy and Chemotherapy. Pharmaceutics 2020, 12, 317. [Google Scholar] [CrossRef]
- Quirk, B.; Olasz, E.; Kumar, S.; Basel, D.; Whelan, H. Photodynamic Therapy for Benign Cutaneous Neurofibromas Using Aminolevulinic Acid Topical Application and 633 nm Red Light Illumination. Photobiomodul. Photomed. Laser Surg. 2021, 39, 411–417. [Google Scholar] [CrossRef]
- Allison, R.R.; Moghissi, K. Photodynamic Therapy (PDT): PDT Mechanisms. Clin. Endosc. 2013, 46, 24–29. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Oxidative Effect | Molecular Mechanisms | Reference |
|---|---|---|---|
| Drosophila melanogaster | ↑ ROS | ↑ Adenylyl cyclase ↑ cAMP | [93] |
| MPNST cell lines | ↑ ER stress levels | ↑ Ras pathway ↑ mTOR pathway | [7] |
| MEFs MPNST cell lines | Proteotoxic stress tolerance | ↑ Ras/MAPK pathway ↑ HSF1 | [89] |
| Mouse oligodendrocytes | ↑ ROS | ↑Ras pathway ↑NO synthase | [94] |
| Mouse macrophages Human monocytic cells | ↑ ROS | ↑Ras pathway | [95] |
| MEFs U87 glioblastoma cell line | ↑ Glycolysis | ↓ SDH ↑ ERK-dependent TRAP1 ↑ HIF1α | [96] |
| MEFs MPNST cell lines | ↑ ROS | ↓ ERK-dependent NADPH dehydrogenase ↓ NAD+-dependent SIRT3 ↑ ERK-dependent TRAP1 | [97] |
| Compounds | Mechanism of Action | Setting | Reference |
|---|---|---|---|
| Curcumin | Oxidative stress | MPSNT cell lines | [87] |
| Lipoamide | Antioxidant | Mouse Schwann cells Mouse model | [92] |
| Mediterranean diet and curcumin | Antioxidant | Clinical study (case series, cutaneous neurofibromas) | [111] |
| Metalloporphyrin catalytic | Antioxidant | Drosophila melanogaster model | [93] |
| mTOR inhibitor + HDAC inhibitor | Catastrophic oxidative stress | MPNST cell lines NSCLC cell line Mouse model | [112] |
| NOS inhibitors | Antioxidant | Mouse model | [113] |
| PDT with ALA + doxycycline | Oxidative stress | MPNST cell line | [114] |
| PDT + cisplatin | Oxidative stress | MPNST cell line MPNST mouse xenograft | [115] |
| PDT with ALA | Oxidative stress | Phase I (cutaneous neurofibromas) | [116] |
| PI-504 | HSP90inhibitors | MPNST cell lines | [7] |
| Selumetinib | MEK inhibitor | FDA approved for plexiform neurofibromas | [108] |
| Thapsigargin | ER calcium-ATPase inhibitor | MPNST cell lines | [7] |
| TRAIL | ROS-influenced Proapoptotic | MPNST cell lines | [87] |
| Tunicamycin | Glycosylation inhibitor | MPNST cell lines | [7] |
| Y100 | Oxidative stress | [106] | |
| Ongoing clinical trials | |||
| Binimetinib | MEK inhibitor | Phase 1 (plexiform neurofibromas) | NCT03231306 |
| FCN-159 | MEK inhibitor | Phase 3 (inoperable plexiform neurofibromas) | NCT05913037 |
| Mediterranean diet with curcumin | Antioxidant | Phase 1 (NF1-related neurofibromas) | NCT05363267 |
| Mirdametinib | MEK inhibitor | Phase 2b (inoperable plexiform neurofibromas) | NCT03962543 |
| NAC | Antioxidant | Phase 2a (learning and motor behavior in children with NF1) Phase 2 (learning and motor behavior in children with NF1) | NCT04481035 NCT04481048 |
| NFX-179 | Topical MEK inhibitor | Phase 2a (NF1-related cutaneous neurofibromas | NCT04435665 NCT05005845 |
| TQ-B3234 | MEK inhibitor | Phase 1 (NF1-related neurofibromas and MPSNT) | NCT05107037 |
| Trametinib | MEK inhibitor | Single arm, open-label plexiform neurofibromas) | NCT03741101 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuhn, E.; Natacci, F.; Corbo, M.; Pisani, L.; Ferrero, S.; Bulfamante, G.; Gambini, D. The Contribution of Oxidative Stress to NF1-Altered Tumors. Antioxidants 2023, 12, 1557. https://doi.org/10.3390/antiox12081557
Kuhn E, Natacci F, Corbo M, Pisani L, Ferrero S, Bulfamante G, Gambini D. The Contribution of Oxidative Stress to NF1-Altered Tumors. Antioxidants. 2023; 12(8):1557. https://doi.org/10.3390/antiox12081557
Chicago/Turabian StyleKuhn, Elisabetta, Federica Natacci, Massimo Corbo, Luigi Pisani, Stefano Ferrero, Gaetano Bulfamante, and Donatella Gambini. 2023. "The Contribution of Oxidative Stress to NF1-Altered Tumors" Antioxidants 12, no. 8: 1557. https://doi.org/10.3390/antiox12081557
APA StyleKuhn, E., Natacci, F., Corbo, M., Pisani, L., Ferrero, S., Bulfamante, G., & Gambini, D. (2023). The Contribution of Oxidative Stress to NF1-Altered Tumors. Antioxidants, 12(8), 1557. https://doi.org/10.3390/antiox12081557