
Caffeine Inhibits Oxidative Stress- and Low Dose Endotoxemia-Induced Senescence—Role of Thioredoxin-1
 ,
,  ,
, _Haendeler.png)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentiviral Production and Transduction
2.3. Transient Transfection of ECs
2.4. Migration Assay
2.5. Immunoblotting
2.6. Immunostaining of ECs
2.7. Measurement of Intracellular ROS by Fluorescence Microscopy
2.8. Total Cellular RNA Isolation
2.9. cDNA Synthesis
2.10. Polymerase Chain Reaction (PCR)
2.11. Statistics
3. Results
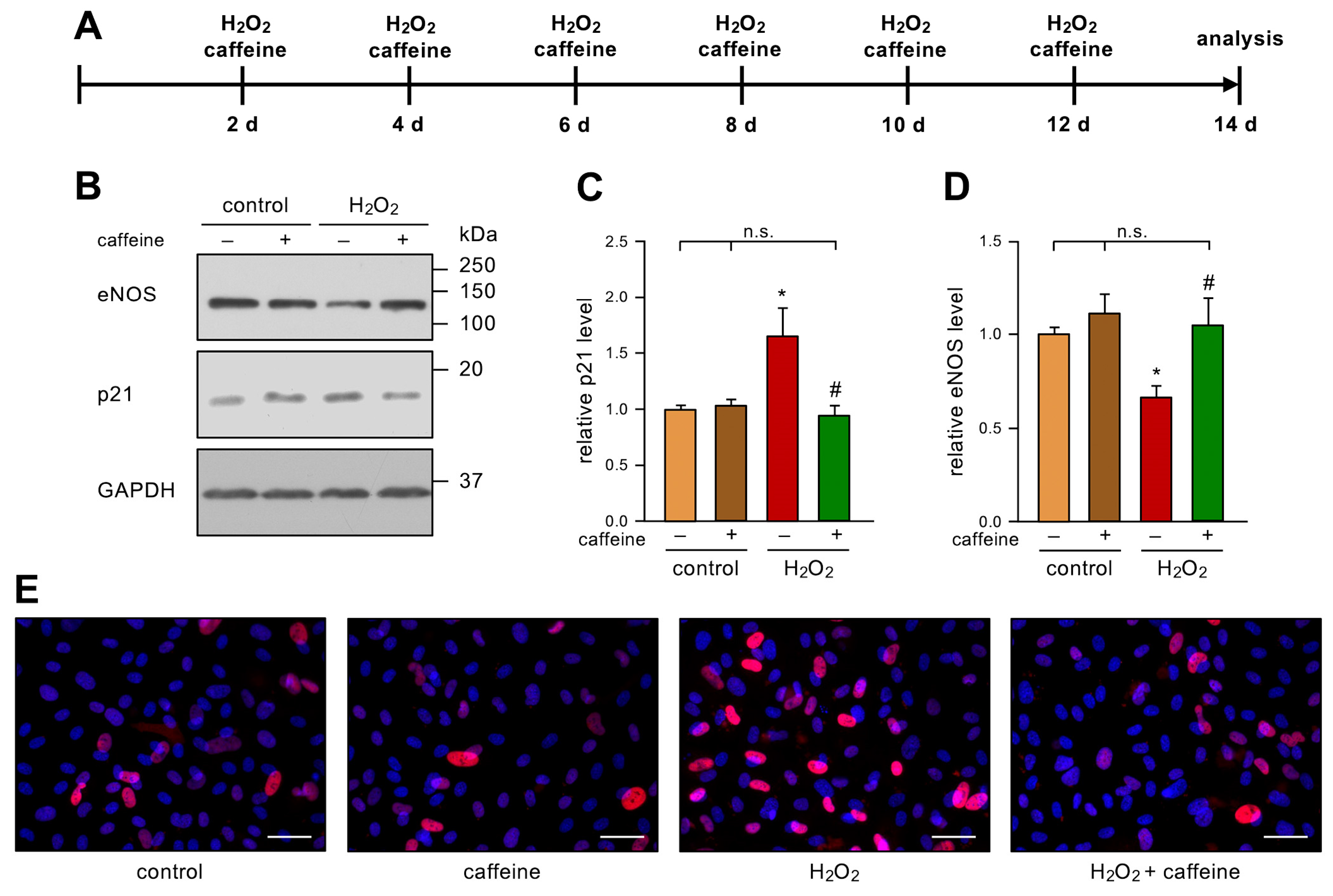
3.1. Caffeine Prevents Stress-Induced Senescence in Endothelial Cells
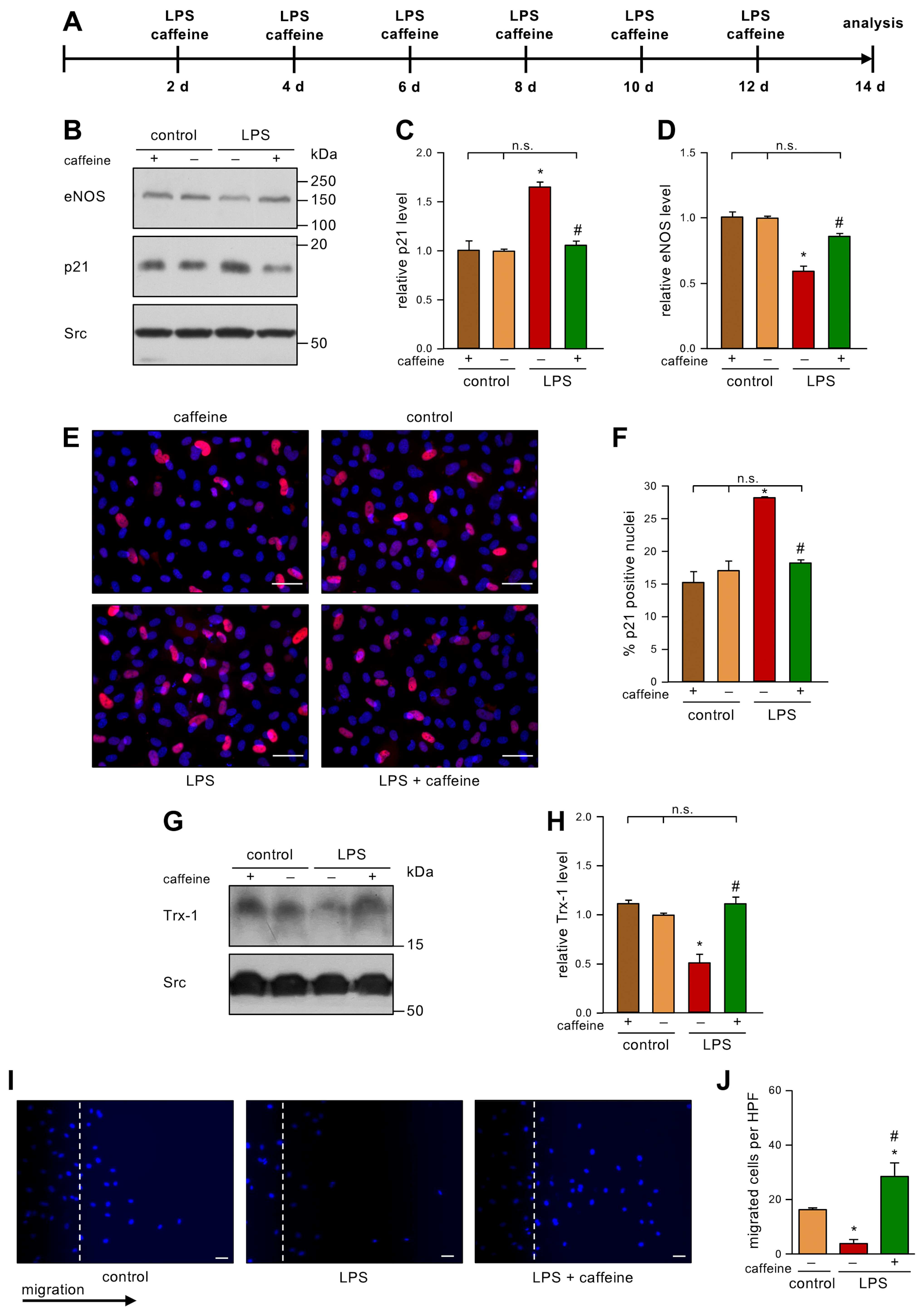
3.2. Caffeine Counteracts Low Dose Endotoxemia-Induced Senescence in Endothelial Cells
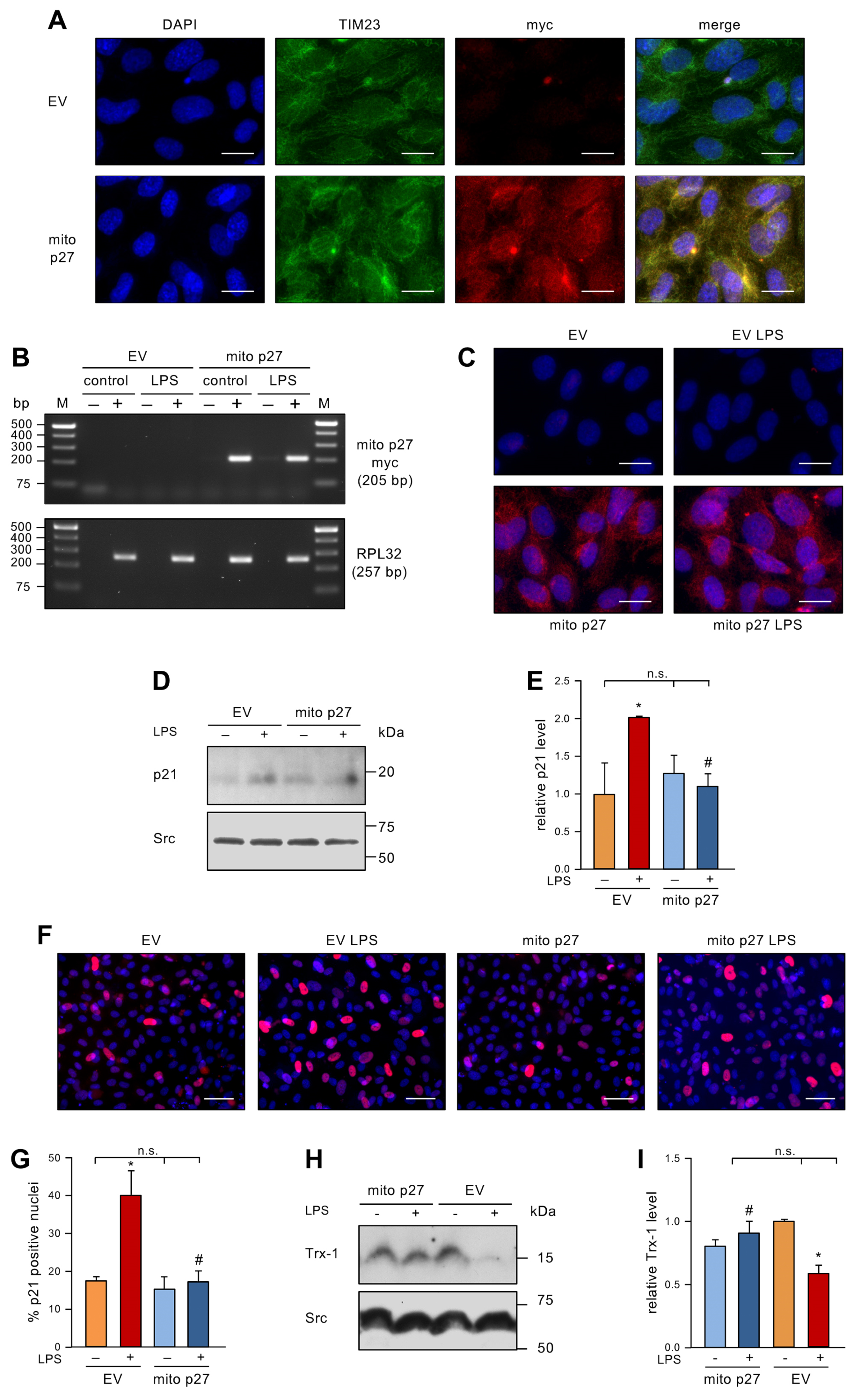
3.3. Permanent Expression of Mitochondrial p27 in Endothelial Cells Inhibits Senescence Induction by Low dose Endotoxemia
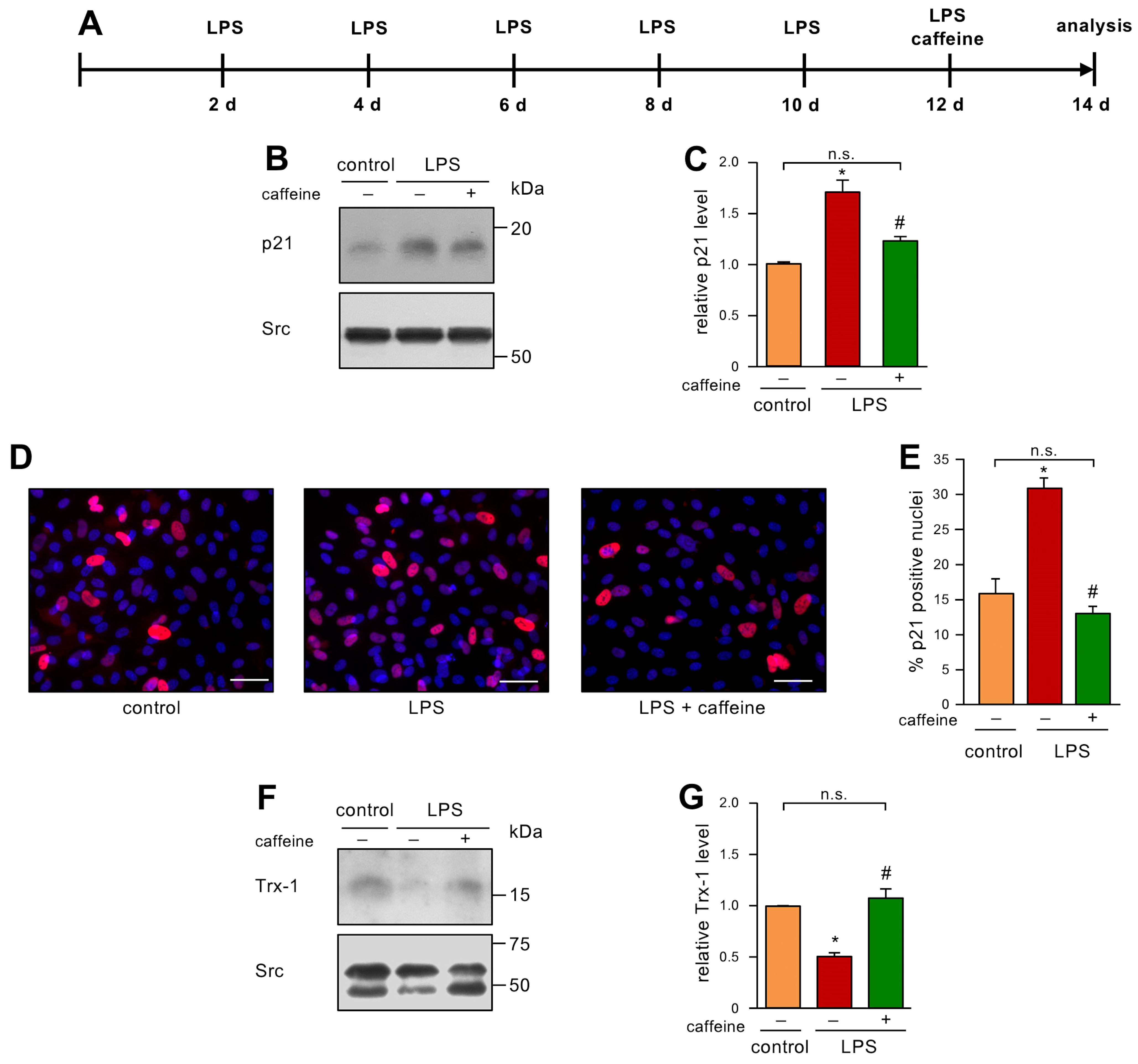
3.4. Caffeine Can Reverse Low dose endotoxemia-Induced Senescence in Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef]
- Zschauer, T.C.; Matsushima, S.; Altschmied, J.; Shao, D.; Sadoshima, J.; Haendeler, J. Interacting with thioredoxin-1--disease or no disease? Antioxid. Redox Signal. 2013, 18, 1053–1062. [Google Scholar] [CrossRef]
- Altschmied, J.; Haendeler, J. Thioredoxin-1 and endothelial cell aging: Role in cardiovascular diseases. Antioxid. Redox Signal. 2009, 11, 1733–1740. [Google Scholar] [CrossRef]
- Haendeler, J. Thioredoxin-1 and posttranslational modifications. Antioxid. Redox Signal. 2006, 8, 1723–1728. [Google Scholar] [CrossRef]
- Goy, C.; Czypiorski, P.; Altschmied, J.; Jakob, S.; Rabanter, L.L.; Brewer, A.C.; Ale-Agha, N.; Dyballa-Rukes, N.; Shah, A.M.; Haendeler, J. The imbalanced redox status in senescent endothelial cells is due to dysregulated Thioredoxin-1 and NADPH oxidase 4. Exp. Gerontol. 2014, 56, 45–52. [Google Scholar] [CrossRef]
- Buchner, N.; Ale-Agha, N.; Jakob, S.; Sydlik, U.; Kunze, K.; Unfried, K.; Altschmied, J.; Haendeler, J. Unhealthy diet and ultrafine carbon black particles induce senescence and disease associated phenotypic changes. Exp. Gerontol. 2013, 48, 8–16. [Google Scholar] [CrossRef]
- Gonnissen, S.; Ptok, J.; Goy, C.; Jander, K.; Jakobs, P.; Eckermann, O.; Kaisers, W.; von Ameln, F.; Timm, J.; Ale-Agha, N.; et al. High Concentration of Low-Density Lipoprotein Results in Disturbances in Mitochondrial Transcription and Functionality in Endothelial Cells. Oxid. Med. Cell Longev. 2019, 2019, 7976382. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Fichtlscherer, S.; Popp, R.; Toennes, S.W.; Fisslthaler, B.; Trepels, T.; Zernecke, A.; Liehn, E.A.; Weber, C.; Zeiher, A.M.; et al. Caffeine enhances endothelial repair by an AMPK-dependent mechanism. Arter. Thromb. Vasc. Biol. 2008, 28, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Ohmichi, T.; Kasai, T.; Shinomoto, M.; Matsuura, J.; Koizumi, T.; Kitani-Morii, F.; Tatebe, H.; Sasaki, H.; Mizuno, T.; Tokuda, T. Quantification of Blood Caffeine Levels in Patients With Parkinson’s Disease and Multiple System Atrophy by Caffeine ELISA. Front. Neurol. 2020, 11, 580127. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.D.; Park, Y.; Abnet, C.C.; Hollenbeck, A.R.; Sinha, R. Association of coffee drinking with total and cause-specific mortality. N Engl. J. Med. 2012, 366, 1891–1904. [Google Scholar] [CrossRef]
- Ale-Agha, N.; Goy, C.; Jakobs, P.; Spyridopoulos, I.; Gonnissen, S.; Dyballa-Rukes, N.; Aufenvenne, K.; von Ameln, F.; Zurek, M.; Spannbrucker, T.; et al. CDKN1B/p27 is localized in mitochondria and improves respiration-dependent processes in the cardiovascular system-New mode of action for caffeine. PLoS Biol. 2018, 16, e2004408. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Tischler, V.; Berk, B.C.; Zeiher, A.M.; Dimmeler, S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat. Cell Biol. 2002, 4, 743–749. [Google Scholar] [CrossRef]
- Jakob, S.; Schroeder, P.; Lukosz, M.; Buchner, N.; Spyridopoulos, I.; Altschmied, J.; Haendeler, J. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J. Biol. Chem. 2008, 283, 33155–33161. [Google Scholar] [CrossRef] [PubMed]
- Jander, K.; Greulich, J.; Gonnissen, S.; Ale-Agha, N.; Goy, C.; Jakobs, P.; Farrokh, S.; Marziano, C.; Sonkusare, S.K.; Haendeler, J.; et al. Extra-Nuclear Functions of the Transcription Factor Grainyhead-Like 3 in the Endothelium-Interaction with Endothelial Nitric Oxide Synthase. Antioxidants 2021, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Ptok, J.; Jakobs, P.; von Ameln, F.; Greulich, J.; Kluge, P.; Semperowitsch, K.; Eckermann, O.; Schaal, H.; Ale-Agha, N.; et al. Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis. Antioxidants 2021, 10, 1427. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic. Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.S.; Qi, B.; Park, Y.C.; Irani, K. Constitutive activation of rac1 results in mitochondrial oxidative stress and induces premature endothelial cell senescence. Arterioscler. Thromb. Vasc. Biol. 2003, 23, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Dyballa-Rukes, N.; Jakobs, P.; Eckers, A.; Ale-Agha, N.; Serbulea, V.; Aufenvenne, K.; Zschauer, T.C.; Rabanter, L.L.; Jakob, S.; von Ameln, F.; et al. The Anti-Apoptotic Properties of APEX1 in the Endothelium Require the First 20 Amino Acids and Converge on Thioredoxin-1. Antioxid. Redox Signal. 2017, 26, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Popp, R.; Goy, C.; Tischler, V.; Zeiher, A.M.; Dimmeler, S. Cathepsin D and H2O2 stimulate degradation of thioredoxin-1: Implication for endothelial cell apoptosis. J. Biol. Chem. 2005, 280, 42945–42951. [Google Scholar] [CrossRef]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Greenberg, J.A.; Chow, G.; Ziegelstein, R.C. Caffeinated coffee consumption, cardiovascular disease, and heart valve disease in the elderly (from the Framingham Study). Am. J. Cardiol. 2008, 102, 1502–1508. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merk, D.; Greulich, J.; Vierkant, A.; Cox, F.; Eckermann, O.; von Ameln, F.; Dyballa-Rukes, N.; Altschmied, J.; Ale-Agha, N.; Jakobs, P.; et al. Caffeine Inhibits Oxidative Stress- and Low Dose Endotoxemia-Induced Senescence—Role of Thioredoxin-1. Antioxidants 2023, 12, 1244. https://doi.org/10.3390/antiox12061244
Merk D, Greulich J, Vierkant A, Cox F, Eckermann O, von Ameln F, Dyballa-Rukes N, Altschmied J, Ale-Agha N, Jakobs P, et al. Caffeine Inhibits Oxidative Stress- and Low Dose Endotoxemia-Induced Senescence—Role of Thioredoxin-1. Antioxidants. 2023; 12(6):1244. https://doi.org/10.3390/antiox12061244
Chicago/Turabian StyleMerk, Dennis, Jan Greulich, Annika Vierkant, Fiona Cox, Olaf Eckermann, Florian von Ameln, Nadine Dyballa-Rukes, Joachim Altschmied, Niloofar Ale-Agha, Philipp Jakobs, and et al. 2023. "Caffeine Inhibits Oxidative Stress- and Low Dose Endotoxemia-Induced Senescence—Role of Thioredoxin-1" Antioxidants 12, no. 6: 1244. https://doi.org/10.3390/antiox12061244
APA StyleMerk, D., Greulich, J., Vierkant, A., Cox, F., Eckermann, O., von Ameln, F., Dyballa-Rukes, N., Altschmied, J., Ale-Agha, N., Jakobs, P., & Haendeler, J. (2023). Caffeine Inhibits Oxidative Stress- and Low Dose Endotoxemia-Induced Senescence—Role of Thioredoxin-1. Antioxidants, 12(6), 1244. https://doi.org/10.3390/antiox12061244