


The Role of Pyrazolo[3,4-d]pyrimidine-Based Kinase Inhibitors in The Attenuation of CCl4-Induced Liver Fibrosis in Rats
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Chemistry
2.2. In Vitro COX-1/COX-2 Inhibition Assay
2.3. Cytotoxicity MTT Assay
2.4. Animals Experimental Design
2.5. Biochemical Tests
2.6. Liver Tissue Specimens
2.7. Histopathological and Immunohistochemistry
2.8. Protein Determination
2.9. Assessment of Oxidative Stress Markers
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. RT PCR
2.12. Molecular Docking
2.13. Statistical Analysis
3. Results
3.1. D1-3 In Vitro COX-1/COX-2 Inhibition Ability with 100% Cell Viability
3.2. In Vivo Toxicity Study
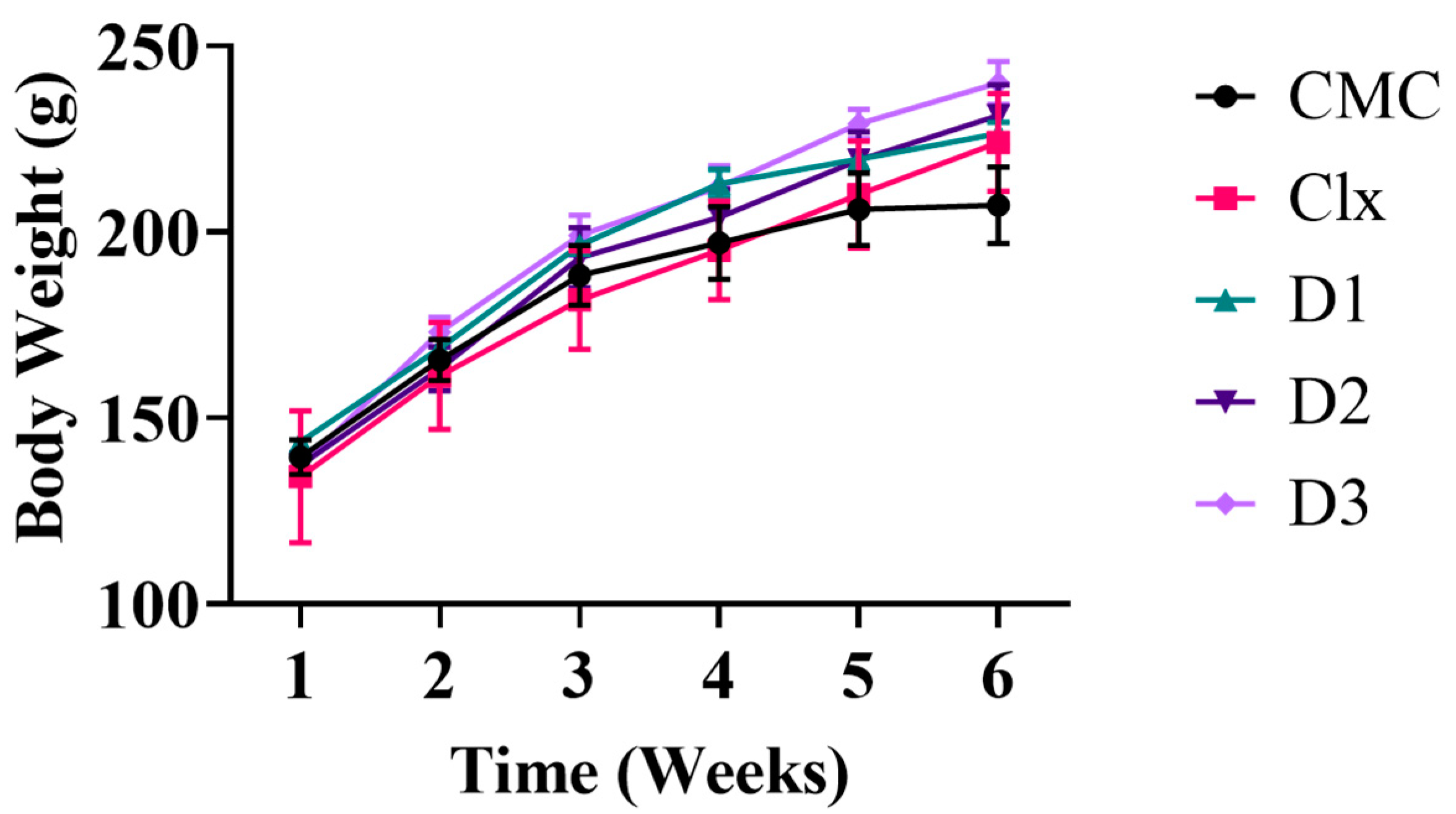
3.2.1. Effect of Celecoxib-Based Fused Ring Derivatives on Rats’ Vitals and Body Weight
3.2.2. Hepatic and Renal Safety of the Compounds
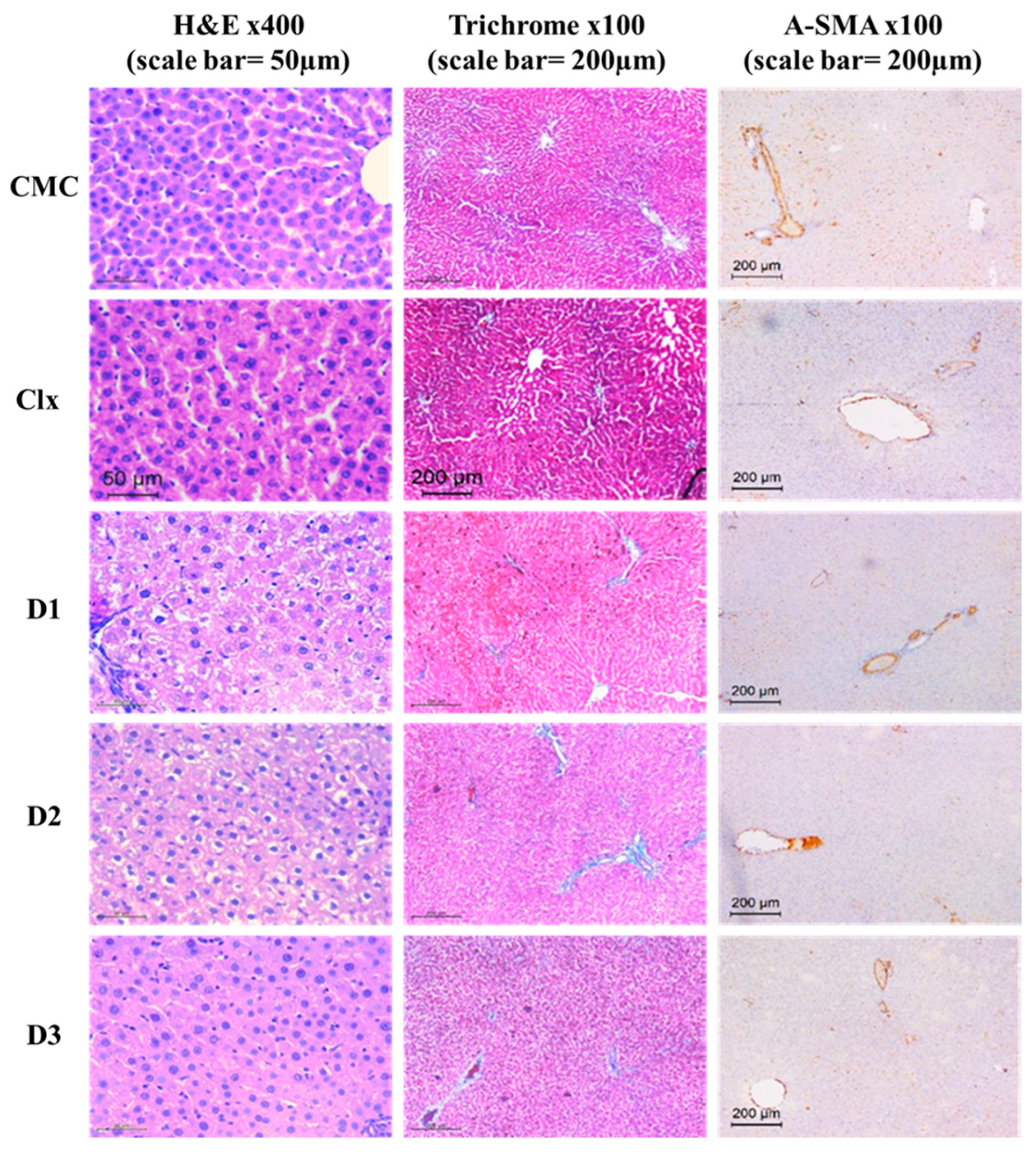
3.2.3. D1-3 Preserved Normal Cellular Architecture of the Liver
3.3. Therapeutic Model Treated after Induction of Fibrosis
3.3.1. D1-3 Suppressed the Severity of CCl4-Induced Fibrosis
3.3.2. Antioxidant Properties of D1-3 in Reducing Hepatic Fibrosis
3.3.3. D1-3 Enhanced Liver Enzymes Balance after CCl4-Induced Fibrosis
3.3.4. D1-3 Extracellular Matrix Protein Remodeling Capability
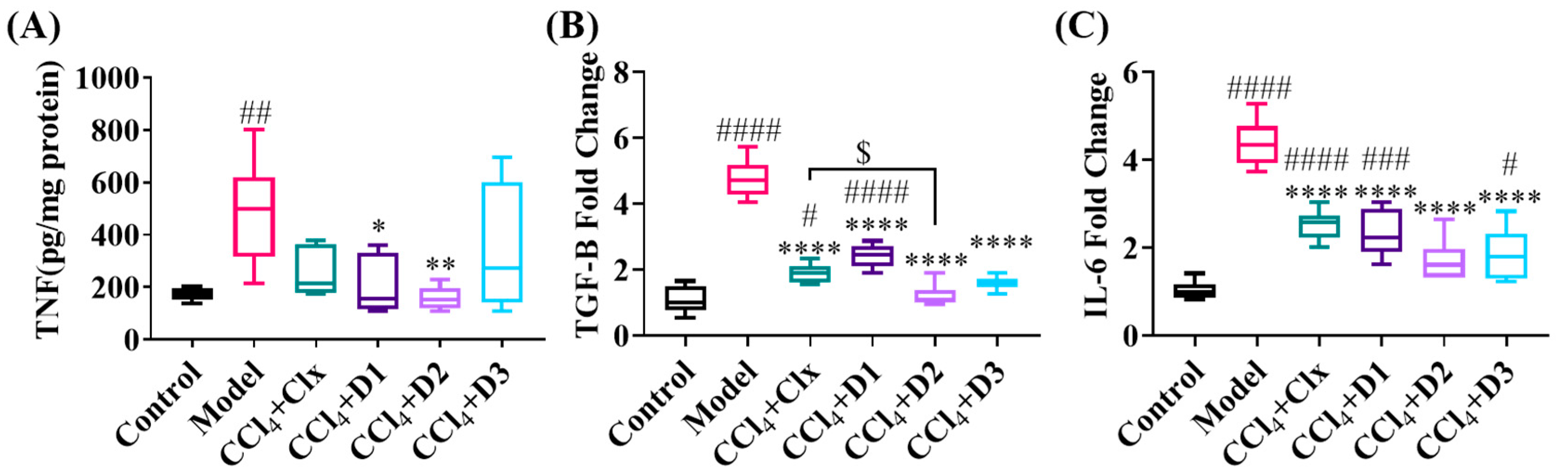
3.3.5. D1-3 Reduced Proinflammatory and Profibrogenic Cytokines in Induced Liver Injury
3.4. In Silico Prediction of D1-3 Possible Mechanism of Action
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Iwakiri, Y. Nitric oxide in liver fibrosis: The role of inducible nitric oxide synthase. Clin. Mol. Hepatol. 2015, 21, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.-Y.; Wang, N.; Zhang, Z.-J.; Lao, L.; Wong, C.-W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/gene/4323 (accessed on 1 September 2020).
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Rice, A.; Pinato, D.; Rombouts, K.; Del Rio Hernandez, A. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci. Rep. 2019, 9, 7299. [Google Scholar] [CrossRef]
- Roderfeld, M.; Weiskirchen, R.; Wagner, S.; Berres, M.L.; Henkel, C.; Grötzinger, J.; Gressner, A.M.; Matern, S.; Roeb, E. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J. 2006, 20, 444–454. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Yang, Y.M.; Seki, E. TNFα in Liver Fibrosis. Curr. Pathobiol. Rep. 2015, 3, 253–261. [Google Scholar] [CrossRef]
- Seki, E.; Brenner, D. Recent advancement of molecular mechanisms of liver fibrosis. J. Hepato-Biliary-Pancreat. Sci. 2015, 22, 512–518. [Google Scholar] [CrossRef]
- Li, J.; Tuo, B. Current and Emerging Approaches for Hepatic Fibrosis Treatment. Gastroenterol. Res. Pract. 2021, 2021, 6612892. [Google Scholar] [CrossRef]
- Jeong, S.W.; Jang, J.Y.; Lee, S.H.; Kim, S.G.; Cheon, Y.K.; Kim, Y.S.; Cho, Y.D.; Kim, H.S.; Lee, J.S.; Jin, S.-Y.; et al. Increased Expression of Cyclooxygenase-2 is Associated with the Progression to Cirrhosis. Korean J. Intern. Med. 2010, 25, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Chávez, E.; Segovia, J.; Shibayama, M.; Tsutsumi, V.; Vergara, P.; Castro-Sánchez, L.; Salazar, E.P.; Moreno, M.G.; Muriel, P. Antifibrotic and fibrolytic properties of celecoxib in liver damage induced by carbon tetrachloride in the rat. Liver Int. 2010, 30, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Ftahy, M.M.; Latif, N.S.A.; Alalkamy, E.F.; El-Batrawi, F.A.; Galal, A.H.; Khatab, H.M. Antifibrotic potential of a selective COX-2 inhibitor (celecoxib) on liver fibrosis in rats. Comp. Clin. Pathol. 2012, 22, 425–430. [Google Scholar] [CrossRef]
- Paik, Y.-H.; Kim, J.K.; I Lee, J.; Kang, S.H.; Kim, D.Y.; An, S.H.; Lee, S.J.; Lee, D.K.; Han, K.-H.; Chon, C.Y.; et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut 2009, 58, 1517–1527. [Google Scholar] [CrossRef]
- Moon, S.H.; Lee, S.J.; Lee, S.C.; Bae, Y.S. Therapeutic Agent for Liver Diseases. U.S. Patent 10,966,969 B2, 6 April 2021. [Google Scholar]
- Zheng, G.-H.; Liu, J.; Guo, F.Y.; Zhang, Z.-H.; Jiang, Y.-J.; Lin, Y.-C.; Lan, X.-Q.; Ren, J.; Wu, Y.-L.; Nan, J.-X.; et al. The in vitro and in vivo study of a pyrazole derivative, J-1063, as a novel anti-liver fibrosis agent: Synthesis, biological evaluation, and mechanistic analysis. Bioorganic Chem. 2022, 122, 105715. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- El Razik, H.A.A.; Mroueh, M.; Faour, W.H.; Shebaby, W.N.; Daher, C.F.; Ashour, H.M.A.; Ragab, H.M. Synthesis of new pyrazolo[3,4-d]pyrimidine derivatives and evaluation of their anti-inflammatory and anticancer activities. Chem. Biol. Drug Des. 2017, 90, 83–96. [Google Scholar] [CrossRef]
- Othman, E.; Bekhit, A.; Anany, M.; Dandekar, T.; Ragab, H.; Wahid, A. Design, Synthesis, and Anticancer Screening for Repurposed Pyrazolo[3,4-d]pyrimidine Derivatives on Four Mammalian Cancer Cell Lines. Molecules 2021, 26, 2961. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Waly, O.M.; Saad, K.M.; El-Subbagh, H.I.; Bayomi, S.M.; Ghaly, M.A. Synthesis, biological evaluation, and molecular modeling simulations of new heterocyclic hybrids as multi-targeted anti-Alzheimer’s agents. Eur. J. Med. Chem. 2022, 231, 114152. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Son, J.Y.; Kim, K.S.; Park, Y.J.; Kim, H.R.; Park, J.H.; Kim, K.-B.; Lee, K.Y.; Kang, K.W.; Kim, I.S.; et al. Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats. Int. J. Mol. Sci. 2019, 20, 3709. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-W.; Gong, J.; Chang, X.M.; Luo, J.Y.; Dong, L.; Hao, Z.M.; Jia, A.; Xu, G.P. Estrogen reduces CCL4- induced liver fibrosis in rats. World J. Gastroenterol. 2002, 8, 883–887. [Google Scholar] [CrossRef]
- Vogler, G.A. Chapter 19—Anesthesia and Analgesia. In The Laboratory Rat, 2nd ed.; Suckow, M.A., Weisbroth, S.H., Franklin, C.L., Eds.; Academic Press: Burlington, NJ, USA, 2006; pp. 627–664. [Google Scholar] [CrossRef]
- Jung, Y.; Lee, Y.; Kim, J.; Kim, W.; Nam, J.; Jeong, S.; Lee, S.; Yoo, J.-W.; Kim, M.-S. Celecoxib coupled to dextran via a glutamic acid linker yields a polymeric prodrug suitable for colonic delivery. Drug Des. Dev. Ther. 2015, 9, 4105–4113. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Wang, T.; You, H.; Jia, J. N-methyl-4-isoleucine cyclosporine attenuates CCl4-induced liver fibrosis in rats by interacting with cyclophilin B and D. J. Gastroenterol. Hepatol. 2010, 26, 558–567. [Google Scholar] [CrossRef]
- Tammen, H.; Schulte, I.; Hess, R.; Menzel, C.; Kellmann, M.; Mohring, T.; Schulz-Knappe, P. Peptidomic analysis of human blood specimens: Comparison between plasma specimens and serum by differential peptide display. Proteomics 2005, 5, 3414–3422. [Google Scholar] [CrossRef]
- Zhao, X.-Y.; Wang, B.-E.; Li, X.-M.; Wang, T.-L. Newly proposed fibrosis staging criterion for assessing carbon tetrachloride- and albumin complex-induced liver fibrosis in rodents. Pathol. Int. 2008, 58, 580–588. [Google Scholar] [CrossRef]
- Hamza, A.A.; Lashin, F.M.; Gamel, M.; Hassanin, S.O.; Abdalla, Y.; Amin, A. Hawthorn Herbal Preparation from Crataegus oxyacantha Attenuates In Vivo Carbon Tetrachloride -Induced Hepatic Fibrosis via Modulating Oxidative Stress and Inflammation. Antioxidants 2020, 9, 1173. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Beutler, E.; Duron, O.; Kelly, B.M. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–888. [Google Scholar]
- Lee, P.Y.; Costumbrado, J.; Hsu, C.Y.; Kim, Y.H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. 2012, 62, e3923. [Google Scholar] [CrossRef]
- Rashad, A.Y.; Kassab, S.E.; Daabees, H.G.; Moneim, A.E.A.; Rostom, S.A. Febuxostat-based amides and some derived heterocycles targeting xanthine oxidase and COX inhibition. Synthesis, in vitro and in vivo biological evaluation, molecular modeling and in silico ADMET studies. Bioorg. Chem. 2021, 113, 104948. [Google Scholar] [CrossRef]
- Gellibert, F.; Woolven, J.; Fouchet, M.-H.; Mathews, N.; Goodland, H.; Lovegrove, V.; Laroze, A.; Nguyen, V.-L.; Sautet, S.; Wang, R.; et al. Identification of 1,5-Naphthyridine Derivatives as a Novel Series of Potent and Selective TGF-β Type I Receptor Inhibitors. J. Med. Chem. 2004, 47, 4494–4506. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.Y.; Milton, M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xuefeng, Y.; Shandong, W.; Jianhua, X. COX-2 in liver fibrosis. Clin. Chim. Acta 2020, 506, 196–203. [Google Scholar] [CrossRef]
- El-Din, M.M.M.; Senbel, A.M.; Bistawroos, A.A.; El-Mallah, A.; El-Din, N.A.N.; Bekhit, A.A.; El Razik, H.A.A. A Novel COX-2 Inhibitor Pyrazole Derivative Proven Effective as an Anti-Inflammatory and Analgesic Drug. Basic Clin. Pharmacol. Toxicol. 2010, 108, 263–273. [Google Scholar] [CrossRef]
- A Willoughby, D.; Moore, A.R.; Colville-Nash, P.R. COX-1, COX-2, and COX-3 and the future treatment of chronic inflammatory disease. Lancet 2000, 355, 646–648. [Google Scholar] [CrossRef]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General Cytotoxicity Assessment by Means of the MTT Assay. Methods Mol. Biol. 2014, 1250, 333–348. [Google Scholar] [CrossRef]
- Bampidis, V.; Azimonti, G.; Bastos, M.L.; Christensen, H.; Dusemund, B.; Kos Durjava, M.; Kouba, M.; López-Alonso, M.; López Puente, S.; Marcon, F.; et al. Safety and efficacy of sodium carboxymethyl cellulose for all animal species. EFSA J. 2020, 18, e06211. [Google Scholar] [CrossRef]
- Mondal, M.I.; Yeasmin, M.S. Toxicity study of food-grade carboxymethyl cellulose synthesized from maize husk in Swiss albino mice. Int. J. Biol. Macromol. 2016, 92, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Hou, L.; Che, G.; Shi, Y.; Liu, X.; Sun, L.; Jia, W.; Zhu, F.; Zhao, Z.; Xu, M.; et al. Dose- and time-dependent systemic adverse reactions of sodium carboxy methyl cellulose after intraperitoneal application in rats. J. Toxicol. Sci. 2021, 46, 223–234. [Google Scholar] [CrossRef]
- Jothy, S.L.; Zakaria, Z.; Chen, Y.; Lau, Y.L.; Latha, L.Y.; Sasidharan, S. Acute Oral Toxicity of Methanolic Seed Extract of Cassia fistula in Mice. Molecules 2011, 16, 5268–5282. [Google Scholar] [CrossRef]
- Lee, J.-M.; Lee, M.-A.; Do, H.-N.; Song, Y.-I.; Bae, R.-J.; Lee, H.-Y.; Park, S.H.; Kang, J.S.; Kang, J.-K. Historical control data from 13-week repeated toxicity studies in Crj:CD (SD) rats. Lab. Anim. Res. 2012, 28, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Dzoyem, J.P.; Kuete, V.; Eloff, J.N. 23—Biochemical Parameters in Toxicological Studies in Africa: Significance, Principle of Methods, Data Interpretation, and Use in Plant Screenings. In Toxicological Survey of African Medicinal Plants; Kuete, V., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 659–715. [Google Scholar] [CrossRef]
- Sauer, J.-M.; Stine, E.R.; Gunawardhana, L.; Hill, D.A.; Sipes, I.G. The Liver as a Target for Chemical-Chemical Interactions. In Advances in Pharmacology; Li, A.P., Ed.; Academic Press: Cambridge, MA, USA, 1997; Volume 43, pp. 37–63. [Google Scholar]
- Gurina, T.S.; Simms, L. Histology, Staining. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Medina, J.; Moreno-Otero, R. Pathophysiological Basis for Antioxidant Therapy in Chronic Liver Disease. Drugs 2005, 65, 2445–2461. [Google Scholar] [CrossRef]
- Sanchez-Valle, V.; Chavez-Tapia, N.; Uribe, M.; Mendez-Sanchez, N. Role of Oxidative Stress and Molecular Changes in Liver Fibrosis: A Review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Niederreiter, L.; Tilg, H. Cytokines and fatty liver diseases. Liver Res. 2018, 2, 14–20. [Google Scholar] [CrossRef]
- Tirani, M.M.; Haghjou, M.M. Reactive oxygen species (ROS), total antioxidant capacity (AOC) and malondialdehyde (MDA) make a triangle in evaluation of zinc stress extension. J. Anim. Plant Sci. 2019, 29, 1100–1111. [Google Scholar]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 2020, 11, 574276. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.; Schierwagen, R.; Schaefer, L.; Klein, S.; Trepat, X.; Trebicka, J. Extracellular Matrix Remodeling in Chronic Liver Disease. Curr. Tissue Microenviron. Rep. 2021, 2, 41–52. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Zhang, J.; Lu, L.; Feng, M.; Wang, J. Dynamic features of liver fibrogenesis and fibrosis resolution in the absence of matrix metalloproteinase-9. Mol. Med. Rep. 2019, 20, 5239–5248. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Jiang, T.; Qi, X.; Zhao, Z.; Li, B.; Aibibula, M.; Min, H.; Zhang, J.; Liu, Y.; Ma, X. Role of Cytokines on the Progression of Liver Fibrosis in Mice Infected with Echinococcus multilocularis. Infect. Drug Resist. 2021, 14, 5651–5660. [Google Scholar] [CrossRef]
- Nie, Q.-H.; Zhang, Y.-F.; Xie, Y.-M.; Luo, X.-D.; Shao, B.; Li, J.; Zhou, Y.-X. Correlation between TIMP-1 expression and liver fibrosis in two rat liver fibrosis models. World J. Gastroenterol. 2006, 12, 3044–3049. [Google Scholar] [CrossRef] [PubMed]
- El-Baz, F.K.; Salama, A.; Salama, R.A.A. Therapeutic Effect of Dunaliella salina Microalgae on Thioacetamide- (TAA-) Induced Hepatic Liver Fibrosis in Rats: Role of TGF-β and MMP9. BioMed Res. Int. 2019, 2019, 7028314. [Google Scholar] [CrossRef]
- Kakino, S.; Ohki, T.; Nakayama, H.; Yuan, X.; Otabe, S.; Hashinaga, T.; Wada, N.; Kurita, Y.; Tanaka, K.; Hara, K.; et al. Pivotal Role of TNF-α in the Development and Progression of Nonalcoholic Fatty Liver Disease in a Murine Model. Horm. Metab. Res. 2017, 50, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Kang, H.; Yang, Y.; Pyun, K. IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin. Exp. Immunol. 1994, 95, 530–535. [Google Scholar] [CrossRef]
- Tacke, F.; Hammerich, L. Interleukins in chronic liver disease: Lessons learned from experimental mouse models. Clin. Exp. Gastroenterol. 2014, 7, 297–306. [Google Scholar] [CrossRef]
- Hafez, M.M.; Al-Harbi, N.O.; Al-Hoshani, A.R.; Al-Hosaini, K.A.; Al Shrari, S.D.; Al Rejaie, S.S.; Sayed-Ahmed, M.M.; Al-Shabanah, O.A. Hepato-protective effect of rutin via IL-6/STAT3 pathway in CCl4-induced hepatotoxicity in rats. Biol. Res. 2015, 48, 30. [Google Scholar] [CrossRef]
- Mair, M.; Zollner, G.; Schneller, D.; Musteanu, M.; Fickert, P.; Gumhold, J.; Schuster, C.; Fuchsbichler, A.; Bilban, M.; Tauber, S.; et al. Signal Transducer and Activator of Transcription 3 Protects From Liver Injury and Fibrosis in a Mouse Model of Sclerosing Cholangitis. Gastroenterology 2010, 138, 2499–2508. [Google Scholar] [CrossRef]
- Wu, Y.; Min, J.; Ge, C.; Shu, J.; Tian, D.; Yuan, Y.; Zhou, D. Interleukin 22 in liver injury, inflammation and cancer. Int. J. Biol. Sci. 2020, 16, 2405. [Google Scholar] [CrossRef] [PubMed]
- An, S.Y.; Petrescu, A.D.; DeMorrow, S. Targeting Certain Interleukins as Novel Treatment Options for Liver Fibrosis. Front. Pharmacol. 2021, 12, 645703. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef]
- Li, M.; Hou, Q.; Zhong, L.; Zhao, Y.; Fu, X. Macrophage Related Chronic Inflammation in Non-Healing Wounds. Front. Immunol. 2021, 12, 681710. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Kopp, J.B. TGF-β and fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef]
- Lo, R.C.; Kim, H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin. Mol. Hepatol. 2017, 23, 302–307. [Google Scholar] [CrossRef]
- Ishibashi, H.; Nakamura, M.; Komori, A.; Migita, K.; Shimoda, S. Liver architecture, cell function, and disease. Semin. Immunopathol. 2009, 31, 399–409. [Google Scholar] [CrossRef]
- Nouchi, T.; Tanaka, Y.; Tsukada, T.; Sato, C.; Marumo, F. Appearance of α-smooth-muscle-actin-positive cells in hepatic fibrosis. Liver Int. 1991, 11, 100–105. [Google Scholar] [CrossRef]
- Gitiara, A.; Tokhanbigli, S.; Mazhari, S.; Baghaei, K.; Hatami, B.; Hashemi, S.M.; Rad, A.A.; Moradi, A.; Nasiri, M.; Ahrabi, N.Z.; et al. Development of experimental fibrotic liver diseases animal model by Carbon Tetracholoride. Gastroenterol. Hepatol. Bed Bench 2017, 10, S122–S128. [Google Scholar]
- Ayatollahi, M.; Hesami, Z.; Jamshidzadeh, A.; Gramizadeh, B. Antioxidant Effects of Bone Marrow Mesenchymal Stem Cell against Carbon Tetrachloride-Induced Oxidative Damage in Rat Livers. Int. J. Organ Transplant. Med. 2014, 5, 166–173. [Google Scholar] [PubMed]
- Bao, Y.-L.; Wang, L.; Pan, H.-T.; Zhang, T.-R.; Chen, Y.-H.; Xu, S.-J.; Mao, X.-L.; Li, S.-W. Animal and Organoid Models of Liver Fibrosis. Front. Physiol. 2021, 12, 666138. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Chen, Q.-L.; Song, Y.-N.; Sun, Y.; Wei, B.; Li, X.-Y.; Hu, Y.-Y.; Liu, P.; Su, S.-B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef]
- Sayed, H.E.S.A.E.; Morsy, L.E.S.; Emara, T.M.A.; Galhom, R.A. Effect of Carbon Tetrachloride (CCL4) on Liver in Adult Albino Rats: Histological study. Egypt. J. Hosp. Med. 2019, 76, 4254–4261. [Google Scholar] [CrossRef]
- Su, W.; Tai, Y.; Tang, S.-H.; Ye, Y.-T.; Zhao, C.; Gao, J.-H.; Tuo, B.-G.; Tang, C.-W. Celecoxib attenuates hepatocyte apoptosis by inhibiting endoplasmic reticulum stress in thioacetamide-induced cirrhotic rats. World J. Gastroenterol. 2020, 26, 4094–4107. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Huang, Z.; Jiang, J.; Gao, J.; Zhao, C.; Tai, Y.; Ma, X.; Zhang, L.; Ye, Y.; Gan, C.; et al. Celecoxib ameliorates liver cirrhosis via reducing inflammation and oxidative stress along spleen-liver axis in rats. Life Sci. 2021, 272, 119203. [Google Scholar] [CrossRef]
- Zhang, L.; Tai, Y.; Zhao, C.; Ma, X.; Tang, S.; Tong, H.; Tang, C.; Gao, J. Inhibition of cyclooxygenase-2 enhanced intestinal epithelial homeostasis via suppressing β-catenin signalling pathway in experimental liver fibrosis. J. Cell. Mol. Med. 2021, 25, 7993–8005. [Google Scholar] [CrossRef]
- Tai, Y.; Zhao, C.; Zhang, L.; Tang, S.; Jia, X.; Tong, H.; Liu, R.; Tang, C.; Gao, J. Celecoxib reduces hepatic vascular resistance in portal hypertension by amelioration of endothelial oxidative stress. J. Cell. Mol. Med. 2021, 25, 10389–10402. [Google Scholar] [CrossRef]
- Hui, A.Y.; Leung, W.K.; Chan, H.L.Y.; Chan, F.K.; Go, M.Y.Y.; Chan, K.K.; Tang, B.D.; Chu, E.S.H.; Sung, J.J.Y. Effect of celecoxib on experimental liver fibrosis in rat. Liver Int. 2005, 26, 125–136. [Google Scholar] [CrossRef]
- Liu, H.; Wei, W.; Li, X. Celecoxib exacerbates hepatic fibrosis and induces hepatocellular necrosis in rats treated with porcine serum. Prostaglandins Other Lipid Mediat. 2009, 88, 63–67. [Google Scholar] [CrossRef]
- Yu, J.; Hui, A.Y.; Chu, E.S.H.; Go, M.Y.Y.; Cheung, K.F.; Wu, C.W.; Chan, H.L.Y.; Sung, J.J.Y. The anti-inflammatory effect of celecoxib does not prevent liver fibrosis in bile duct-ligated rats. Liver Int. 2009, 29, 25–36. [Google Scholar] [CrossRef]
- Harris, T.R.; Kodani, S.; Rand, A.A.; Yang, J.; Imai, D.M.; Hwang, S.H.; Hammock, B.D. Celecoxib Does Not Protect against Fibrosis and Inflammation in a Carbon Tetrachloride–Induced Model of Liver Injury. Mol. Pharmacol. 2018, 94, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Huang, Z.; Lin, T.; Liu, S.; Chang, H.; Yan, Z.; Zhang, H.; Liu, C. New Insight into the Anti-liver Fibrosis Effect of Multitargeted Tyrosine Kinase Inhibitors: From Molecular Target to Clinical Trials. Front. Pharmacol. 2016, 6, 300. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yang, X.; Duan, Y.; Han, J.; Liao, C. Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. J. Med. Chem. 2021, 64, 1283–1345. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; An, H.-T.; Park, J.-S.; Park, O.; Duh, A.J.; Kim, K.; Chung, K.H.; Lee, K.C.; Oh, Y.; Lee, S. Tyrosine kinase inhibitor neratinib attenuates liver fibrosis by targeting activated hepatic stellate cells. Sci. Rep. 2020, 10, 14756. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2017, 67, 549–559. [Google Scholar] [CrossRef]
- Gonzalo, T.; Beljaars, L.; van de Bovenkamp, M.; Temming, K.; van Loenen, A.-M.; Reker-Smit, C.; Meijer, D.K.F.; Lacombe, M.; Opdam, F.; Kéri, G.; et al. Local Inhibition of Liver Fibrosis by Specific Delivery of a Platelet-Derived Growth Factor Kinase Inhibitor to Hepatic Stellate Cells. J. Pharmacol. Exp. Ther. 2007, 321, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-R.; Ma, H.-D.; Tsuneyama, K.; Yang, W.; Wang, Y.-H.; Lu, F.-T.; Liu, C.-H.; Liu, P.; He, X.-S.; Diehl, A.M.; et al. STAT3-mediated attenuation of CCl4-induced mouse liver fibrosis by the protein kinase inhibitor sorafenib. J. Autoimmun. 2013, 46, 25–34. [Google Scholar] [CrossRef]
- Brown, M.; Cheung, M.; Dickerson, S.; Drewry, D.; Lackey, K.; Peat, A.; Thomson, S.; Veal, J.; Wilson, J. Pyrazolopyrimidines as Kinase Inhibitors. U.S. Patent 2005/0267133 A1, 1 December 2005. [Google Scholar]
- Rostom, S.A.F.; Hassan, G.; El-Subbagh, H. Synthesis and Biological Evaluation of Some Polymethoxylated Fused Pyridine Ring Systems as Antitumor Agents. Arch. Pharm. 2009, 342, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.; Badr, M.H.; El Razik, H.A.A.; Ashour, H.M.; Wahab, A.E.A. Synthesis of Some Pyrazolines and Pyrimidines Derived from Polymethoxy Chalcones as Anticancer and Antimicrobial Agents. Arch. Pharm. 2011, 344, 572–587. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Rostom, S.A.F.; Badr, M.H.; Ismail, A.E.; Almohammadi, A.M. Synthesis of Some 1,4,6-Trisubstituted-2-oxo-1,2-dihydropyridine-3-carbonitriles and Their Biological Evaluation as Cytotoxic and Antimicrobial Agents. Arch. Pharm. 2015, 348, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.; Badr, M.H.; El Razik, H.A.A.; Ashour, H.M. Structure-based development of novel triazoles and related thiazolotriazoles as anticancer agents and Cdc25A/B phosphatase inhibitors. Synthesis, in vitro biological evaluation, molecular docking and in silico ADME-T studies. Eur. J. Med. Chem. 2017, 139, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Taguchi, K.; Moroi, Y.; Enoki, Y.; Tokuda, R.; Yamasaki, K.; Imoto, S.; Matsumoto, K. Trimethoxy Trityl Groups as a Potent Substituent for Anti-cancer Cytidine Analog Prodrugs. J. Pharm. Sci. 2022, 111, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.K.; Malagobadan, S.; Arshad, N.M.; Nagoor, N.H. A Review of the Structure—Activity Relationship of Natural and Synthetic Antimetastatic Compounds. Biomolecules 2020, 10, 138. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Group | Weeks | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Toxicity study (n = 40) | CMC (n = 8) | 2.4 mL/kg of CMC (0.5%) in deionized water, orally, daily | - | ||||||||||
| Clx (n = 8) | 20 mg/kg of celecoxib (8.33 mg/mL) in 0.5%CMC, orally, daily [15,27] | - | |||||||||||
| D1 (n = 8) | D1 was given orally in a dose equimolar to celecoxib;18.058 mg/kg/day | - | |||||||||||
| D2 (n = 8) | D2 was given orally in a dose equimolar to celecoxib;18.18 mg/kg/day | - | |||||||||||
| D3 (n = 8) | D3 was given orally in a dose equimolar to celecoxib;21.2 mg/kg/day | - | |||||||||||
| Therapeutic model (n = 58) | Control (n = 8) | 1.2 mL/kg of corn oil, IP, twice a week | CMC was given orally (same dose as CMC group) | ||||||||||
| Model (n = 10) | 2 mL/kg of CCl4 (40%) in corn oil, IP, twice a week [28] | CMC was given orally (same dose as CMC group) | |||||||||||
| CCl4 + Clx (n = 10) | CCl4 was injected (same dose as model group) | Clx was given orally (same dose as Clx group) | |||||||||||
| CCl4 + D1 (n = 10) | CCl4 was injected (same dose as model group) | D1 was given orally (same dose as D1 group) | |||||||||||
| CCl4 + D2 (n = 10) | CCl4 was injected (same dose as model group) | D2 was given orally (same dose as D2 group) | |||||||||||
| CCl4 + D3 (n = 10) | CCl4 was injected (same dose as model group) | D3 was given orally (same dose as D3 group) | |||||||||||
| Gene Symbol | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| GAPDH | GTA TTG GGC GCC TGG TCA CC | CGC TCC TGG AAG ATG GTG ATG G |
| TGF-β | ATC CCT GCG ACC CAC ACA AG | CAA CTG CTT TGG AAG GAC TCG |
| MMP-9 | CAATCCTTGCAATGTGGATG | TAAGGAAGGGGCCCTGTAAT |
| Il-6 | TGA TGG ATG CTT CCA AAC TG | GAG CAT TGG AAG TTG GGG TA |
| α-SMA | CGA TAG AAC ACG GCA TCA TCA C | GCA TAG CCC TCA TAG ATA GGC A |
| COL1A1 | CAT GTT CAG CTT TGT GGA CCT | GCA GCT GAC TTC AGG GAT GT |
| Compound | In Vitro Inhibition IC50 ± SEM (µM) | Selectivity Ratio COX-1/COX-2 | Cytotoxicity IC50 ± SEM (µM) | Safety Index (SI) * | |
|---|---|---|---|---|---|
| COX-1 | COX_2 | ||||
| D1 | 7.5 ± 0.09 | 0.135 ± 0.0004 | 55.5 | 370.7 ± 0.089 | 2745.9 |
| D2 | 8.5 ± 0.04 | 0.103 ± 0.028 | 82.5 | 344.95 ± 0.30 | 3349.0 |
| D3 | 11.5 ± 0.05 | 0.089 ± 0.0004 | 129.2 | 356.5 ± 2.9 | 4042.7 |
| Celecoxib | 14.5 ± 0.047 | 0.046 ± 0.001 | 312.9 | 144.63 ± 1.08 | 3144.1 |
| Indomethacin | 0.099 ± 0.0004 | 0.079 ± 0.0004 | 1.25 | ||
| Vehicle Control (CMC) | Clx | D1 | D2 | D3 | |
|---|---|---|---|---|---|
| Cholesterol (mg/dL) | 64.5 ± 4.98 | 52.5 ± 2.21 | 71.66 ± 7 | 67 ± 6.47 | 62.33 ± 1.68 |
| Triglycerides (mg/dL) | 49.16 ± 9.18 | 53 ± 7.11 | 67 ± 5.42 | 61.83 ± 7.11 | 64 ± 8.01 |
| ALT (U/L) | 37.33 ± 2.99 | 42 ± 3.33 | 49.66 ± 4.99 | 39.66 ± 2.13 | 37.83 ± 2.45 |
| AST (U/L) | 156.16 ± 9.68 | 147.5 ± 18.65 | 139.5 ± 7.78 | 156.16 ± 9.68 | 147.33 ± 9.81 |
| ALP (U/L) | 133.5 ± 6.67 | 138.66 ± 13.40 | 159.5 ± 7.48 | 154.16 ± 10.92 | 123.16 ± 4.12 |
| Total Bilirubin (mg/dL) | 0.081 ± 0.01 | 0.084 ± 0.006 | 0.073 ± 0.004 | 0.071 ± 0.01 | 0.076 ± 0.006 |
| Albumin (g/dL) | 3.78 ± 0.09 | 3.73 ± 0.06 | 3.9 ± 0.12 | 3.78 ± 0.12 | 3.86 ± 0.12 |
| Creatinine (mg/dL) | 0.40 ± 0.05 | 0.35 ± 0.01 | 0.36 ± 0.03 | 0.37 ± 0.01 | 0.42 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghobrial, D.K.; El-Nikhely, N.; Sheta, E.; Ragab, H.M.; Rostom, S.A.F.; Saeed, H.; Wahid, A. The Role of Pyrazolo[3,4-d]pyrimidine-Based Kinase Inhibitors in The Attenuation of CCl4-Induced Liver Fibrosis in Rats. Antioxidants 2023, 12, 637. https://doi.org/10.3390/antiox12030637
Ghobrial DK, El-Nikhely N, Sheta E, Ragab HM, Rostom SAF, Saeed H, Wahid A. The Role of Pyrazolo[3,4-d]pyrimidine-Based Kinase Inhibitors in The Attenuation of CCl4-Induced Liver Fibrosis in Rats. Antioxidants. 2023; 12(3):637. https://doi.org/10.3390/antiox12030637
Chicago/Turabian StyleGhobrial, Diana K., Nefertiti El-Nikhely, Eman Sheta, Hanan M. Ragab, Sherif A. F. Rostom, Hesham Saeed, and Ahmed Wahid. 2023. "The Role of Pyrazolo[3,4-d]pyrimidine-Based Kinase Inhibitors in The Attenuation of CCl4-Induced Liver Fibrosis in Rats" Antioxidants 12, no. 3: 637. https://doi.org/10.3390/antiox12030637
APA StyleGhobrial, D. K., El-Nikhely, N., Sheta, E., Ragab, H. M., Rostom, S. A. F., Saeed, H., & Wahid, A. (2023). The Role of Pyrazolo[3,4-d]pyrimidine-Based Kinase Inhibitors in The Attenuation of CCl4-Induced Liver Fibrosis in Rats. Antioxidants, 12(3), 637. https://doi.org/10.3390/antiox12030637