

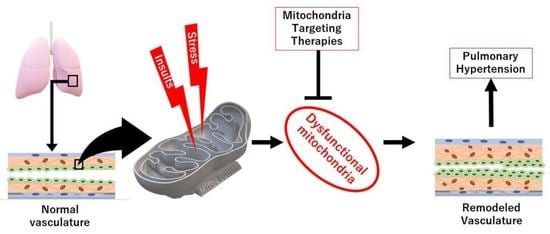
Mitochondrial Dysfunction in Pulmonary Hypertension

Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
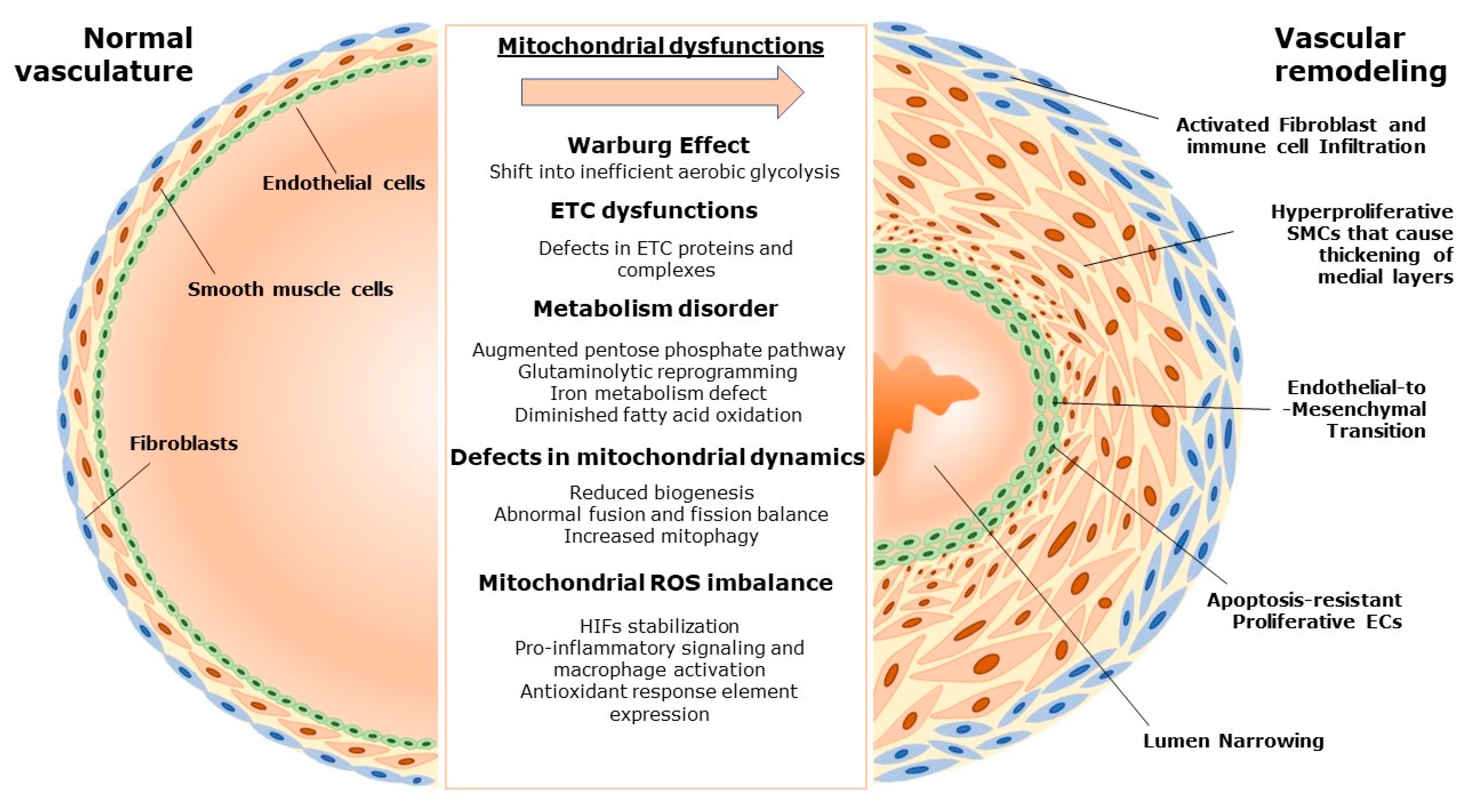
2. Pulmonary Vascular Remodeling and the Role of Mitochondria
2.1. Mitochondria and Vascular Homeostasis
2.2. Mitochondria-Driven Changes in Pulmonary Vascular Remodeling
3. Mitochondrial Dysfunction in Various Forms of Pulmonary Hypertension
4. Targeting Mitochondrial Dysfunction to Treat PH
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endor. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Bazan, I.; Zhang, Y.; Fares, W.H.; Lee, P.J. Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L782–L796. [Google Scholar] [CrossRef]
- Liang, S.; Yegambaram, M.; Wang, T.; Wang, J.; Black, S.M.; Tang, H. Mitochondrial metabolism, redox, and calcium homeostasis in pulmonary arterial hypertension. Biomedicines 2022, 10, 341. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Girerd, B.; Ricard, N.; Huertas, A.; Montani, D.; Humbert, M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: Importance of endothelial communication. Chest 2015, 147, 529–537. [Google Scholar] [CrossRef]
- Tobal, R.; Potjewijd, J.; van Empel, V.P.M.; Ysermans, R.; Schurgers, L.J.; Reutelingsperger, C.P.; Damoiseaux, J.G.M.C.; van Paassen, P. Vascular remodeling in pulmonary arterial hypertension: The potential involvement of innate and adaptive immunity. Front. Med. 2021, 8, 2732. [Google Scholar] [CrossRef]
- Cussac, L.-A.; Cardouat, G.; Tiruchellvam Pillai, N.; Campagnac, M.; Robillard, P.; Montillaud, A.; Guibert, C.; Gailly, P.; Marthan, R.; Quignard, J.-F.; et al. TRPV4 channel mediates adventitial fibroblast activation and adventitial remodeling in pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L135–L146. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Khoyrattee, N.; Savineau, J.-P.; Muller, B.; Guibert, C. Mitochondria: Roles in pulmonary hypertension. Int. J. Biochem. Cell Biol. 2014, 55, 93–97. [Google Scholar] [CrossRef]
- Suliman, H.B.; Nozik-Grayck, E. Mitochondrial dysfunction: Metabolic drivers of pulmonary hypertension. Antioxid. Redox Signal. 2019, 31, 843–857. [Google Scholar] [CrossRef]
- Dromparis, P.; Sutendra, G.; Michelakis, E.D. The role of mitochondria in pulmonary vascular remodeling. J. Mol. Med. 2010, 88, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Xiao, Y.; Deng, X.; Luo, J.; Hong, C.; Qin, X. The Warburg effect: A new story in pulmonary arterial hypertension. Clin. Chim. Acta 2016, 461, 53–58. [Google Scholar] [CrossRef]
- Mick, E.; Titov, D.V.; Skinner, O.S.; Sharma, R.; Jourdain, A.A.; Mootha, V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. eLife 2020, 9, e49178. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Invest. 2018, 128, 3704–3715. [Google Scholar] [CrossRef]
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.-H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol. Med. 2015, 93, 229–242. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef]
- Nisoli, E.; Carruba, M.O. Nitric oxide and mitochondrial biogenesis. J. Cell Sci. 2006, 119, 2855–2862. [Google Scholar] [CrossRef]
- Yeligar, S.M.; Kang, B.-Y.; Bijli, K.M.; Kleinhenz, J.M.; Murphy, T.C.; Torres, G.; San Martin, A.; Sutliff, R.L.; Hart, C.M. PPARγ Regulates mitochondrial structure and function and human pulmonary artery smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 648–657. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Bhandari, V. Targeting mitochondrial dysfunction in lung diseases: Emphasis on mitophagy. Front. Physiol. 2013, 4, 384. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1–mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Dasgupta, A.; Chen, K.; Lima, P.D.A.; Mewburn, J.; Wu, D.; Al-Qazazi, R.; Jones, O.; Tian, L.; Potus, F.; Bonnet, S.; et al. PINK1-induced phosphorylation of mitofusin 2 at serine 442 causes its proteasomal degradation and promotes cell proliferation in lung cancer and pulmonary arterial hypertension. FASEB J. 2021, 35, e21771. [Google Scholar] [CrossRef]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26, 170094. [Google Scholar] [CrossRef]
- Gillespie, M.N.; Al-Mehdi, A.-B.; McMurtry, I.F. Mitochondria in hypoxic pulmonary vasoconstriction. Am. J. Respir. Crit. Care Med. 2013, 187, 338–340. [Google Scholar] [CrossRef]
- He, S.; Zhu, T.; Fang, Z. The role and regulation of pulmonary artery smooth muscle cells in pulmonary hypertension. Int. J. Hypertens. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Li, M.; Riddle, S.; Zhang, H.; D’Alessandro, A.; Flockton, A.; Serkova, N.J.; Hansen, K.C.; Moldovan, R.; McKeon, B.A.; Frid, M.; et al. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor C-terminal binding protein-1. Circulation 2016, 134, 1105–1121. [Google Scholar] [CrossRef]
- Sommer, N.; Theine, F.F.; Pak, O.; Tello, K.; Richter, M.; Gall, H.; Wilhelm, J.; Savai, R.; Weissmann, N.; Seeger, W.; et al. Mitochondrial respiration in peripheral blood mononuclear cells negatively correlates with disease severity in pulmonary arterial hypertension. J. Clin. Med. 2022, 11, 4132. [Google Scholar] [CrossRef] [PubMed]
- Lteif, C.; Ataya, A.; Duarte, J.D. Therapeutic challenges and emerging treatment targets for pulmonary hypertension in left heart disease. J. Am. Heart Assoc. 2021, 10, e020633. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Michael Hart, C.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Bretón-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef]
- Archer, S.L. Pyruvate kinase and warburg metabolism in pulmonary arterial hypertension. Circulation 2017, 136, 2486–2490. [Google Scholar] [CrossRef]
- Colon Hidalgo, D.; Elajaili, H.; Suliman, H.; George, M.P.; Delaney, C.; Nozik, E. Metabolism, mitochondrial dysfunction, and redox homeostasis in pulmonary hypertension. Antioxidants 2022, 11, 428. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Wang, L.; Qi, X.-M.; Sha, Y.-H.; Yang, T. 3-Bromopyruvate alleviates the development of monocrotaline-induced rat pulmonary arterial hypertension by decreasing aerobic glycolysis, inducing apoptosis, and suppressing inflammation. Chin. Med. J. 2020, 133, 49–60. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Tuder, R.M.; El Kasmi, K.C. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J. Appl. Physiol. 2015, 119, 1164–1172. [Google Scholar] [CrossRef]
- Archer, S.L.; Weir, E.K.; Wilkins, M.R. Basic science of pulmonary arterial hypertension for clinicians. Circulation 2010, 121, 2045–2066. [Google Scholar] [CrossRef]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Lloyd Jones, P.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef]
- Rafikova, O.; Srivastava, A.; Desai, A.A.; Rafikov, R.; Tofovic, S.P. Recurrent inhibition of mitochondrial complex III induces chronic pulmonary vasoconstriction and glycolytic switch in the rat lung. Respir. Res. 2018, 19, 69. [Google Scholar] [CrossRef]
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in pulmonary hypertension. Annu. Rev. Physiol. 2021, 83, 551–576. [Google Scholar] [CrossRef]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia inducible-factor1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 2010, 176, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yan, J.; Liu, P.; Wang, Z.; Wang, C.; Zhong, W.; Xu, L. The role of nuclear factor of activated T cells in pulmonary arterial hypertension. Cell Cycle 2017, 16, 508–514. [Google Scholar] [CrossRef]
- DeMarco, V.G. Contribution of oxidative stress to pulmonary arterial hypertension. World J. Cardiol. 2010, 2, 316. [Google Scholar] [CrossRef]
- Xu, D.; Hu, Y.-H.; Gou, X.; Li, F.-Y.; Yang, X.-Y.-C.; Li, Y.-M.; Chen, F. Oxidative stress and antioxidative therapy in pulmonary arterial hypertension. Molecules 2022, 27, 3724. [Google Scholar] [CrossRef]
- Dorfmüller, P.; Chaumais, M.-C.; Giannakouli, M.; Durand-Gasselin, I.; Raymond, N.; Fadel, E.; Mercier, O.; Charlotte, F.; Montani, D.; Simonneau, G.; et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir. Res. 2011, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, Y.; Ono, S.; Suzuki, S.; Tanita, T.; Chida, M.; Song, C.; Noda, M.; Tabata, T.; Voelkel, N.F.; Fujimura, S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Physiol. 2001, 90, 1299–1306. [Google Scholar] [CrossRef]
- Jiang, W.-L.; Han, X.; Zhang, Y.-F.; Xia, Q.-Q.; Zhang, J.-M.; Wang, F. Arctigenin prevents monocrotaline-induced pulmonary arterial hypertension in rats. RSC Adv. 2019, 9, 552–559. [Google Scholar] [CrossRef]
- Park, W.H. Exogenous H2O2 induces growth inhibition and cell death of human pulmonary artery smooth muscle cells via glutathione depletion. Mol. Med. Rep. 2016, 14, 936–942. [Google Scholar] [CrossRef]
- Bonnet, S.; Boucherat, O. The ROS controversy in hypoxic pulmonary hypertension revisited. Eur. Respir. J. 2018, 51, 1800276. [Google Scholar] [CrossRef]
- Morten, K.J.; Ackrell, B.A.C.; Melov, S. Mitochondrial reactive oxygen species in mice lacking superoxide dismutase 2. J. Biol. Chem. 2006, 281, 3354–3359. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Yang, K.; Cang, H.; Huang, X.; Li, H.; Yi, J. The biphasic redox sensing of SENP3 accounts for the HIF-1 transcriptional activity shift by oxidative stress. Acta Pharmacol. Sin. 2012, 33, 953–963. [Google Scholar] [CrossRef]
- Liu, R.; Xu, C.; Zhang, W.; Cao, Y.; Ye, J.; Li, B.; Jia, S.; Weng, L.; Liu, Y.; Liu, L.; et al. FUNDC1-mediated mitophagy and HIF1α activation drives pulmonary hypertension during hypoxia. Cell Death Dis. 2022, 13, 634. [Google Scholar] [CrossRef]
- Kitagawa, A.; Jacob, C.; Jordan, A.; Waddell, I.; McMurtry, I.F.; Gupte, S.A. Inhibition of glucose-6-phosphate dehydrogenase activity attenuates right ventricle pressure and hypertrophy elicited by VEGFR inhibitor + hypoxia. J. Pharmacol. Exp. Ther. 2021, 377, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Mprah, R.; Adzika, G.K.; Gyasi, Y.I.; Ndzie Noah, M.L.; Adu-Amankwaah, J.; Adekunle, A.O.; Duah, M.; Wowui, P.I.; Weili, Q. Glutaminolysis: A driver of vascular and cardiac remodeling in pulmonary arterial hypertension. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Niihori, M.; Eccles, C.A.; Kurdyukov, S.; Zemskova, M.; Varghese, M.V.; Stepanova, A.A.; Galkin, A.; Rafikov, R.; Rafikova, O. Rats with a human mutation of NFU1 develop pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 62, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L. Ironing out pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 12604–12606. [Google Scholar] [CrossRef]
- Talati, M.; Hemnes, A. Fatty acid metabolism in pulmonary arterial hypertension: Role in right ventricular dysfunction and hypertrophy. Pulm. Circ. 2015, 5, 269–278. [Google Scholar] [CrossRef]
- Feng, W.; Wang, J.; Yan, X.; Zhang, Q.; Chai, L.; Wang, Q.; Shi, W.; Chen, Y.; Liu, J.; Qu, Z.; et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 2021, 54, e13048. [Google Scholar] [CrossRef]
- Ryan, J.J.; Marsboom, G.; Fang, Y.-H.; Toth, P.T.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878. [Google Scholar] [CrossRef]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Ohtsuki, T.; Al-Mamun, M.E.; Siddique, M.A.H.; Yaoita, N.; et al. ADAMTS8 promotes the development of pulmonary arterial hypertension and right ventricular failure. Circ. Res. 2019, 125, 884–906. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.-Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef]
- Fan, Y.; Hao, Y.; Gao, D.; Gao, L.; Li, G.; Zhang, Z. Phenotype and function of macrophage polarization in monocrotaline-induced pulmonary arterial hypertension rat model. Physiol. Res. 2021, 70, 213. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; D’alessandro, A.; El Kasmi, K.; Li, M.; Zhang, H.; Ježek, P.; Stenmark, K.R. Metabolic reprogramming and redox signaling in pulmonary hypertension. Pulm. Vasc. Redox Signal. Health Dis. 2017, 967, 241–260. [Google Scholar]
- Kojima, H.; Tokunou, T.; Takahara, Y.; Sunagawa, K.; Hirooka, Y.; Ichiki, T.; Tsutsui, H. Hypoxia-inducible factor-1 α deletion in myeloid lineage attenuates hypoxia-induced pulmonary hypertension. Physiol. Rep. 2019, 7, e14025. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Nozik-Grayck, E.; Gerasimovskaya, E.; Anwar, A.; Li, M.; Riddle, S.; Frid, M. The adventitia: Essential role in pulmonary vascular remodeling. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2010; pp. 141–161. [Google Scholar]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, D.; Li, M.; Plecitá-Hlavatá, L.; D’Alessandro, A.; Tauber, J.; Riddle, S.; Kumar, S.; Flockton, A.; McKeon, B.A.; et al. Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/pyruvate kinase muscle axis. Circulation 2017, 136, 2468–2485. [Google Scholar] [CrossRef]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Naik, J.S.; Weise-Cross, L.; Detweiler, N.D.; Herbert, L.M.; Yellowhair, T.R.; Resta, T.C. Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLoS ONE 2017, 12, e0180455. [Google Scholar] [CrossRef] [PubMed]
- Coskun, F.Y.; Taysı, S.; Kayıkçıoğlu, M. Can serum 8-hydroxy-2′-deoxyguanosine levels reflect the severity of pulmonary arterial hypertension? Rev. Assoc. Med. Bras. 2021, 67, 1437–1442. [Google Scholar] [CrossRef]
- Wong, C.-M.; Bansal, G.; Pavlickova, L.; Marcocci, L.; Suzuki, Y.J. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid. Redox Signal. 2013, 18, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 Overexpression Induces Pulmonary Hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Chen, X.; Andresen, B.; Hill, M.; Zhang, J.; Booth, F.; Zhang, C. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr. Hypertens. Rev. 2008, 4, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Bru-Mercier, G.; Courboulin, A.; Quatredeniers, M.; Rücker-Martin, C.; Antigny, F.; Nakhleh, M.K.; Ranchoux, B.; Gouadon, E.; Vinhas, M.-C.; et al. NMDA-type glutamate receptor activation promotes vascular remodeling and pulmonary arterial hypertension. Circulation 2018, 137, 2371–2389. [Google Scholar] [CrossRef]
- Quatredeniers, M.; Mendes-Ferreira, P.; Santos-Ribeiro, D.; Nakhleh, M.K.; Ghigna, M.-R.; Cohen-Kaminsky, S.; Perros, F. Iron deficiency in pulmonary arterial hypertension: A deep dive into the mechanisms. Cells 2021, 10, 477. [Google Scholar] [CrossRef]
- Mathew, R. Pulmonary hypertension and metabolic syndrome: Possible connection, PPARγ and Caveolin-1. World J. Cardiol. 2014, 6, 692. [Google Scholar] [CrossRef]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol. Hear. Circ. Physiol. 2015, 309, H1375–H1389. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.B.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Tu, L.; Izikki, M.; Dewachter, L.; Zadigue, P.; Humbert, M.; Adnot, S.; Fadel, E.; Eddahibi, S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22α-targeted overexpression of the serotonin transporter. FASEB J. 2009, 23, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Cuttica, M.J.; Beussink-Nelson, L.; Kozyleva, A.; Sanchez, C.; Mkrdichian, H.; Selvaraj, S.; Dematte, J.E.; Lee, D.C.; Shah, S.J. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: A pilot study. Pulm. Circ. 2015, 5, 547–556. [Google Scholar] [CrossRef]
- Liu, F.; Yin, L.; Zhang, L.; Liu, W.; Liu, J.; Wang, Y.; Yu, B. Trimetazidine improves right ventricular function by increasing miR-21 expression. Int. J. Mol. Med. 2012, 30, 849–855. [Google Scholar] [CrossRef]
- Prins, K.W.; Thenappan, T.; Weir, E.K.; Kalra, R.; Pritzker, M.; Archer, S.L. Repurposing medications for treatment of pulmonary arterial hypertension: What’s old is new again. J. Am. Heart Assoc. 2019, 8, e011343. [Google Scholar] [CrossRef]
- Koulmann, N.; Novel-Chaté, V.; Peinnequin, A.; Chapot, R.; Serrurier, B.; Simler, N.; Richard, H.; Ventura-Clapier, R.; Bigard, X. Cyclosporin a inhibits hypoxia-induced pulmonary hypertension and right ventricle hypertrophy. Am. J. Respir. Crit. Care Med. 2006, 174, 699–705. [Google Scholar] [CrossRef]
- Lee, D.S.; Jung, Y.W. Protective effect of right ventricular mitochondrial damage by cyclosporine a in monocrotaline-induced pulmonary hypertension. Korean Circ. J. 2018, 48, 1135. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, H.; Porvasnik, S.L.; Terada, N.; Agarwal, A.; Cheng, Y.; Visner, G.A. Heme oxygenase-1 mediates the protective effects of rapamycin in monocrotaline-induced pulmonary hypertension. Lab. Investig. 2006, 86, 62–71. [Google Scholar] [CrossRef]
- Zhao, Q.; Song, P.; Zou, M.-H. AMPK and pulmonary hypertension: Crossroads between vasoconstriction and vascular remodeling. Front. Cell Dev. Biol. 2021, 9, 691585. [Google Scholar] [CrossRef]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Walker, B.R.; Jernigan, N.L.; Resta, T.C. Contribution of mitochondrial reactive oxygen species to chronic hypoxia-induced pulmonary hypertension. FASEB J. 2020, 34, 642–653. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryanto, G.R.T.; Suraya, R.; Nagano, T. Mitochondrial Dysfunction in Pulmonary Hypertension. Antioxidants 2023, 12, 372. https://doi.org/10.3390/antiox12020372
Ryanto GRT, Suraya R, Nagano T. Mitochondrial Dysfunction in Pulmonary Hypertension. Antioxidants. 2023; 12(2):372. https://doi.org/10.3390/antiox12020372
Chicago/Turabian StyleRyanto, Gusty Rizky Teguh, Ratoe Suraya, and Tatsuya Nagano. 2023. "Mitochondrial Dysfunction in Pulmonary Hypertension" Antioxidants 12, no. 2: 372. https://doi.org/10.3390/antiox12020372
APA StyleRyanto, G. R. T., Suraya, R., & Nagano, T. (2023). Mitochondrial Dysfunction in Pulmonary Hypertension. Antioxidants, 12(2), 372. https://doi.org/10.3390/antiox12020372