
Amyloid β-Oligomers Inhibit the Nuclear Ca2+ Signals and the Neuroprotective Gene Expression Induced by Gabazine in Hippocampal Neurons

 , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
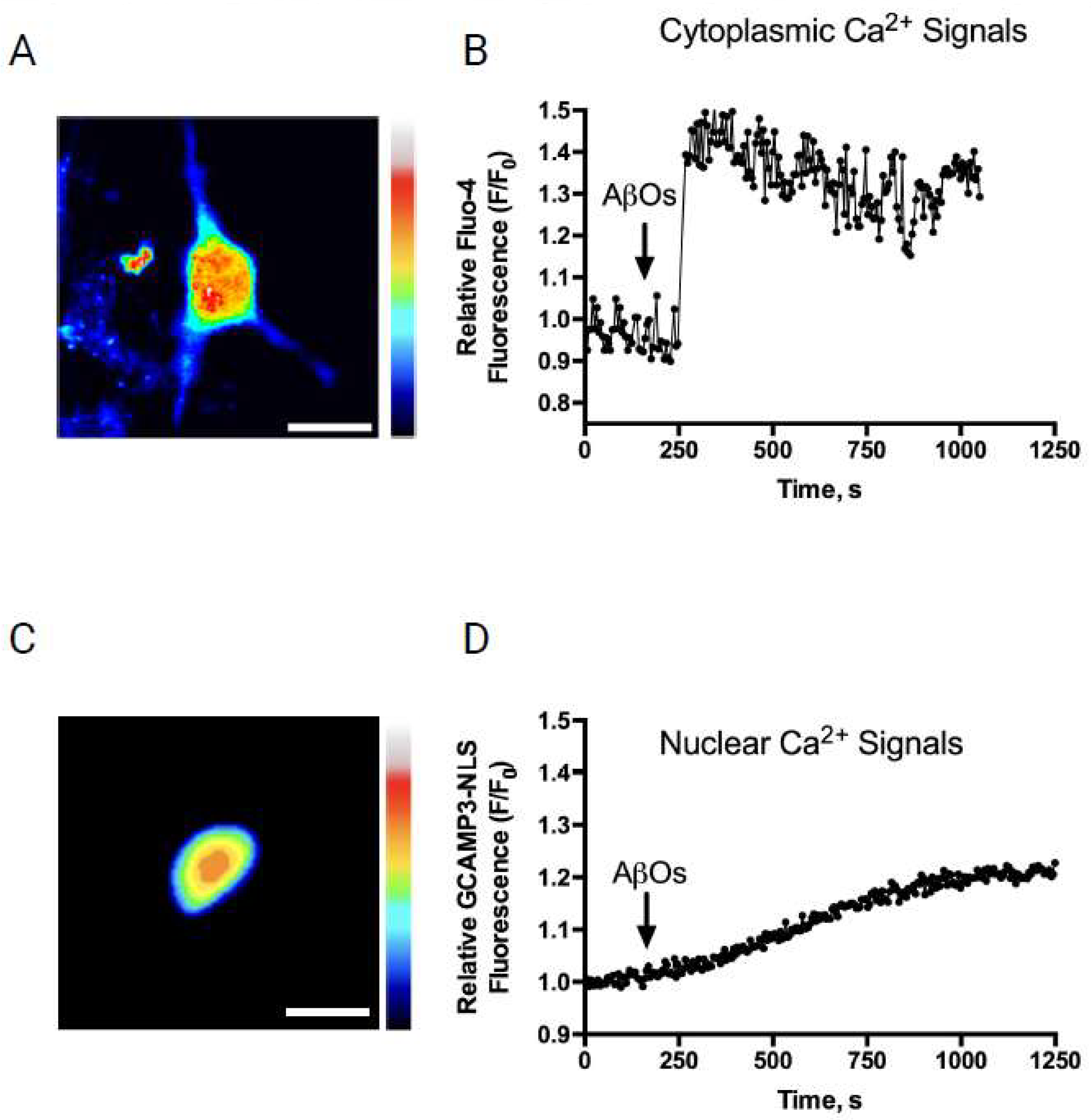
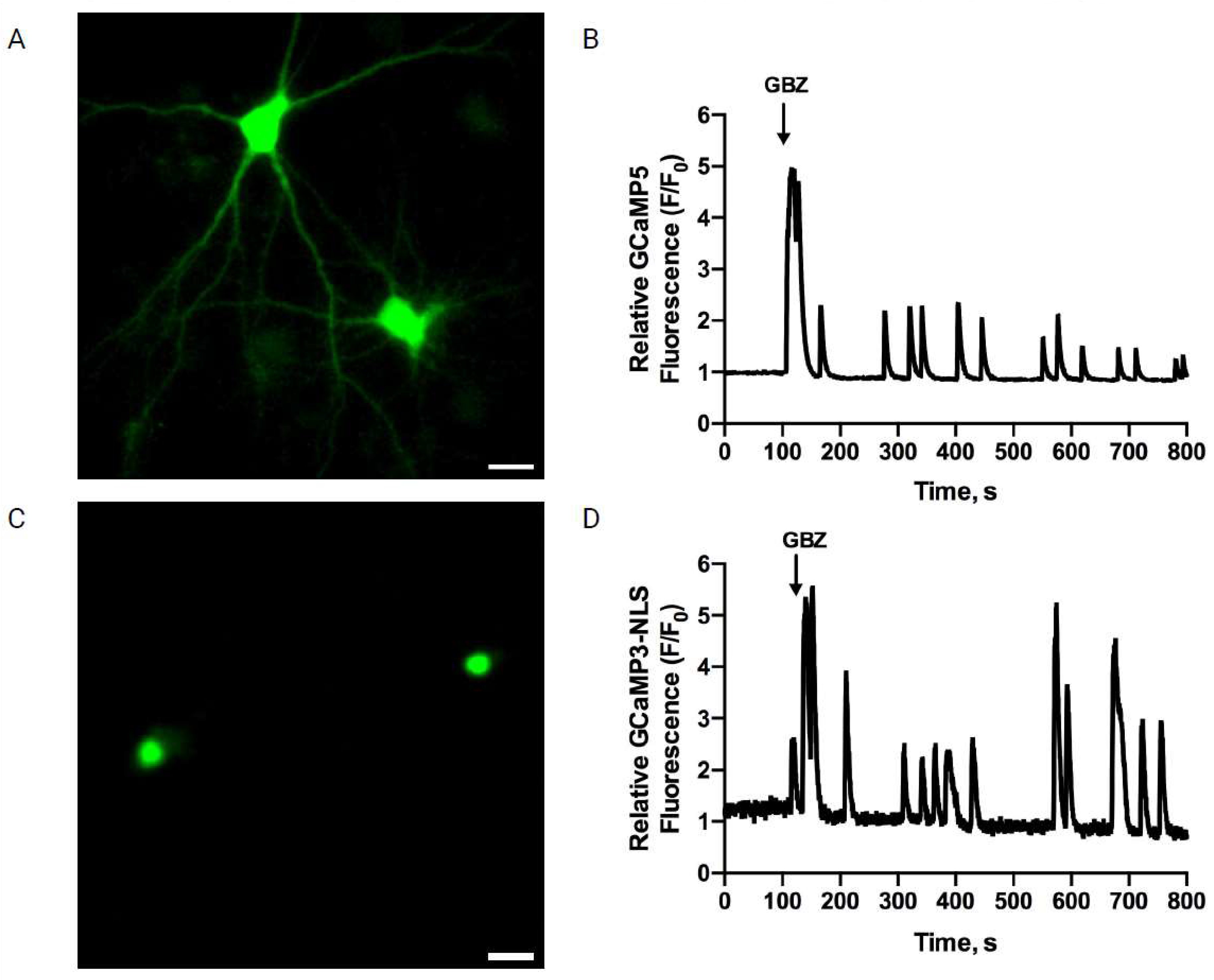
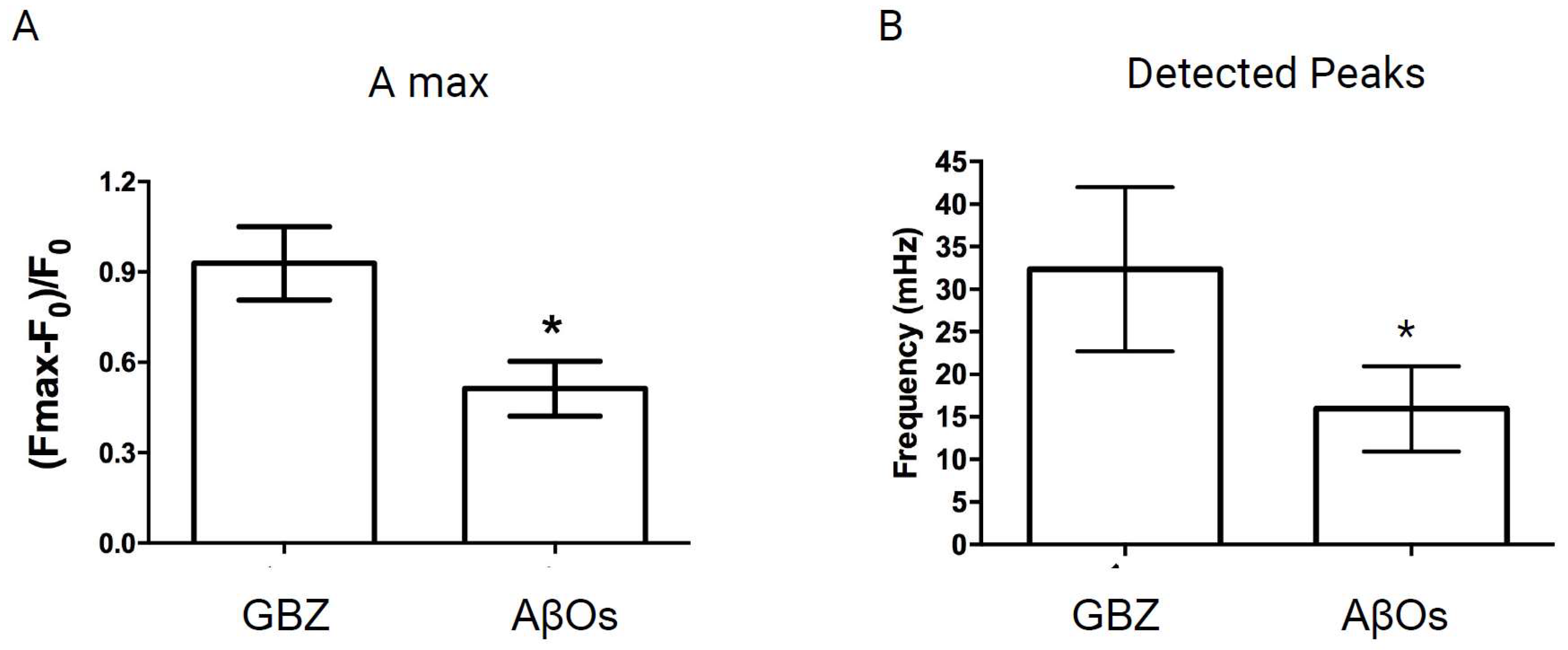
3.1. AβOs Disrupt the Nuclear Ca2+ Transients Induced by Gabazine
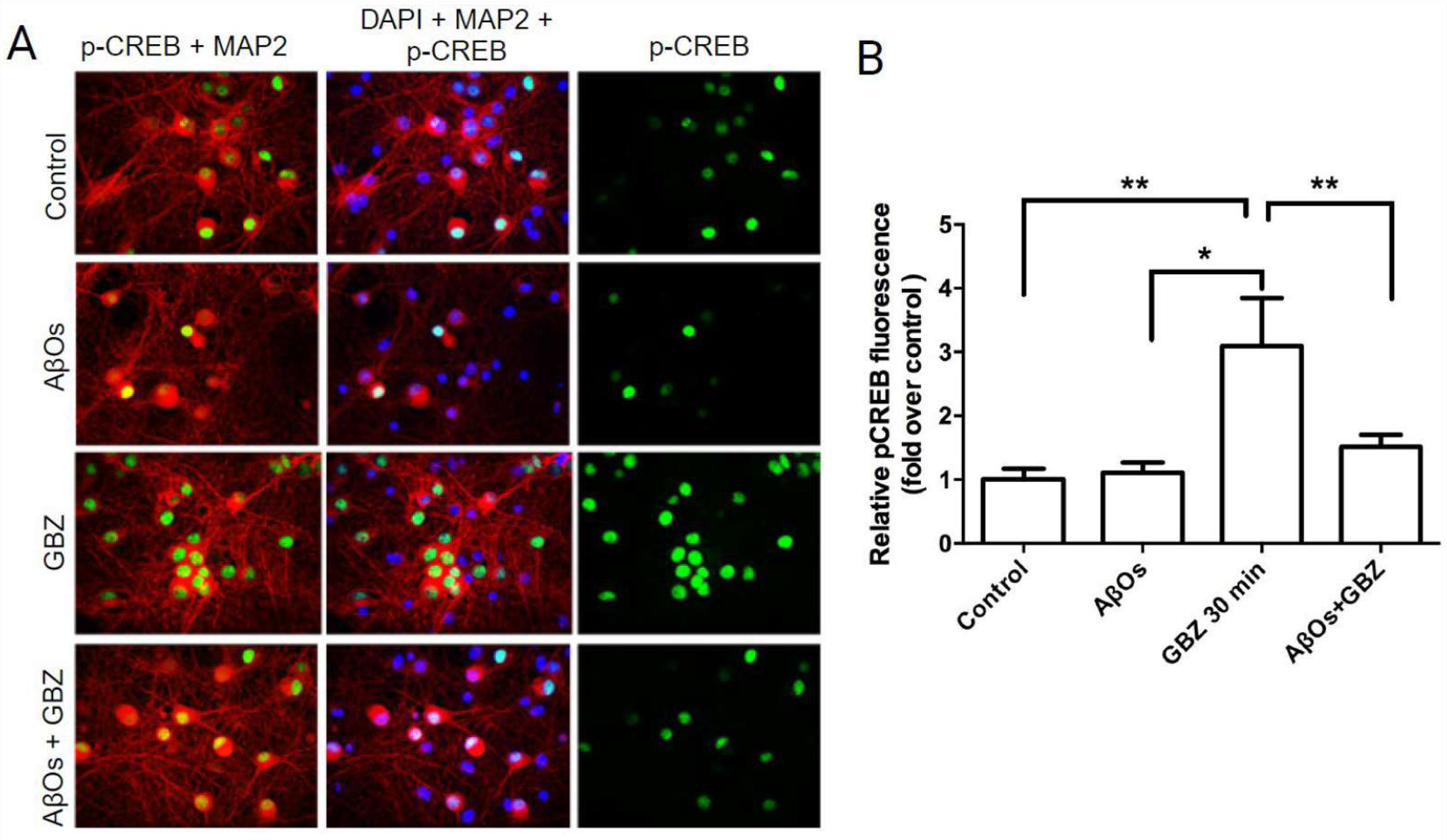
3.2. AβOs Prevent the CREB Phosphorylation Increase Induced by Gabazine
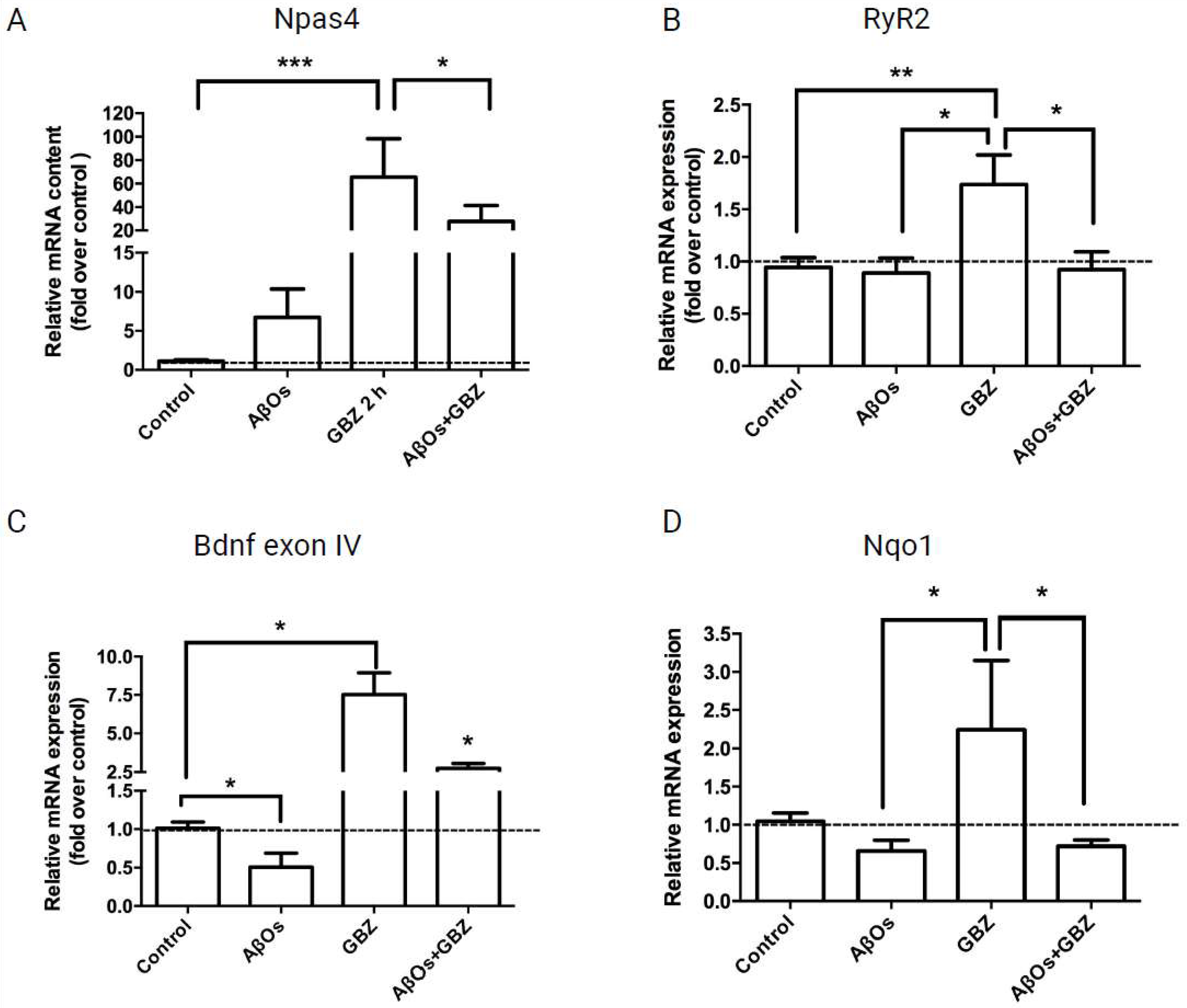
3.3. AβOs Inhibit the Increase in Npas4, RyR2, BDNF and Nqo1 mRNA Levels Induced by Gabazine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Alzheimer’s Association. 2023 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-Associated Protein Tau. A Component of Alzheimer Paired Helical Filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Hibar, D.P.; Adams, H.H.H.; Jahanshad, N.; Chauhan, G.; Stein, J.L.; Hofer, E.; Renteria, M.E.; Bis, J.C.; Arias-Vasquez, A.; Ikram, M.K.; et al. Novel Genetic Loci Associated with Hippocampal Volume. Nat. Commun. 2017, 8, 13624. [Google Scholar] [CrossRef]
- Fotuhi, M.; Do, D.; Jack, C. Modifiable Factors That Alter the Size of the Hippocampus with Ageing. Nat. Rev. Neurol. 2012, 8, 189–202. [Google Scholar] [CrossRef]
- Younan, D.; Petkus, A.J.; Widaman, K.F.; Wang, X.; Casanova, R.; Espeland, M.A.; Gatz, M.; Henderson, V.W.; Manson, J.E.; Rapp, S.R.; et al. Particulate Matter and Episodic Memory Decline Mediated by Early Neuroanatomic Biomarkers of Alzheimer’s Disease. Brain 2020, 143, 289–302. [Google Scholar] [CrossRef]
- Leal, S.L.; Yassa, M.A. Neurocognitive Aging and the Hippocampus across Species. Trends Neurosci. 2015, 38, 800–812. [Google Scholar] [CrossRef]
- Adler, D.H.; Wisse, L.E.M.; Ittyerah, R.; Pluta, J.B.; Ding, S.-L.; Xie, L.; Wang, J.; Kadivar, S.; Robinson, J.L.; Schuck, T.; et al. Characterizing the Human Hippocampus in Aging and Alzheimer’s Disease Using a Computational Atlas Derived from Ex Vivo MRI and Histology. Proc. Natl. Acad. Sci. USA 2018, 115, 4252–4257. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, Nonfibrillar Ligands Derived from Aβ 1–42 Are Potent Central Nervous System Neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ Oligomers Induce Neuronal Oxidative Stress through an N-Methyl-D-Aspartate Receptor-Dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Adasme, T.; SanMartin, C.; Sebollela, A.; Hetz, C.; Carrasco, M.A.; Ferreira, S.T.; Hidalgo, C. Amyloid Beta-Peptide Oligomers Stimulate RyR-Mediated Ca2+ Release Inducing Mitochondrial Fragmentation in Hippocampal Neurons and Prevent RyR-Mediated Dendritic Spine Remodeling Produced by BDNF. Antioxid. Redox Signal 2011, 14, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Lobos, P.; Bruna, B.; Cordova, A.; Barattini, P.; Galáz, J.L.; Adasme, T.; Hidalgo, C.; Muñoz, P.; Paula-Lima, A. Astaxanthin Protects Primary Hippocampal Neurons against Noxious Effects of Aβ-Oligomers. Neural Plast. 2016, 2016, 3456783. [Google Scholar] [CrossRef]
- Ortiz-Sanz, C.; Gaminde-Blasco, A.; Valero, J.; Bakota, L.; Brandt, R.; Zugaza, J.L.; Matute, C.; Alberdi, E. Early Effects of Aβ Oligomers on Dendritic Spine Dynamics and Arborization in Hippocampal Neurons. Front. Synaptic Neurosci. 2020, 12, 2. [Google Scholar] [CrossRef]
- SanMartin, C.D.; Veloso, P.; Adasme, T.; Lobos, P.; Bruna, B.; Galaz, J.; Garcia, A.; Hartel, S.; Hidalgo, C.; Paula-Lima, A.C. RyR2-Mediated Ca2+ Release and Mitochondrial ROS Generation Partake in the Synaptic Dysfunction Caused by Amyloid Beta Peptide Oligomers. Front. Mol. Neurosci. 2017, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Frere, S.; Slutsky, I. Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef]
- Tse and Herrup, K. Re-imagining Alzheimer’s Disease—The Diminishing Importance of Amyloid and a Glimpse of What Lies Ahead. ARPN J. Eng. Appl. Sci. 2017, 12, 3218–3221. [Google Scholar] [CrossRef]
- Fani, G.; La Torre, C.E.; Cascella, R.; Cecchi, C.; Vendruscolo, M.; Chiti, F. Misfolded Protein Oligomers Induce an Increase of Intracellular Ca2+ Causing an Escalation of Reactive Oxidative Species. Cell. Mol. Life Sci. 2022, 79, 500. [Google Scholar] [CrossRef] [PubMed]
- Rummel, N.G.; Butterfield, D.A. Altered Metabolism in Alzheimer Disease Brain: Role of Oxidative Stress. Antioxid. Redox Signal 2022, 36, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative Stress in the Brain: Novel Cellular Targets That Govern Survival during Neurodegenerative Disease. Prog. Neurobiol. 2005, 75, 207–246. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid. Redox Signal 2021, 34, 591–610. [Google Scholar] [CrossRef]
- SanMartin, C.D.; Adasme, T.; Hidalgo, C.; Paula-Lima, A.C. The Antioxidant N-Acetylcysteine Prevents the Mitochondrial Fragmentation Induced by Soluble Amyloid-Beta Peptide Oligomers. Neurodegener. Dis. 2012, 10, 34–37. [Google Scholar] [CrossRef]
- Hagenston, A.M.; Bading, H. Calcium Signaling in Synapse-to-Nucleus Communication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004564. [Google Scholar] [CrossRef]
- Lobos, P.; Córdova, A.; Vega-Vásquez, I.; Ramírez, O.A.; Adasme, T.; Toledo, J.; Cerda, M.; Härtel, S.; Paula-Lima, A.; Hidalgo, C. RyR-Mediated Ca2+ Release Elicited by Neuronal Activity Induces Nuclear Ca2+ Signals, CREB Phosphorylation, and Npas4/RyR2 Expression. Proc. Natl. Acad. Sci. USA 2021, 118, e2102265118. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bading, H. Nuclear Calcium Signalling in the Regulation of Brain Function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Brito-Moreira, J.; Ferreira, S.T. Deregulation of Excitatory Neurotransmission Underlying Synapse Failure in Alzheimer’s Disease. J. Neurochem. 2013, 126, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Brito-Moreira, J.; Paula-Lima, A.C.; Bomfim, T.R.; Oliveira, F.F.; Sepúlveda, F.J.; de Mello, F.G.; Aguayo, L.G.; Panizzutti, R.; Ferreira, S.T. Aβ Oligomers Induce Glutamate Release from Hippocampal Neurons. Curr. Alzheimer Res. 2011, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Karpova, A.; Samer, S.; Turacak, R.; Yuanxiang, P.; Kreutz, M.R. Integration of Nuclear Ca2+ Transients and Subnuclear Protein Shuttling Provides a Novel Mechanism for the Regulation of CREB-Dependent Gene Expression. Cell. Mol. Life Sci. 2023, 80, 228. [Google Scholar] [CrossRef]
- Kim, J.; Seo, S.; Park, J.H.Y.; Lee, K.W.; Kim, J.; Kim, J.-C. Ca2+ -Permeable TRPV1 Receptor Mediates Neuroprotective Effects in a Mouse Model of Alzheimer’s Disease via BDNF/CREB Signaling Pathway. Mol. Cells 2023, 46, 319–328. [Google Scholar] [CrossRef]
- Chiantia, G.; Hidisoglu, E.; Marcantoni, A. The Role of Ryanodine Receptors in Regulating Neuronal Activity and Its Connection to the Development of Alzheimer’s Disease. Cells 2023, 12, 1236. [Google Scholar] [CrossRef]
- Marcello, E.; Di Luca, M.; Gardoni, F. Synapse-to-Nucleus Communication: From Developmental Disorders to Alzheimer’s Disease. Curr. Opin. Neurobiol. 2018, 48, 160–166. [Google Scholar] [CrossRef]
- Brini, M.; Murgia, M.; Pasti, L.; Picard, D.; Pozzan, T.; Rizzuto, R. Nuclear Ca2+ Concentration Measured with Specifically Targeted Recombinant Aequorin. EMBO J. 1993, 12, 4813–4819. [Google Scholar] [CrossRef] [PubMed]
- Eder, A.; Bading, H. Calcium Signals Can Freely Cross the Nuclear Envelope in Hippocampal Neurons: Somatic Calcium Increases Generate Nuclear Calcium Transients. BMC Neurosci. 2007, 8, 57. [Google Scholar] [CrossRef]
- Allbritton, N.L.; Meyer, T.; Stryer, L. Range of Messenger Action of Calcium Ion and Inositol 1,4,5-Trisphosphate. Science 1992, 258, 1812–1815. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Chawla, S.; Hardingham, G.E.; Quinn, D.R.; Bading, H. CBP: A Signal-Regulated Transcriptional Coactivator Controlled by Nuclear Calcium and CaM Kinase IV. Science 1998, 281, 1505–1509. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Arnold, F.J.L.; Bading, H. Nuclear Calcium Signaling Controls CREB-Mediated Gene Expression Triggered by Synaptic Activity. Nat. Neurosci. 2001, 4, 261–267. [Google Scholar] [CrossRef]
- Pegoraro, S.; Broccard, F.D.; Ruaro, M.E.; Bianchini, D.; Avossa, D.; Pastore, G.; Bisson, G.; Altafini, C.; Torre, V. Sequential Steps Underlying Neuronal Plasticity Induced by a Transient Exposure to Gabazine. J. Cell Physiol. 2009, 222, 713–728. [Google Scholar] [CrossRef]
- Sokal, D.M.; Mason, R.; Parker, T.L. Multi-Neuronal Recordings Reveal a Differential Effect of Thapsigargin on Bicuculline- or Gabazine-Induced Epileptiform Excitability in Rat Hippocampal Neuronal Networks. Neuropharmacology 2000, 39, 2408–2417. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Esvald, E.-E.; Tuvikene, J.; Sirp, A.; Patil, S.; Bramham, C.R.; Timmusk, T. CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. J. Neurosci. 2020, 40, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Adasme, T.; Haeger, P.; Paula-Lima, A.C.; Espinoza, I.; Casas-Alarcón, M.M.; Carrasco, M.A.; Hidalgo, C. Involvement of Ryanodine Receptors in Neurotrophin-Induced Hippocampal Synaptic Plasticity and Spatial Memory Formation. Proc. Natl. Acad. Sci. USA 2011, 108, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- More, J.Y.; Bruna, B.A.; Lobos, P.E.; Galaz, J.L.; Figueroa, P.L.; Namias, S.; Sánchez, G.L.; Barrientos, G.C.; Valdés, J.L.; Paula-Lima, A.C.; et al. Calcium Release Mediated by Redox-Sensitive RyR2 Channels Has a Central Role in Hippocampal Structural Plasticity and Spatial Memory. Antioxid. Redox Signal 2018, 29, 1125–1146. [Google Scholar] [CrossRef] [PubMed]
- Weng, F.-J.; Garcia, R.I.; Lutzu, S.; Alviña, K.; Zhang, Y.; Dushko, M.; Ku, T.; Zemoura, K.; Rich, D.; Garcia-Dominguez, D.; et al. Npas4 Is a Critical Regulator of Learning-Induced Plasticity at Mossy Fiber-CA3 Synapses during Contextual Memory Formation. Neuron 2018, 97, 1137–1152.e5. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, C.M.; McFeeley, B.M.; Simon, S.S.; Ledreux, A.; Håkansson, K.; Granholm, A.; Mohammed, A.H.; Daffner, K.R. BDNF Mediates Improvement in Cognitive Performance after Computerized Cognitive Training in Healthy Older Adults. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12337. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. The Diverse Functionality of NQO1 and Its Roles in Redox Control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Sharma, V.; Kaur, A.; Singh, T.G. Counteracting Role of Nuclear Factor Erythroid 2-Related Factor 2 Pathway in Alzheimer’s Disease. Biomed. Pharmacother. 2020, 129, 110373. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Ueno, S.; Bracamontes, J.; Zorumski, C.; Weiss, D.S.; Steinbach, J.H. Bicuculline and Gabazine Are Allosteric Inhibitors of Channel Opening of the GABA A Receptor. J. Neurosci. 1997, 17, 625–634. [Google Scholar] [CrossRef]
- Tong, L.; Balazs, R.; Thornton, P.L.; Cotman, C.W. β-Amyloid Peptide at Sublethal Concentrations Downregulates Brain-Derived Neurotrophic Factor Functions in Cultured Cortical Neurons. J. Neurosci. 2004, 24, 6799–6809. [Google Scholar] [CrossRef]
- Ross, W.N. Understanding Calcium Waves and Sparks in Central Neurons. Nat. Rev. Neurosci. 2012, 13, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Mozolewski, P.; Jeziorek, M.; Schuster, C.M.; Bading, H.; Frost, B.; Dobrowolski, R. The Role of Nuclear Ca2+ in Maintaining Neuronal Homeostasis and Brain Health. J. Cell Sci. 2021, 134, jcs254904. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, J.K.; Gonzalez, K.C.; Herrlinger, S.A.; Hirabayashi, Y.; Hewitt, V.L.; Blockus, H.; Szoboszlay, M.; Rolotti, S.V.; Geiller, T.C.; Negrean, A.; et al. Compartment-Specific Tuning of Dendritic Feature Selectivity by Intracellular Ca2+ Release. Science 2022, 375, eabm1670. [Google Scholar] [CrossRef]
- Marengo, J.J.; Bull, R.; Hidalgo, C. Calcium Dependence of Ryanodine-Sensitive Calcium Channels from Brain Cortex Endoplasmic Reticulum. FEBS Lett. 1996, 383, 59–62. [Google Scholar] [CrossRef]
- Marengo, J.J.; Hidalgo, C.; Bull, R. Sulfhydryl Oxidation Modifies the Calcium Dependence of Ryanodine-Sensitive Calcium Channels of Excitable Cells. Biophys. J. 1998, 74, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.; Marengo, J.J.; Finkelstein, J.P.; Behrens, M.I.; Alvarez, O. SH Oxidation Coordinates Subunits of Rat Brain Ryanodine Receptor Channels Activated by Calcium and ATP. Am. J. Physiol. Cell Physiol. 2003, 285, C119–C128. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, D.; Alvarez, A.; Leal, N.; Adasme, T.; Espinoza, I.; Valdes, J.A.; Troncoso, N.; Hartel, S.; Hidalgo, J.; Hidalgo, C.; et al. High-Frequency Field Stimulation of Primary Neurons Enhances Ryanodine Receptor-Mediated Ca2+ Release and Generates Hydrogen Peroxide, Which Jointly Stimulate NF-KappaB Activity. Antioxid. Redox Signal 2011, 14, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Paula-Lima, A.C.; Adasme, T.; Hidalgo, C. Contribution of Ca2+ Release Channels to Hippocampal Synaptic Plasticity and Spatial Memory: Potential Redox Modulation. Antioxid. Redox Signal 2014, 21, 892–914. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Undurraga, I.; Lobos, P.; Sánchez-Robledo, V.; Arias-Cavieres, A.; SanMartín, C.D.; Barrientos, G.; More, J.; Muñoz, P.; Paula-Lima, A.C.; Hidalgo, C.; et al. Long-Term Potentiation and Spatial Memory Training Stimulate the Hippocampal Expression of RyR2 Calcium Release Channels. Front. Cell Neurosci. 2023, 17, 1132121. [Google Scholar] [CrossRef]
- Datta, D.; Leslie, S.N.; Wang, M.; Morozov, Y.M.; Yang, S.; Mentone, S.; Zeiss, C.; Duque, A.; Rakic, P.; Horvath, T.L.; et al. Age-related Calcium Dysregulation Linked with Tau Pathology and Impaired Cognition in Non-human Primates. Alzheimer’s Dement. 2021, 17, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Liu, Y.; Sun, B.; Zhan, X.; Estillore, J.P.; Turner, R.W.; Chen, S.R.W. Increased RyR2 Open Probability Induces Neuronal Hyperactivity and Memory Loss with or without Alzheimer’s Disease–Causing Gene Mutations. Alzheimer’s Dement. 2022, 18, 2088–2098. [Google Scholar] [CrossRef]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M.F. An Endoplasmic-Reticulum-Specific Apoptotic Pathway Is Involved in Prion and Amyloid-Beta Peptides Neurotoxicity. Neurobiol. Dis. 2006, 23, 669–678. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-Translational Remodeling of Ryanodine Receptor Induces Calcium Leak Leading to Alzheimer’s Disease-like Pathologies and Cognitive Deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Sipilä, S.T.; Huttu, K.; Soltesz, I.; Voipio, J.; Kaila, K. Depolarizing GABA Acts on Intrinsically Bursting Pyramidal Neurons to Drive Giant Depolarizing Potentials in the Immature Hippocampus. J. Neurosci. 2005, 25, 5280–5289. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A.R. Advantages of an Antagonist: Bicuculline and Other GABA Antagonists. Br. J. Pharmacol. 2013, 169, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble Amyloid-β Oligomers as Synaptotoxins Leading to Cognitive Impairment in Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Kharitonova, E.K.; Bacskai, B.J. Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells 2020, 9, 2513. [Google Scholar] [CrossRef]
- Schrank, S.; McDaid, J.; Briggs, C.A.; Mustaly-Kalimi, S.; Brinks, D.; Houcek, A.; Singer, O.; Bottero, V.; Marr, R.A.; Stutzmann, G.E. Human-Induced Neurons from Presenilin 1 Mutant Patients Model Aspects of Alzheimer’s Disease Pathology. Int. J. Mol. Sci. 2020, 21, 1030. [Google Scholar] [CrossRef]
- Aloni, E.; Oni-Biton, E.; Tsoory, M.; Moallem, D.H.; Segal, M. Synaptopodin Deficiency Ameliorates Symptoms in the 3xTg Mouse Model of Alzheimer’s Disease. J. Neurosci. 2019, 39, 3983–3992. [Google Scholar] [CrossRef]
- Dridi, H.; Liu, Y.; Reiken, S.; Liu, X.; Argyrousi, E.K.; Yuan, Q.; Miotto, M.C.; Sittenfeld, L.; Meddar, A.; Soni, R.K.; et al. Heart Failure-Induced Cognitive Dysfunction Is Mediated by Intracellular Ca2+ Leak through Ryanodine Receptor Type 2. Nat. Neurosci. 2023, 26, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Webber, E.K.; Fivaz, M.; Stutzmann, G.E.; Griffioen, G. Cytosolic Calcium: Judge, Jury and Executioner of Neurodegeneration in Alzheimer’s Disease and Beyond. Alzheimer’s Dement. 2023, 19, 3701–3717. [Google Scholar] [CrossRef]
- Sun, X.; Lin, Y. Npas4: Linking Neuronal Activity to Memory. Trends Neurosci. 2016, 39, 264–275. [Google Scholar] [CrossRef]
- Brigidi, G.S.; Hayes, M.G.B.; Delos Santos, N.P.; Hartzell, A.L.; Texari, L.; Lin, P.-A.; Bartlett, A.; Ecker, J.R.; Benner, C.; Heinz, S.; et al. Genomic Decoding of Neuronal Depolarization by Stimulus-Specific NPAS4 Heterodimers. Cell 2019, 179, 373–391.e27. [Google Scholar] [CrossRef]
- Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.-K.; Hu, L.S.; Malik, A.N.; Greenberg, M.E. Activity-Dependent Regulation of Inhibitory Synapse Development by Npas4. Nature 2008, 455, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A.; Hatsuta, H.; Kikuchi, M.; Nakaya, A.; Saito, Y.; Tsukie, T.; Hara, N.; Ogishima, S.; Kitamura, N.; Akazawa, K.; et al. Genes Associated with the Progression of Neurofibrillary Tangles in Alzheimer’s Disease. Transl. Psychiatry 2014, 4, e396. [Google Scholar] [CrossRef]
- Fan, W.; Long, Y.; Lai, Y.; Wang, X.; Chen, G.; Zhu, B. NPAS4 Facilitates the Autophagic Clearance of Endogenous Tau in Rat Cortical Neurons. J. Mol. Neurosci. 2016, 58, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Opsomer, R.; Contino, S.; Perrin, F.; Gualdani, R.; Tasiaux, B.; Doyen, P.; Vergouts, M.; Vrancx, C.; Doshina, A.; Pierrot, N.; et al. Amyloid Precursor Protein (APP) Controls the Expression of the Transcriptional Activator Neuronal PAS Domain Protein 4 (NPAS4) and Synaptic GABA Release. eNeuro 2020, 7, ENEURO.0322-19.2020. [Google Scholar] [CrossRef] [PubMed]
- Pollina, E.A.; Gilliam, D.T.; Landau, A.T.; Lin, C.; Pajarillo, N.; Davis, C.P.; Harmin, D.A.; Yap, E.-L.; Vogel, I.R.; Griffith, E.C.; et al. A NPAS4–NuA4 Complex Couples Synaptic Activity to DNA Repair. Nature 2023, 614, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.S.; Li, E.; Scharnagl, L.; Poupardin, R.; Altendorfer, B.; Mrowetz, H.; Hutter-Paier, B.; Weiger, T.M.; Heneka, M.T.; Attems, J.; et al. CD8+ T-Cells Infiltrate Alzheimer’s Disease Brains and Regulate Neuronal- and Synapse-Related Gene Expression in APP-PS1 Transgenic Mice. Brain Behav. Immun. 2020, 89, 67–86. [Google Scholar] [CrossRef]
- Louis Sam Titus, A.S.C.; Sharma, D.; Kim, M.S.; D’Mello, S.R. The Bdnf and Npas4 Genes Are Targets of HDAC3-Mediated Transcriptional Repression. BMC Neurosci. 2019, 20, 65. [Google Scholar] [CrossRef]
- Herre, M.; Korb, E. The Chromatin Landscape of Neuronal Plasticity. Curr. Opin. Neurobiol. 2019, 59, 79–86. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Aromolaran, K.A.; Zukin, R.S. The Emerging Field of Epigenetics in Neurodegeneration and Neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.-H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 Reverses Alzheimer’s Disease-Related Pathologies in Vitro and in the 3xTg-AD Mouse Model. Proc. Natl. Acad. Sci. USA 2018, 115, E11148–E11157. [Google Scholar] [CrossRef] [PubMed]
- Mousa, H.H.; Sharawy, M.H.; Nader, M.A. Empagliflozin Enhances Neuroplasticity in Rotenone-Induced Parkinsonism: Role of BDNF, CREB and Npas4. Life Sci. 2023, 312, 121258. [Google Scholar] [CrossRef]
- Burnside, S.W.; Hardingham, G.E. Transcriptional Regulators of Redox Balance and Other Homeostatic Processes with the Potential to Alter Neurodegenerative Disease Trajectory. Biochem. Soc. Trans. 2017, 45, 1295–1303. [Google Scholar] [CrossRef]
- Kuczewski, N.; Porcher, C.; Lessmann, V.; Medina, I.; Gaiarsa, J.-L. Activity-Dependent Dendritic Release of BDNF and Biological Consequences. Mol. Neurobiol. 2009, 39, 37–49. [Google Scholar] [CrossRef]
- Danzer, S.C.; Crooks, K.R.C.; Lo, D.C.; McNamara, J.O. Increased Expression of Brain-Derived Neurotrophic Factor Induces Formation of Basal Dendrites and Axonal Branching in Dentate Granule Cells in Hippocampal Explant Cultures. J. Neurosci. 2002, 22, 9754–9763. [Google Scholar] [CrossRef]
- Tanaka, J.; Horiike, Y.; Matsuzaki, M.; Miyazaki, T.; Ellis-Davies, G.C.R.; Kasai, H. Protein Synthesis and Neurotrophin-Dependent Structural Plasticity of Single Dendritic Spines. Science 2008, 319, 1683–1687. [Google Scholar] [CrossRef]
- Kitanishi, T.; Ikegaya, Y.; Matsuki, N.; Yamada, M.K. Experience-Dependent, Rapid Structural Changes in Hippocampal Pyramidal Cell Spines. Cereb. Cortex 2009, 19, 2572–2578. [Google Scholar] [CrossRef]
- Poo, M. Neurotrophins as Synaptic Modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Esvald, E.-E.; Tuvikene, J.; Moistus, A.; Rannaste, K.; Kõomägi, S.; Timmusk, T. Differential Regulation of the BDNF Gene in Cortical and Hippocampal Neurons. J. Neurosci. 2022, 42, 9110–9128. [Google Scholar] [CrossRef]
- West, A.E.; Pruunsild, P.; Timmusk, T. Neurotrophins: Transcription and Translation. Handb. Exp. Pharmacol. 2014, 220, 67–100. [Google Scholar] [CrossRef] [PubMed]
- Balschun, D. Deletion of the Ryanodine Receptor Type 3 (RyR3) Impairs Forms of Synaptic Plasticity and Spatial Learning. EMBO J. 1999, 18, 5264–5273. [Google Scholar] [CrossRef]
- Galeotti, N.; Quattrone, A.; Vivoli, E.; Norcini, M.; Bartolini, A.; Ghelardini, C. Different Involvement of Type 1, 2, and 3 Ryanodine Receptors in Memory Processes. Learn. Mem. 2008, 15, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Welcher, A.A.; Shelton, D.; Schuman, E.M. Neurotrophins and Time: Different Roles for TrkB Signaling in Hippocampal Long-Term Potentiation. Neuron 1997, 19, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Thornton, P.L.; Balazs, R.; Cotman, C.W. β-Amyloid-(1–42) Impairs Activity-Dependent CAMP-Response Element-Binding Protein Signaling in Neurons at Concentrations in Which Cell Survival Is Not Compromised. J. Biol. Chem. 2001, 276, 17301–17306. [Google Scholar] [CrossRef]
- Garzon, D.J.; Fahnestock, M. Oligomeric Amyloid Decreases Basal Levels of Brain-Derived Neurotrophic Factor (BDNF) MRNA via Specific Downregulation of BDNF Transcripts IV and V in Differentiated Human Neuroblastoma Cells. J. Neurosci. 2007, 27, 2628–2635. [Google Scholar] [CrossRef]
- Bruna, B.; Lobos, P.; Herrera-Molina, R.; Hidalgo, C.; Paula-Lima, A.; Adasme, T. The Signaling Pathways Underlying BDNF-Induced Nrf2 Hippocampal Nuclear Translocation Involve ROS, RyR-Mediated Ca2+ Signals, ERK and PI3K. Biochem. Biophys. Res. Commun. 2018, 505, 201–207. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef]
- Qiu, J.; Dando, O.; Febery, J.A.; Fowler, J.H.; Chandran, S.; Hardingham, G.E. Neuronal Activity and Its Role in Controlling Antioxidant Genes. Int. J. Mol. Sci. 2020, 21, 1933. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive Regulation of the Brain’s Antioxidant Defences by Neurons and Astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Kepa, J.K.; Ross, D. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) Localizes to the Mitotic Spindle in Human Cells. PLoS ONE 2012, 7, e44861. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ali, R.; Verma, S. Aβ-Oligomers: A Potential Therapeutic Target for Alzheimer’s Disease. Int. J. Biol. Macromol. 2023, 239, 124231. [Google Scholar] [CrossRef]
- Perneczky, R.; Jessen, F.; Grimmer, T.; Levin, J.; Flöel, A.; Peters, O.; Froelich, L. Anti-Amyloid Antibody Therapies in Alzheimer’s Disease. Brain 2023, 146, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D. If Amyloid Drives Alzheimer Disease, Why Have Anti-Amyloid Therapies Not yet Slowed Cognitive Decline? PLoS Biol. 2022, 20, e3001694. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobos, P.; Vega-Vásquez, I.; Bruna, B.; Gleitze, S.; Toledo, J.; Härtel, S.; Hidalgo, C.; Paula-Lima, A. Amyloid β-Oligomers Inhibit the Nuclear Ca2+ Signals and the Neuroprotective Gene Expression Induced by Gabazine in Hippocampal Neurons. Antioxidants 2023, 12, 1972. https://doi.org/10.3390/antiox12111972
Lobos P, Vega-Vásquez I, Bruna B, Gleitze S, Toledo J, Härtel S, Hidalgo C, Paula-Lima A. Amyloid β-Oligomers Inhibit the Nuclear Ca2+ Signals and the Neuroprotective Gene Expression Induced by Gabazine in Hippocampal Neurons. Antioxidants. 2023; 12(11):1972. https://doi.org/10.3390/antiox12111972
Chicago/Turabian StyleLobos, Pedro, Ignacio Vega-Vásquez, Barbara Bruna, Silvia Gleitze, Jorge Toledo, Steffen Härtel, Cecilia Hidalgo, and Andrea Paula-Lima. 2023. "Amyloid β-Oligomers Inhibit the Nuclear Ca2+ Signals and the Neuroprotective Gene Expression Induced by Gabazine in Hippocampal Neurons" Antioxidants 12, no. 11: 1972. https://doi.org/10.3390/antiox12111972
APA StyleLobos, P., Vega-Vásquez, I., Bruna, B., Gleitze, S., Toledo, J., Härtel, S., Hidalgo, C., & Paula-Lima, A. (2023). Amyloid β-Oligomers Inhibit the Nuclear Ca2+ Signals and the Neuroprotective Gene Expression Induced by Gabazine in Hippocampal Neurons. Antioxidants, 12(11), 1972. https://doi.org/10.3390/antiox12111972