In Vitro α-Glucosidase and α-Amylase Inhibition, Cytotoxicity and Free Radical Scavenging Profiling of the 6-Halogeno and Mixed 6,8-Dihalogenated 2-Aryl-4-methyl-1,2-dihydroquinazoline 3-Oxides


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
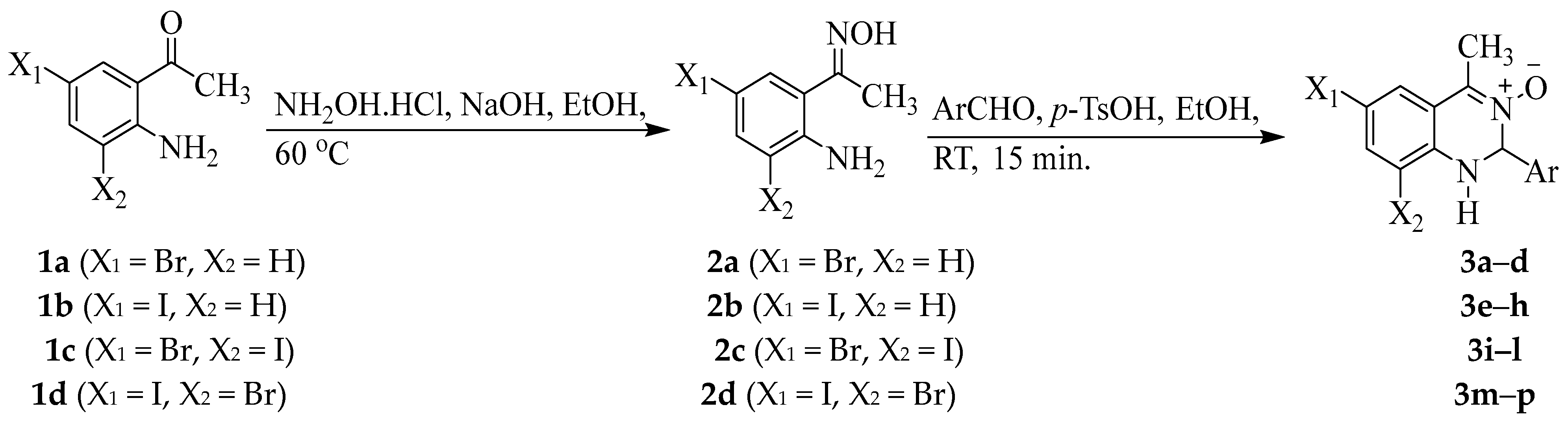
2.2. Typical Procedure for the Oximation of 1a–d
2.3. Typical Procedure for the Synthesis of 3a–p
2.4. Inhibition of α-Glucosidase and α-Amylase Activities by 3a–p
2.4.1. Inhibition of α-Glucosidase
2.4.2. Inhibition of α-Amylase by 3a–p
2.5. In Vitro Cytotoxicity of 3a–p against the MCF-7, A549 and HEK293-T Cell Lines
2.6. Free Radical Scavenging Assays of 3a–p
2.6.1. The 2,2-Diphenyl-1-picrylhydrazyl (DPPH) Radical Scavenging Assay
2.6.2. NO Free Radical Scavenging Assay
2.6.3. Assay on 3a, 3c, 3f, 3i, 3l, 3n and 3p for Inhibitory Activity against NO Generated from NOS
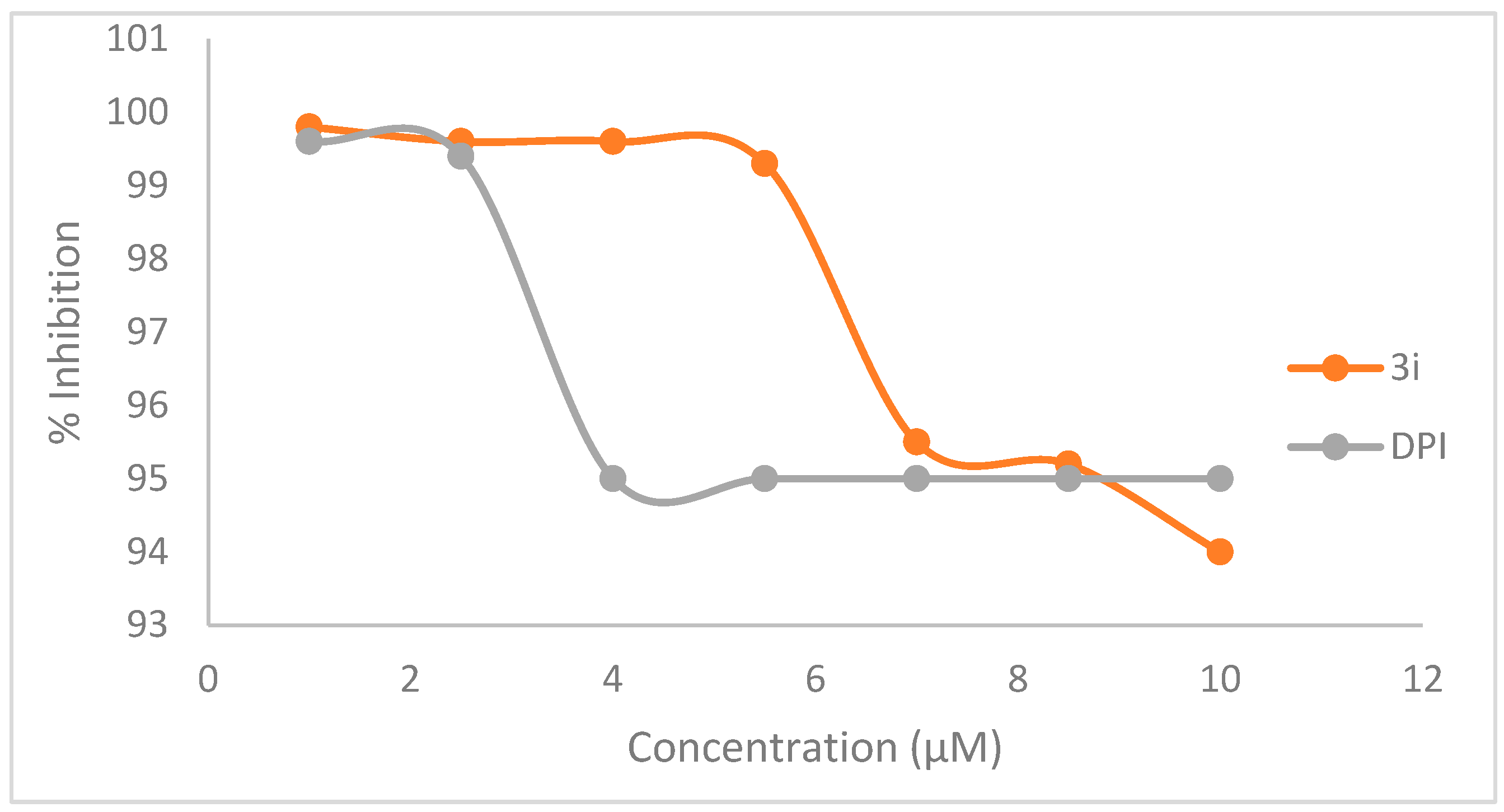
2.6.4. Kinetic Study on Inhibitory Activity of NOS by 3i and Diphenyleneiodonium (DPI)
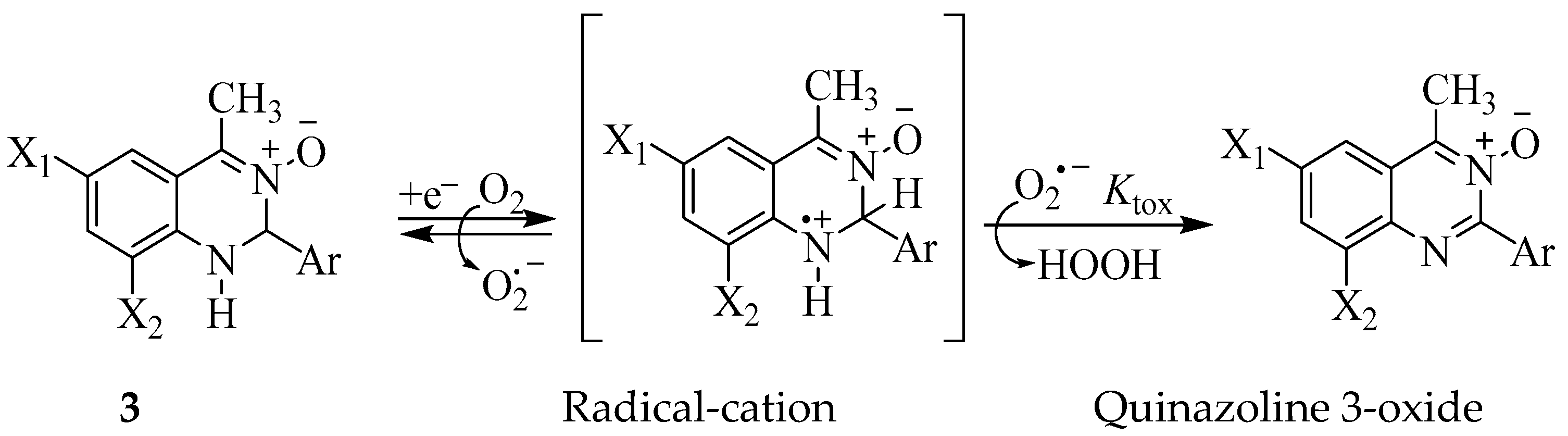
2.7. Superoxide Dismutase (SOD) Inhibitory Assay on 3a, 3c, 3f, 3i, 3l, 3n and 3p
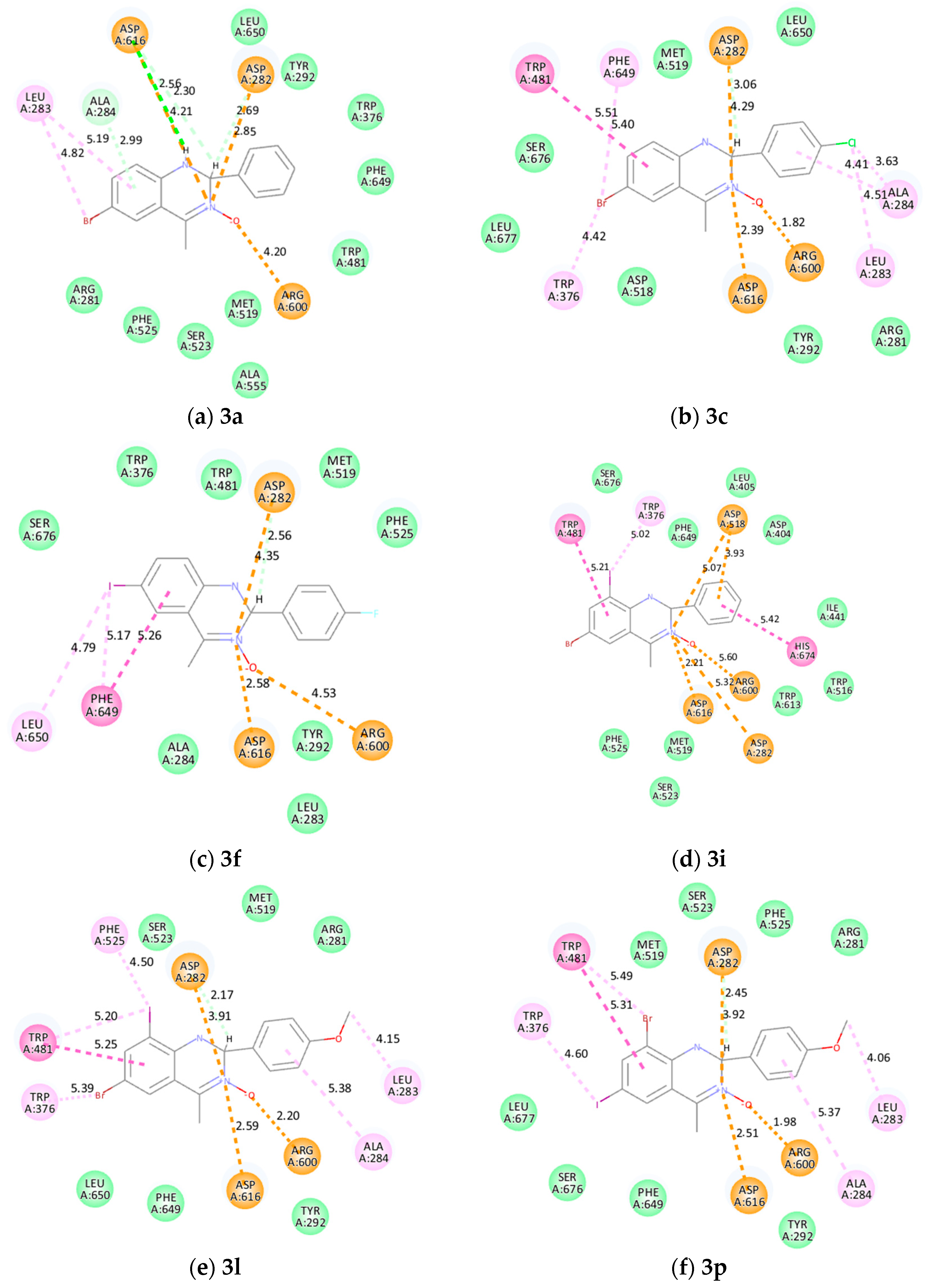
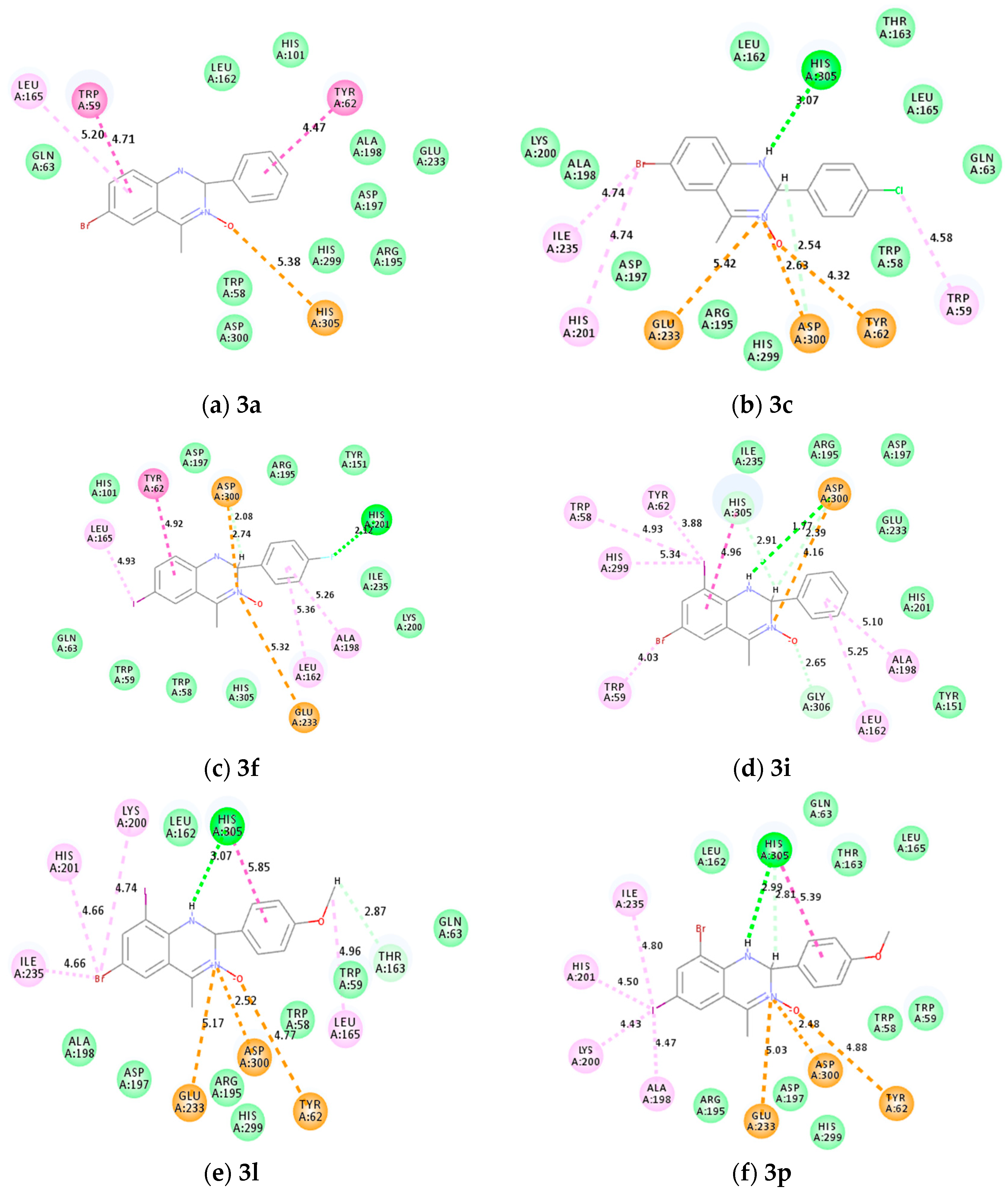
2.8. Molecular Docking of Representative Compounds 3a, 3c, 3f, 3i, 3l and 3p
2.8.1. Molecular Docking into α-Glucosidase
2.8.2. Molecular Docking into α-Amylase
2.9. Drug-Likeness Estimation of Selected Compounds 3a–p
3. Results and Discussion
3.1. Chemistry
3.2. Bio-Evaluation of Compounds 3a–p
Inhibition of α-Glucosidase and α-Amylase and Cytotoxicity Studies of 3a–p
3.3. Computational Studies
3.3.1. Molecular Docking 3a, 3c, 3f, 3i, 3l and 3p into α-Glucosidase and α-Amylase
3.3.2. Pharmacokinetics Properties Prediction of 3a–p
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Budreviciute, A.; Damiati, S.; Sabir, D.K.; Onder, K.; Schuller-Goetzburg, P.; Plakys, G.; Katileviciute, A.; Khoja, S.; Kodzius, R. Management and prevention strategies for non-communicable diseases (NCDs) and their risk factors. Front. Public Health 2020, 8, 574111. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Duvoor, C.; Dendi, V.S.R.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus, management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bai, Y.; Jin, Z.; Svensson, B. Food-derived non-phenolic α-amylase and α-glucosidase inhibitors for controlling starch digestion rate and guiding diabetes-friendly recipes. LWT 2022, 153, 112455. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Büsselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef]
- Pili, R.; Chang, J.; Partis, R.; Mueller, R.A.; Chrest, F.J.; Passaniti, A. The α-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res. 1995, 55, 2920–2926. [Google Scholar] [PubMed]
- Chetan, S.; Amarjeet, K.; Thind, S.S.; Baljit, S.; Shiveta, R. Advanced glycation end-products (AGEs): An emerging concern for processed food industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar]
- Rani, V.; Deep, G.; Signh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; Machado, A.I.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; Gatcia, M.L.; Silva, A.M.; et al. Sugar-lowering drugs for type 2 diabetes mellitus and metabolic syndrome—Review of classical and new compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- She, X.; Gates, K.S. Enzyme-activated generation of reactive oxygen species from heterocyclic N-oxides under aerobic and anaerobic conditions and its relevance to hypoxia-selective prodrugs. Chem. Res. Toxicol. 2019, 32, 348–361. [Google Scholar]
- Mfuh, A.M.; Larionov, O.V. Heterocyclic N-oxides—An emerging class of therapeutic agents. Curr. Med. Chem. 2015, 22, 2819–2857. [Google Scholar] [CrossRef]
- Belova, N.V.; Giricheva, N.I.; Fedorov, M.S. Substituent effect on the properties of pyridine-N-oxides. Struct. Chem. 2015, 26, 1459–1465. [Google Scholar] [CrossRef]
- Combs, D.W.; Rampulla, M.S.; Russell, R.K.; Rampulla, R.; Klaubert, D.H.; Ritchie, D.; Meeks, A.S.; Kirchner, T. Design, synthesis and bronchodilatory activity of a series of quinazoline-3-oxides. Drug Des. Deliv. 1990, 6, 241–254. [Google Scholar] [PubMed]
- Pathare, R.S.; Maurya, A.K.; Kumari, A.; Agnihotri, V.K.; Vermac, V.P.; Sawant, D.M. Synthesis of quinazoline-3-oxides via a Pd(II) catalyzed azide–isocyanide coupling/cyclocondensation reaction. Org. Biomol. Chem. 2019, 17, 363–368. [Google Scholar] [PubMed]
- Mphahlele, M.J.; Onwu, E.E.; Agbo, E.N.; Maluleka, M.M.; More, G.K.; Choong, Y.S. Synthesis, in vitro and in silico enzyme (COX-1/2 & LOX-5), free radical scavenging and cytotoxicity profiling of the 2,4-dicarbo substituted quinazoline 3-oxides. Med. Chem. Res. 2022, 31, 146–164. [Google Scholar]
- Mphahlele, M.J. A review on the synthesis and chemical transformation of quinazoline 3-oxides. Molecules 2022, 27, 7985. [Google Scholar] [CrossRef] [PubMed]
- Mikiciuk-Olasik, E.; Baszczak-Swiatkiewiz, K.; Zurek, E.; Krajewska, U.; Rózalski, M.; Kruszynski, R.; Bartczak, T.J. New derivatives of quinazoline and 1,2-dihydroquinazoline N3-oxide with expected antitumor activity. Arch. Pharm. Pharm. Med. Chem. 2004, 337, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Combs, D.W.; Rampulla, M.S.; Falotico, R. 2-Aryl-l,2-dihydro-6,7-dimethoxyqulnazoline-3-oxides with positive inotropic activity. Bioorg. Med. Chem. Lett. 1991, 1, 133–136. [Google Scholar] [CrossRef]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; De Azevedo Junior, W.F.; Leita, A.C.L. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef]
- Fejzagić, A.V.; Gebauer, J.; Huwa, N.; Classen, T. Halogenating enzymes for active agent synthesis: First steps are done and many have to follow. Molecules 2019, 24, 4008. [Google Scholar] [CrossRef] [PubMed]
- Tiz, D.B.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New halogen-containing drugs approved by FDA in 2021: An overview on their syntheses and pharmaceutical use. Molecules 2022, 27, 1643. [Google Scholar]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ali, M.T.; Shawan, M.M.A.K.; Sarwar, M.G.; Khan, M.A.K.; Halim, M.A. Halogen-directed drug design for Alzheimer’s disease: A combined density functional and molecular docking study. SpringerPlus 2016, 5, 1346–1359. [Google Scholar] [CrossRef]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen bonding—A novel interaction for rational drug design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543. [Google Scholar] [CrossRef]
- Cabrita, M.T.; Vale, C.; Rauter, A.P. Halogenated compounds from marine algae. Mar. Drugs 2010, 8, 2301–2317. [Google Scholar] [CrossRef]
- Petrov, S.A.; Yusubov, M.S.; Beloglazkina, E.K.; Nenajdenko, V.G. Synthesis of radioiodinated compounds. Classical approaches and achievements of recent years. Int. J. Mol. Sci. 2022, 22, 13789. [Google Scholar] [CrossRef]
- Wilcken, R.; Liu, X.; Zimmermann, M.O.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 2012, 134, 6810–6818. [Google Scholar] [CrossRef]
- Cavina, L.; Van der Born, D.; Klaren, P.H.M.; Feiters, M.C.; Boerman, O.C.; Rutjes, F.P.J.T. Design of radioiodinated pharmaceuticals: Structural features affecting metabolic stability towards in vivo deiodination. Eur. J. Med. Chem. 2017, 2017, 3387–3414. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Magwaza, N.M.; More, K.G.; Gildenhuys, S. An in vitro and in silico α-amylase/α-glucosidase/protein tyrosine phosphatase 1 beta & radical scavenging profiling of the 3,5,7-tricarbo substituted 1H-indazoles. Med. Chem. Res. 2022, 31, 2132–2151. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Yang, D.-Y. Visible light-mediated synthesis of quinazolines from 1,2-dihydroquinazoline 3-oxides. Tetrahedron 2013, 69, 10438–10444. [Google Scholar] [CrossRef]
- Butler, M.; Cabrer, G.M. Determination of the position of the N-O function in substituted pyrazine N-oxides by chemometric analysis of carbon-13 nuclear magnetic resonance data. J. Mol. Struct. 2013, 1043, 37–42. [Google Scholar] [CrossRef]
- Yuriev, E.; Chalmers, D.; Capuano, B. Conformational analysis of drug molecules: A practical exercise in the medicinal chemistry course. J. Chem. Educ. 2009, 86, 477–478. [Google Scholar] [CrossRef]
- Fang, Z.; Song, Y.; Zhan, F.; Zhang, Q.; Liu, X. Conformational restriction: An effective tactic in ‘follow-on’-based drug discovery. Future Med. Chem. 2014, 6, 885–901. [Google Scholar] [CrossRef]
- Kessler, H. Differences of the conformation in crystal and solution and methods for the determination of the conformation in solution by NMR spectroscopy. Z. Anal. Chem. 1987, 327, 66–67. [Google Scholar] [CrossRef]
- Łukomska, M.; Rybarczyk-Pirek, A.J.; Jabłoński, M.; Palusiak, M. On the nature of NO-bonding in N-oxide group. Phys. Chem. Chem. Phys. 2015, 17, 16375–16387. [Google Scholar] [CrossRef] [PubMed]
- Naumann, K. Influence of chlorine substituents on biological activity of chemicals: A review. Pest. Manag. Sci. 2000, 56, 3–21. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Nasi, M.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cossarizza, A. Interfering with ROS metabolism in cancer cells: The potential role of quercetin. Cancers 2010, 2, 1288–1311. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-F.; Yin, S.; Chen, Z.-Q.; Li, F.; Zhao, B. High glucose promotes tumor cell proliferation and migration in lung adenocarcinoma via the RAGE NOXs pathway. Mol. Med. Rep. 2018, 17, 8536–8541. [Google Scholar] [PubMed]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Bridges, A.J. Chemical inhibitors of protein kinases. Chem. Rev. 2001, 101, 2541–2571. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Valgimigli, L. Advantages and limitations of common testing methods for antioxidants. Free Radic. Res. 2015, 49, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, A.J.; Higgs, A.; Moncada, S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 191–220. [Google Scholar] [CrossRef]
- Oudemans-van Straaten, H.M.; Spoelstra-de Man, A.M.E.; De Waard, M.C. Vitamin C revisited. Crit. Care 2014, 18, 460. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.N.; Rios, N.; Trujillo, M.; Radi, X.R.; Denicola, A.; Alvarez, B. Detection and quantification of nitric oxide–derived oxidants in biological systems. J. Biol. Chem. 2019, 294, 14776–14802. [Google Scholar] [CrossRef] [PubMed]
- Apak, R.; Calokerinos, A.; Gorinstein, S.; Segundo, M.A.; Hibbert, D.B.; Gülçin, I.; Çekiç, S.D.; Güçlü, K.; Özyürek, M.; Çelik, S.E.; et al. Methods to evaluate the scavenging activity of antioxidants toward reactive oxygen and nitrogen species (IUPAC Technical Report). Pure Appl. Chem. 2022, 94, 87–144. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Fasehun, O.A.; Kwon, N.S.; Gross, S.S.; Gonzalez, J.A.; Levi, R.; Nathan, C.F. Inhibition of macrophage and endothelial nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. 1991, 5, 98–103. [Google Scholar] [CrossRef]
- Giacco, F.B. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 40, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug. Discov. 2012, 7, 217–234. [Google Scholar] [CrossRef]
- Al-Hamdani, Y.S.; Tkatchenko, A. Understanding non-covalent interactions in larger molecular complexes from first principles. Chem. Phys. 2019, 150, 010901. [Google Scholar] [CrossRef]
- Song, F.; Xu, G.; Gaul, M.D.; Zhao, B.; Lu, T.; Zhang, R.L.; DesJarlais, R.; DiLoreto, K.; Huebert, N.; Shook, B.; et al. Design, synthesis and structure activity relationships of indazole and indole derivatives as potent glucagon receptor antagonists. Bioorg. Med. Chem. Lett. 2019, 29, 1974–1980. [Google Scholar] [CrossRef]
- Aispuro-Pérez, A.; López-Ávalos, J.; García-Páez, F.; Montes-Avila, J.; Picos-Corrales, L.A.; Ochoa-Terán, A.; Bastidas, P.; Montaño, S.; Calderón-Zamora, L.; Osuna-Martínez, U.; et al. Synthesis and molecular docking studies of imines as α-glucosidase and α-amylase inhibitors. Bioorg. Chem. 2020, 94, 103491. [Google Scholar] [CrossRef]
- Dai, T.; Chen, J.; Li, Q.; Li, P.; Hu, P.; Liu, C.; Li, T. Investigation the interaction between procyanidin dimer and α-amylase: Spectroscopic analyses and molecular docking simulation. Int. J. Biol. Macromol. 2018, 113, 427–433. [Google Scholar] [CrossRef]
- Patil, M.; Patil, S.; Maheshwari, V.L.; Zawar, L.; Patil, R.H. Recent updates on in silico screening of natural products as potential inhibitors of enzymes of biomedical and pharmaceutical importance. In Natural Products as Enzyme Inhibitors; Maheshwari, V.L., Patil, R.H., Eds.; Springer: Singapore, 2022. [Google Scholar] [CrossRef]
- Hermans, M.M.P.; Kroos, M.A.; Van Beeumen, J.; Oostra, B.A.; Reuser, A.J.J. Human lysosomal α-glucosidase: Characterization of the catalytic site. J. Biol. Chem. 1991, 266, 13507–13512. [Google Scholar] [CrossRef]
- Rydberg, E.H.; Li, C.; Maurus, R.; Overall, C.M.; Brayer, G.D.; Withers, S.G. Mechanistic analyses of catalysis in human pancreatic α-amylase: Detailed kinetic and structural studies of mutants of three conserved carboxylic acids. Biochemistry 2002, 41, 4492–4502. [Google Scholar] [CrossRef]
- Shao, J.; Kuiper, B.P.; Thunnissen, A.M.W.H.; Cool, R.H.; Zhou, L.; Huang, C.; Dijkstra, B.W.; Broos, J. The role of tryptophan in π interactions in proteins: An experimental approach. J. Am. Chem. Soc. 2022, 144, 13815–13822. [Google Scholar] [CrossRef]
- Ye, X.; Chen, Z.; Zhang, Z.; Fu, Y.; Deng, Z.; Peng, Y. An convenient approach to 2,4-disubstituted quinazoline-3-oxides using active MnO2 as oxidant. Canadian J. Chem. 2019, 97, 682–689. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Magwaza, N.M.; Gildenhuys, S.; Setshedi, I.B. Synthesis, Synthesis, α-glucosidase inhibition, and antioxidant activity of the 7-carbo–substituted 5-bromo-3-methylindazoles. Bioorg. Chem. 2020, 97, 103702. [Google Scholar] [CrossRef]
- Bruker, APEX-3, SAINT+, Software (Includes XPREP and SADABS); Version 6.02; Bruker AXS Inc.: Madison, Wisconsin, USA, 2016.
- Farrugia, L.J. WinGX and ORTEP for Windows an update. J. Appl. Crystallogr. 2012, 245, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL-2017/1. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef]
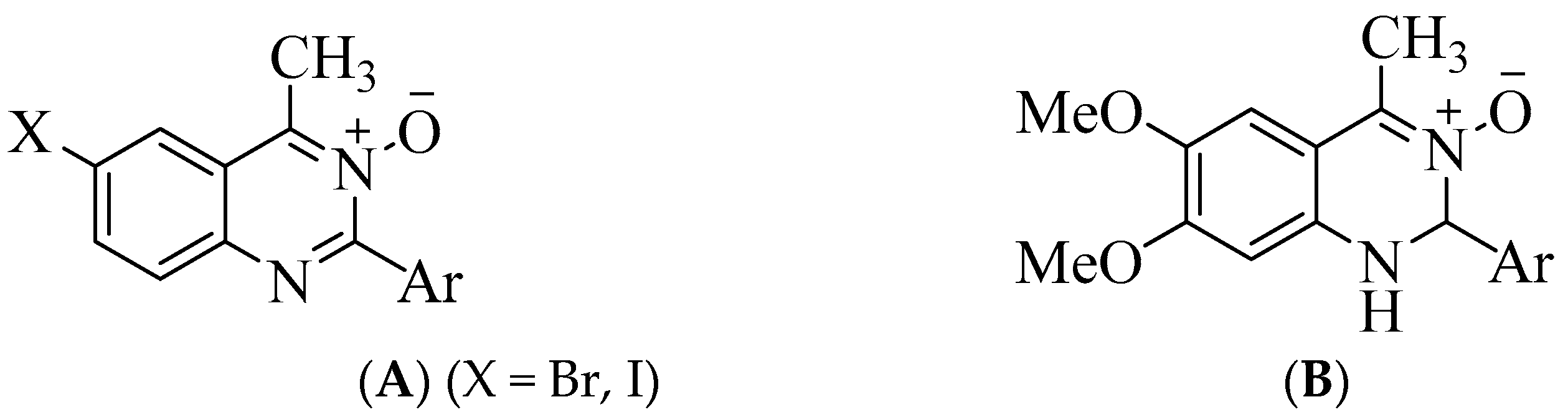


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3a–p | X1 | X2 | Ar | % Yield |
|---|---|---|---|---|
| 3a | Br | H | -C6H5 | 62 |
| 3b | Br | H | -C6H4(4-F) | 50 |
| 3c | Br | H | -C6H4(4-Cl) | 84 |
| 3d | Br | H | -C6H4(4-OCH3) | 90 |
| 3e | I | H | -C6H5 | 68 |
| 3f | I | H | -C6H4(4-F) | 72 |
| 3g | I | H | -C6H4(4-Cl) | 88 |
| 3h | I | H | -C6H4(4-OCH3) | 86 |
| 3i | Br | I | -C6H5 | 74 |
| 3j | Br | I | -C6H4(4-F) | 60 |
| 3k | Br | I | -C6H4(4-Cl) | 80 |
| 3l | Br | I | -C6H4(4-OCH3) | 84 |
| 3m | I | Br | -C6H5 | 76 |
| 3n | I | Br | -C6H4(4-F) | 66 |
| 3o | I | Br | -C6H4(4-Cl) | 98 |
| 3p | I | Br | -C6H4(4-OCH3) | 94 |
| IC50 (µM ± SD) | |||||
|---|---|---|---|---|---|
| 3a–p | α-Glucosidase | α-Amylase | MCF-7 | A549 | Hek293-T |
| 3a | 1.08 ± 0.02 | 5.33 ± 0.01 | 10.83 ± 0.09 | 15.84 ± 0.07 | 40.18 ± 0.02 |
| 3b | 7.47 ± 0.05 | 36.35 ± 0.01 | 12.80 ± 0.07 | 19.31 ± 0.08 | 41.72 ± 0.04 |
| 3c | 0.92 ± 0.01 | 30.48 ± 0.02 | 25.48 ± 0.08 | 15.26 ± 0.09 | 27.54 ± 0.05 |
| 3d | 68.2 ± 0.01 | 54.08 ± 0.03 | 13.68 ± 0.09 | 13.45 ± 0.03 | 37.53 ± 0.18 |
| 3e | 6.04 ± 0.01 | 49.50 ± 0.06 | 15.83 ± 0.14 | 11.39 ± 0.12 | 20.12 ± 0.06 |
| 3f | 9.27 ± 0.02 | 0.64 ± 0.01 | 19.01 ± 0.04 | 13.71 ± 0.11 | 22.32 ± 0.08 |
| 3g | 15.08 ± 0.08 | 53.42 ± 0.01 | 10.38 ± 0.08 | 18.63 ± 0.11 | 21.70 ± 0.13 |
| 3h | 27.18 ± 0.02 | 55.72 ± 0.02 | 22.62 ± 0.12 | 18.71 ± 0.16 | 24.26 ± 0.04 |
| 3i | 1.01 ± 0.05 | 1.18 ± 0.06 | 15.43 ± 0.06 | 16.85 ± 0.05 | 34.90 ± 0.13 |
| 3j | 19.69 ± 0.01 | 17.48 ± 0.03 | 15.06 ± 0.01 | 13.70 ± 0.12 | 32.83 ± 0.06 |
| 3k | 13.47 ± 0.04 | 4.46 ± 0.02 | 16.65 ± 0.05 | 15.37 ± 0.13 | 21.04 ± 0.05 |
| 3l | 1.04 ± 0.03 | 54.18 ± 0.01 | 18.12± 0.10 | 16.34 ± 0.06 | 32.39 ± 0.17 |
| 3m | 9.14 ± 0.03 | 16.73 ± 0.01 | 15.34 ± 0.07 | 16.34 ± 0.06 | 27.97 ± 0.08 |
| 3n | 43.23 ± 0.05 | 4.71 ± 0.01 | 14.79 ± 0.10 | 17.22 ± 0.07 | 32.42 ± 0.06 |
| 3o | 7.07 ± 0.04 | 49.18 ± 0.01 | 16.50 ± 0.01 | 20.00 ± 0.05 | 58.98 ± 0.17 |
| 3p | 0.78 ± 0.05 | 73.66 ± 0.02 | 13.47 ± 0.06 | 12.24 ± 0.02 | 59.02 ± 0.02 |
| Acarbose | 4.40 ± 0.04 | 2.92 ± 0.02 | - | - | - |
| Doxorubicin | - | - | 0.25 ± 0.05 | 0.36 ± 0.07 | 0.87 ± 0.04 |
| Gefitinib | - | - | 0.19 ± 0.04 | 0.25 ± 0.03 | 0.40 ± 0.02 |
| IC50 (µM ± SD) | |||
|---|---|---|---|
| 3a–p | DPPH | NO | NO from NOS |
| 3a | 4.74 ± 0.027 | 27.34 ± 0.04 | 5.24 ± 0.002 |
| 3b | 1.07 ± 0.048 | 15.10 ± 0.029 | - |
| 3c | 33.52 ± 0.025 | 7.45 ± 0.002 | 1.10 ± 0.003 |
| 3d | 9.55 ± 0.006 | 24.42 ± 0.004 | - |
| 3e | 1.10 ± 0.031 | 52.38 ± 0.003 | - |
| 3f | 23.66 ± 0.009 | 6.74 ± 0.002 | 4.70 ± 0.004 |
| 3g | 26.88 ± 0.043 | 4.25 ± 0.005 | - |
| 3h | 0.21 ± 0.057 | 10.05 ± 0.003 | - |
| 3i | 1.04 ± 0.077 | 61.26 ± 0.005 | 29.08 ± 0.004 |
| 3j | 61.01 ± 0.053 | 5.17 ± 0.007 | - |
| 3k | 4.08 ± 0.038 | 15.00 ± 0.005 | - |
| 3l | 27.70 ± 0.040 | 67.81 ± 0.004 | 1.06 ± 0.005 |
| 3m | 8.88 ± 0.011 | 55.39 ± 0.002 | - |
| 3n | 6.84 ± 0.009 | 0.88 ± 0.004 | 3.50 ± 0.01 |
| 3o | 88.45 ± 0.009 | 23.25 ± 0.002 | - |
| 3p | 60.03 ± 0.014 | 5.46 ± 0.004 | 15.51 ± 0.009 |
| Ascorbic acid | 5.02 ± 0.009 | 5.39 ± 0.005 | 0.999 ± 0.006 |
| Property | Compound | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 3d | 3e | 3f | 3g | 3h | 3i | 3j | 3k | 3l | 3m | 3n | 3o | 3p | |
| Lipophilicity (XLogP3) | 2.64 | 2.74 | 3.27 | 2.61 | 2.60 | 2.70 | 3.23 | 2.57 | 3.29 | 3.39 | 3.92 | 3.26 | 3.29 | 3.39 | 3.92 | 3.26 |
| Polarity-TPSA (Å) | 40.78 | 40.78 | 40.78 | 50.01 | 40.78 | 40.78 | 40.78 | 50.01 | 40.78 | 40.78 | 40.78 | 50.01 | 40.78 | 40.78 | 40.78 | 50.01 |
| Solubility (Log S) | −3.87 | −4.02 | −4.46 | −3.93 | −4.14 | −4.29 | −4.72 | −4.19 | −5.04 | −5.19 | −5.63 | −5.10 | −5.04 | −5.19 | −5.63 | −5.10 |
| GI Absorption | High | High | High | High | High | High | High | High | High | High | High | High | High | High | High | High |
| Saturation (fraction Csp3) | 0.13 | 0.13 | 0.13 | 0.19 | 0.13 | 0.13 | 0.13 | 0.19 | 0.13 | 0.13 | 0.13 | 0.19 | 0.13 | 0.13 | 0.13 | 0.19 |
| Molecular weight | 317.18 | 335.17 | 351.63 | 347.21 | 364.18 | 382.17 | 398.63 | 394.21 | 443.08 | 461.07 | 477.52 | 473.10 | 443.08 | 461.07 | 477.52 | 473.10 |
| mLogP | 4.12 | 4.51 | 4.63 | 3.77 | 4.24 | 4.63 | 4.74 | 3.89 | 4.86 | 5.24 | 5.36 | 4.51 | 4.86 | 5.24 | 5.36 | 4.51 |
| Hydrogen bond acceptor | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| Hydrogen bond donor | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Rotatable bonds | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 |
| Lipinski’s violation | None | Yes 1 | Yes 1 | None | Yes 1 | Yes 1 | Yes 1 | None | Yes 1 | Yes 1 | Yes 1 | Yes 1 | Yes 1 | Yes 1 | Yes 1 | Yes 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magwaza, N.M.; More, G.K.; Gildenhuys, S.; Mphahlele, M.J. In Vitro α-Glucosidase and α-Amylase Inhibition, Cytotoxicity and Free Radical Scavenging Profiling of the 6-Halogeno and Mixed 6,8-Dihalogenated 2-Aryl-4-methyl-1,2-dihydroquinazoline 3-Oxides. Antioxidants 2023, 12, 1971. https://doi.org/10.3390/antiox12111971
Magwaza NM, More GK, Gildenhuys S, Mphahlele MJ. In Vitro α-Glucosidase and α-Amylase Inhibition, Cytotoxicity and Free Radical Scavenging Profiling of the 6-Halogeno and Mixed 6,8-Dihalogenated 2-Aryl-4-methyl-1,2-dihydroquinazoline 3-Oxides. Antioxidants. 2023; 12(11):1971. https://doi.org/10.3390/antiox12111971
Chicago/Turabian StyleMagwaza, Nontokozo M., Garland K. More, Samantha Gildenhuys, and Malose J. Mphahlele. 2023. "In Vitro α-Glucosidase and α-Amylase Inhibition, Cytotoxicity and Free Radical Scavenging Profiling of the 6-Halogeno and Mixed 6,8-Dihalogenated 2-Aryl-4-methyl-1,2-dihydroquinazoline 3-Oxides" Antioxidants 12, no. 11: 1971. https://doi.org/10.3390/antiox12111971
APA StyleMagwaza, N. M., More, G. K., Gildenhuys, S., & Mphahlele, M. J. (2023). In Vitro α-Glucosidase and α-Amylase Inhibition, Cytotoxicity and Free Radical Scavenging Profiling of the 6-Halogeno and Mixed 6,8-Dihalogenated 2-Aryl-4-methyl-1,2-dihydroquinazoline 3-Oxides. Antioxidants, 12(11), 1971. https://doi.org/10.3390/antiox12111971