
Neuroprotective Role of α-Lipoic Acid in Iron-Overload-Mediated Toxicity and Inflammation in In Vitro and In Vivo Models

 , ,
, ,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. xCELLigence Real-Time Cell Analysis
2.3. Apoptosis Rate and Reactive Oxygene Species Analysis
2.4. GSH Evaluation
2.5. Mitochondrial Membrane Potential (DiOC2(3)) and Mass
2.6. Real-Time PCR for Gene Expression Analysis
2.7. ELISA
2.8. Immunofluorescence
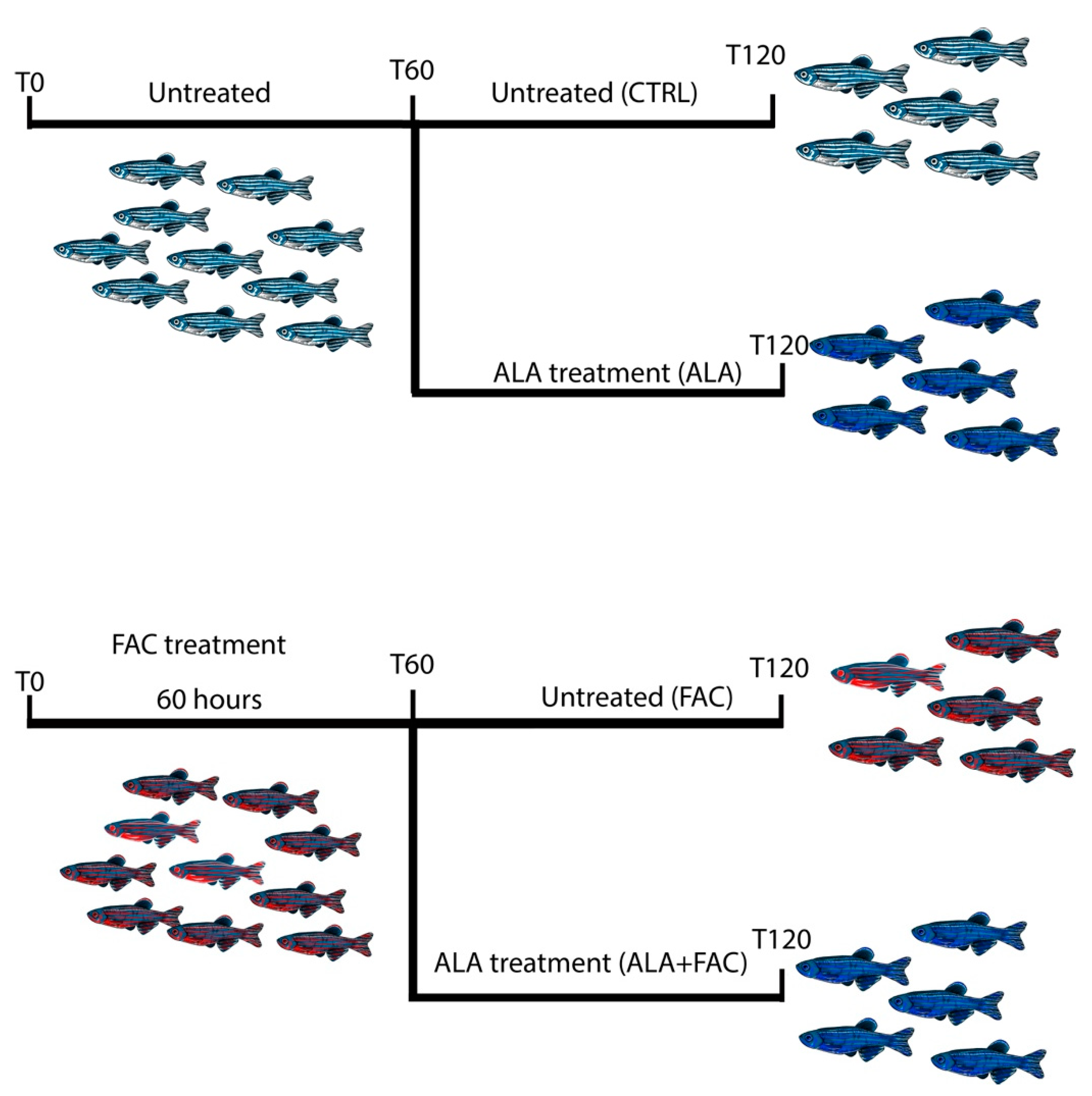
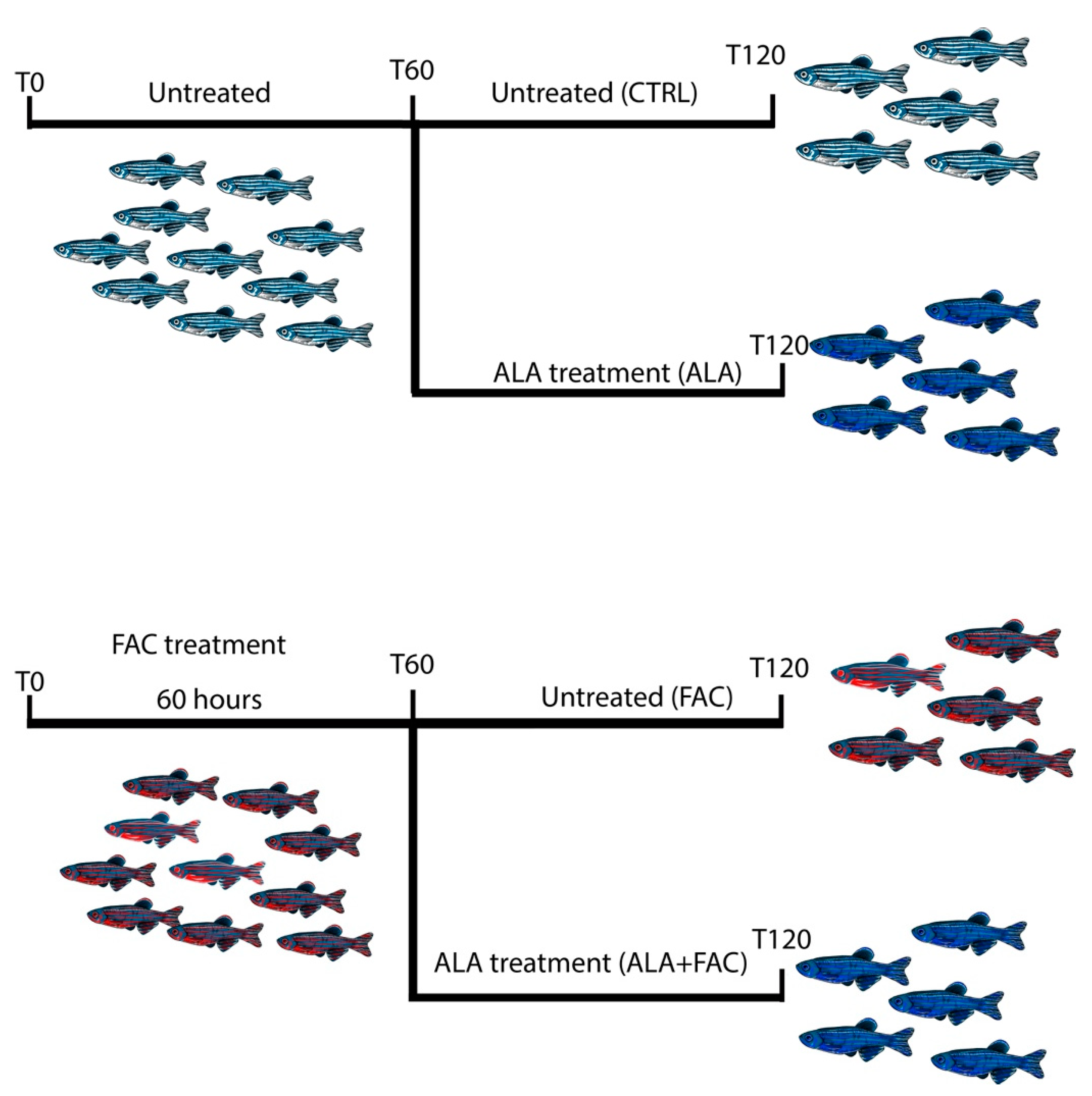
2.9. Iron-Overload Zebrafish Model
2.10. Immunoistochemistry
2.11. Statistical Analysis
3. Results
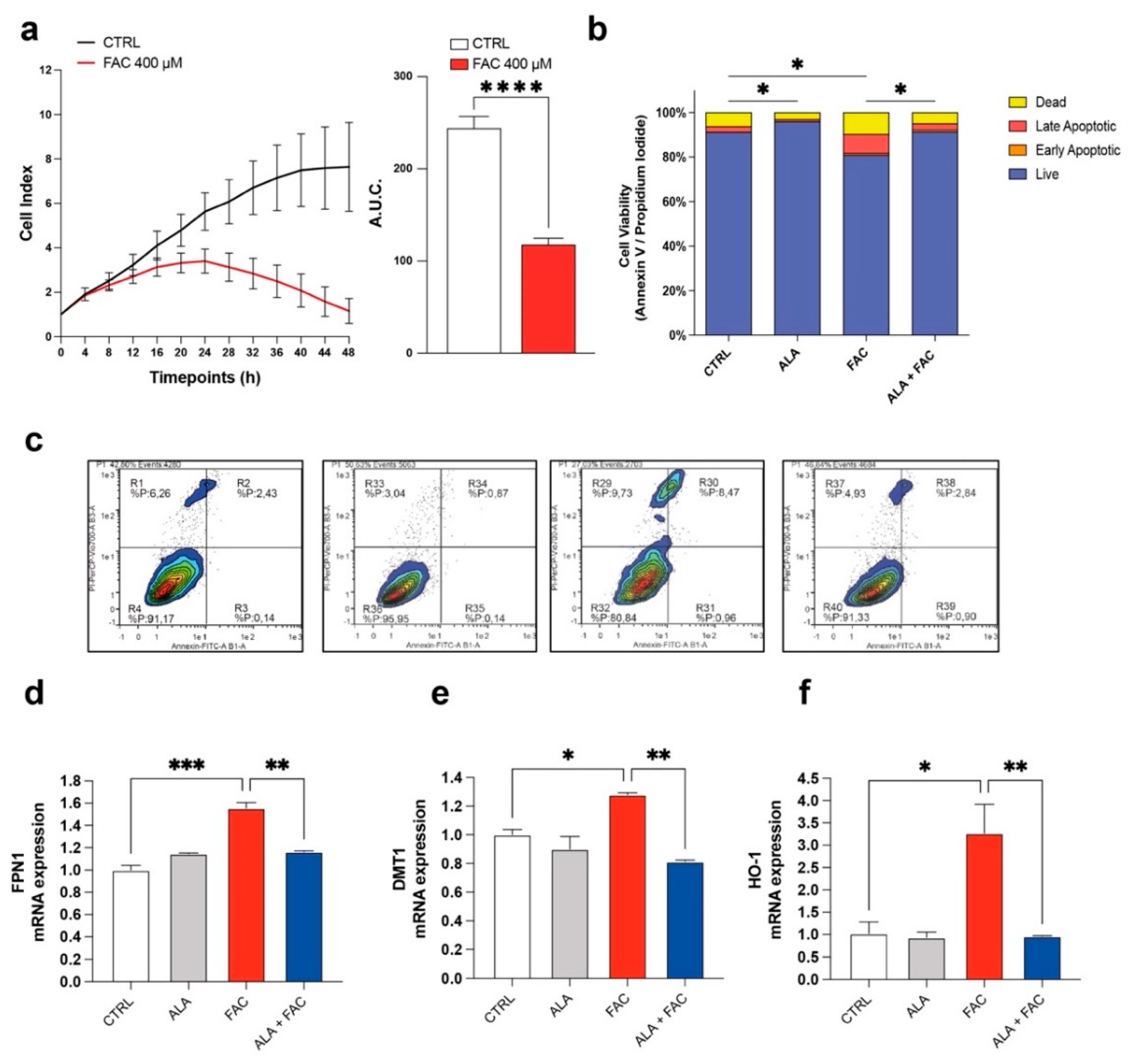
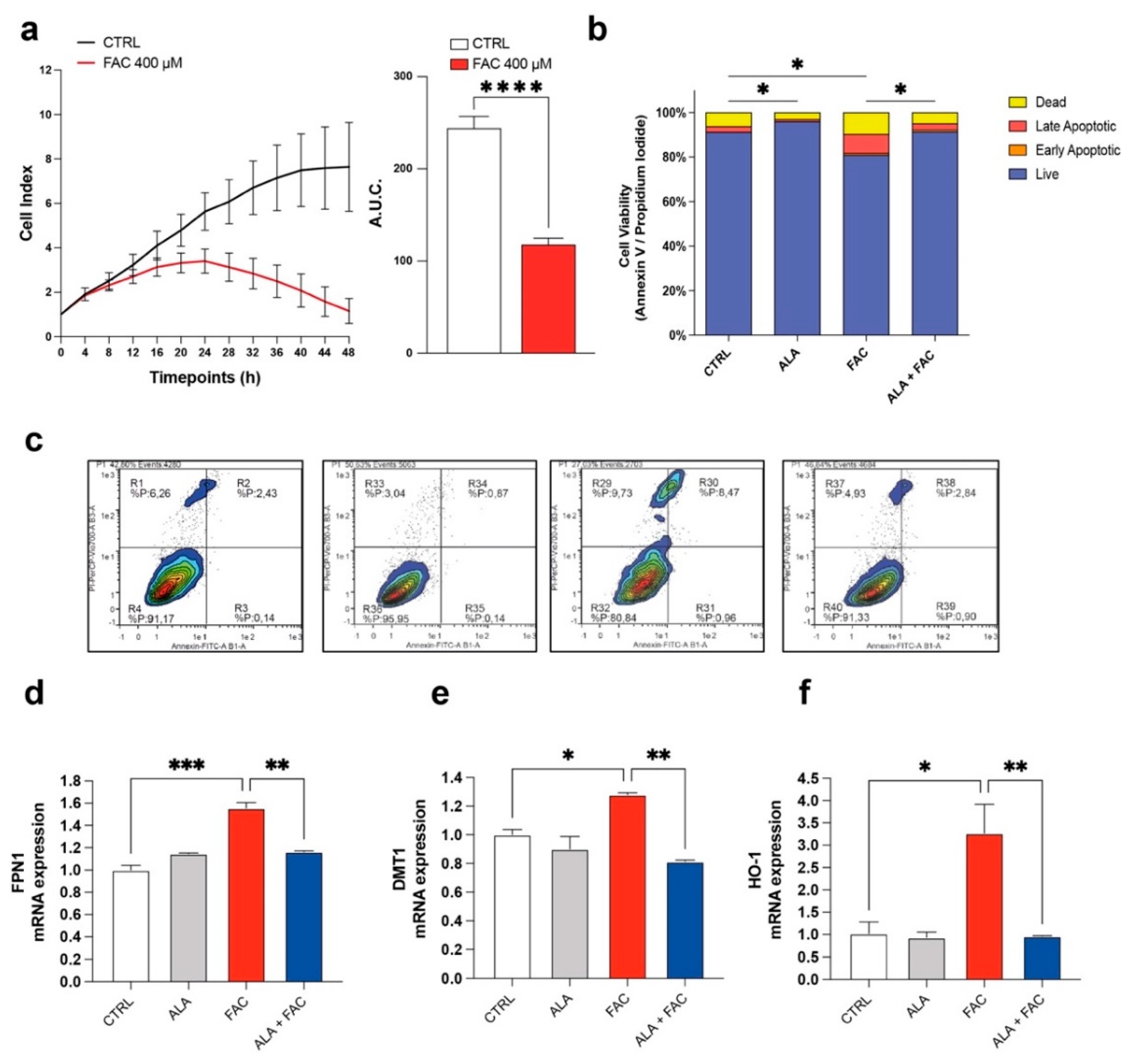
3.1. Iron Overload Affects Cell Survival and Iron-Metabolism-Related Gene Expression
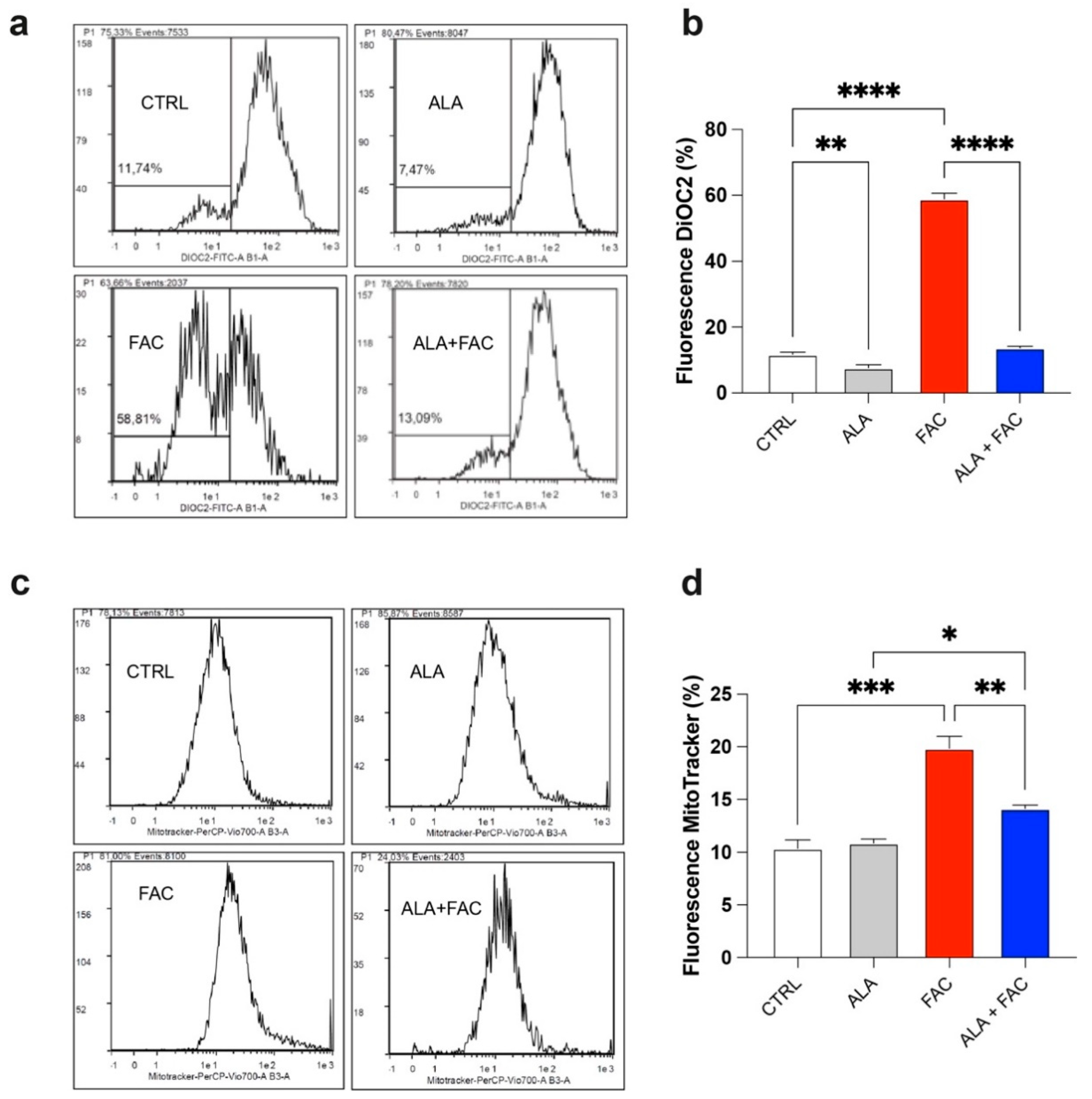
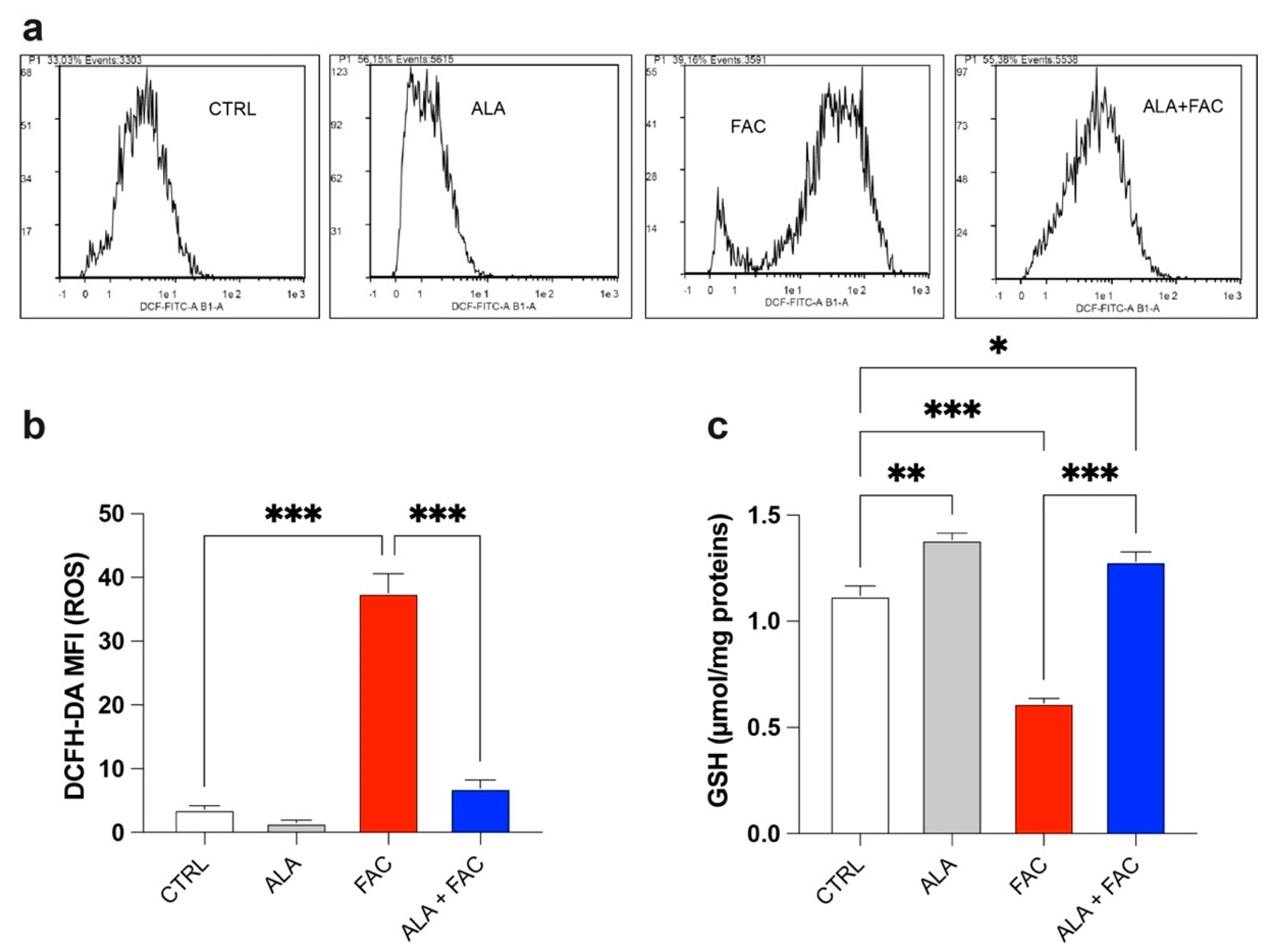
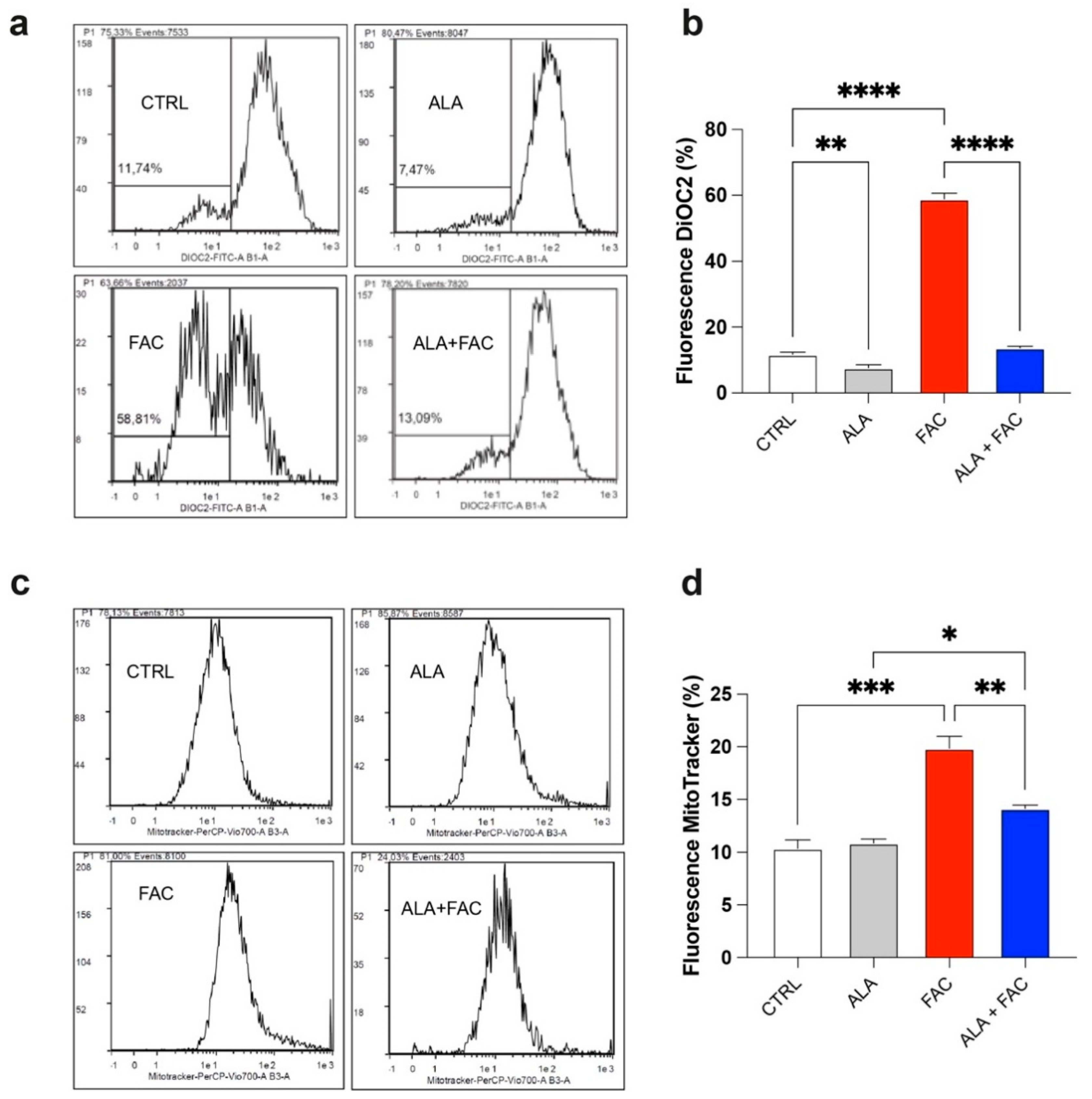
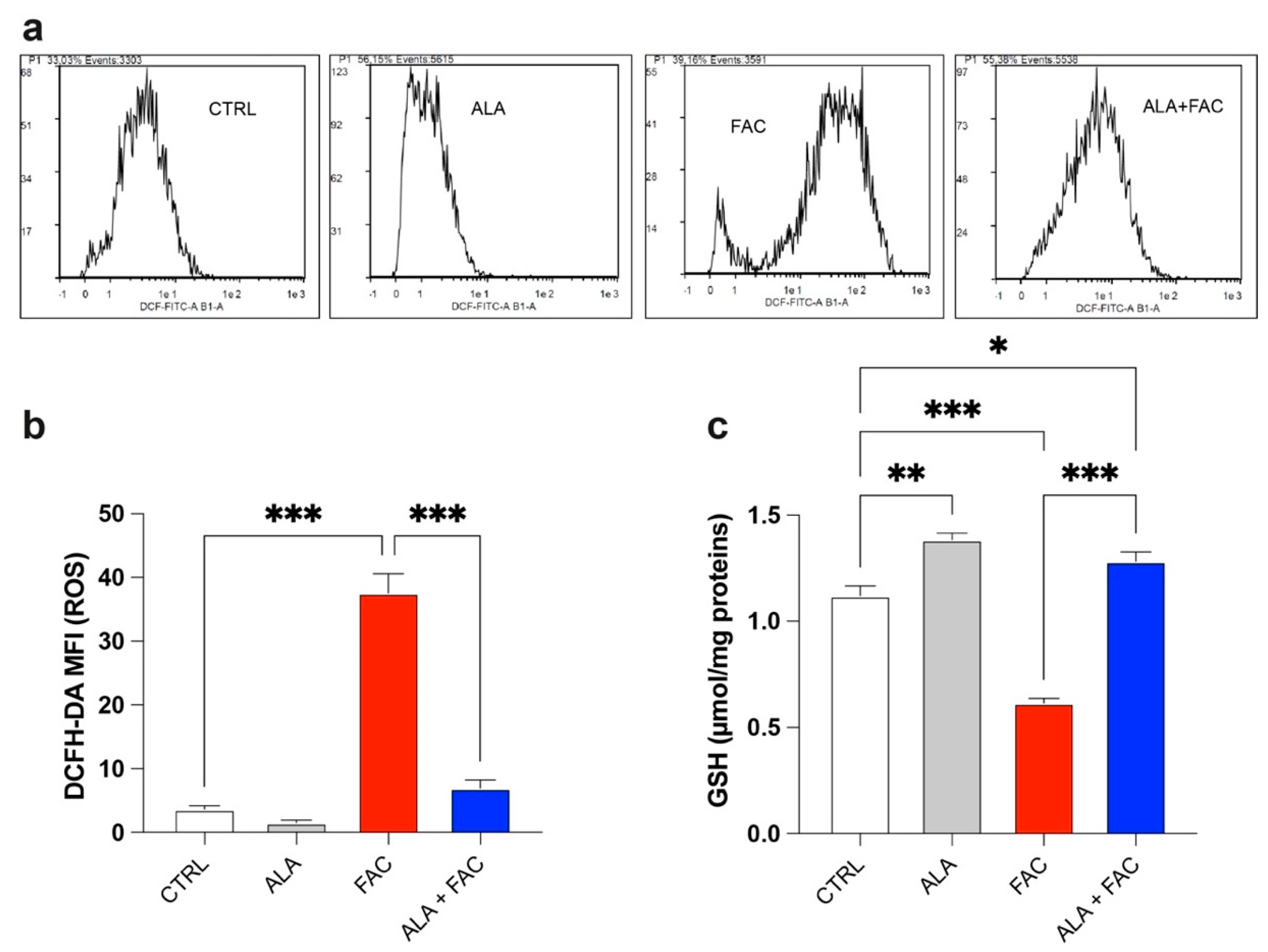
3.2. Iron Overload Affects Mitochondrial Functionality Inducing Oxidative Stress
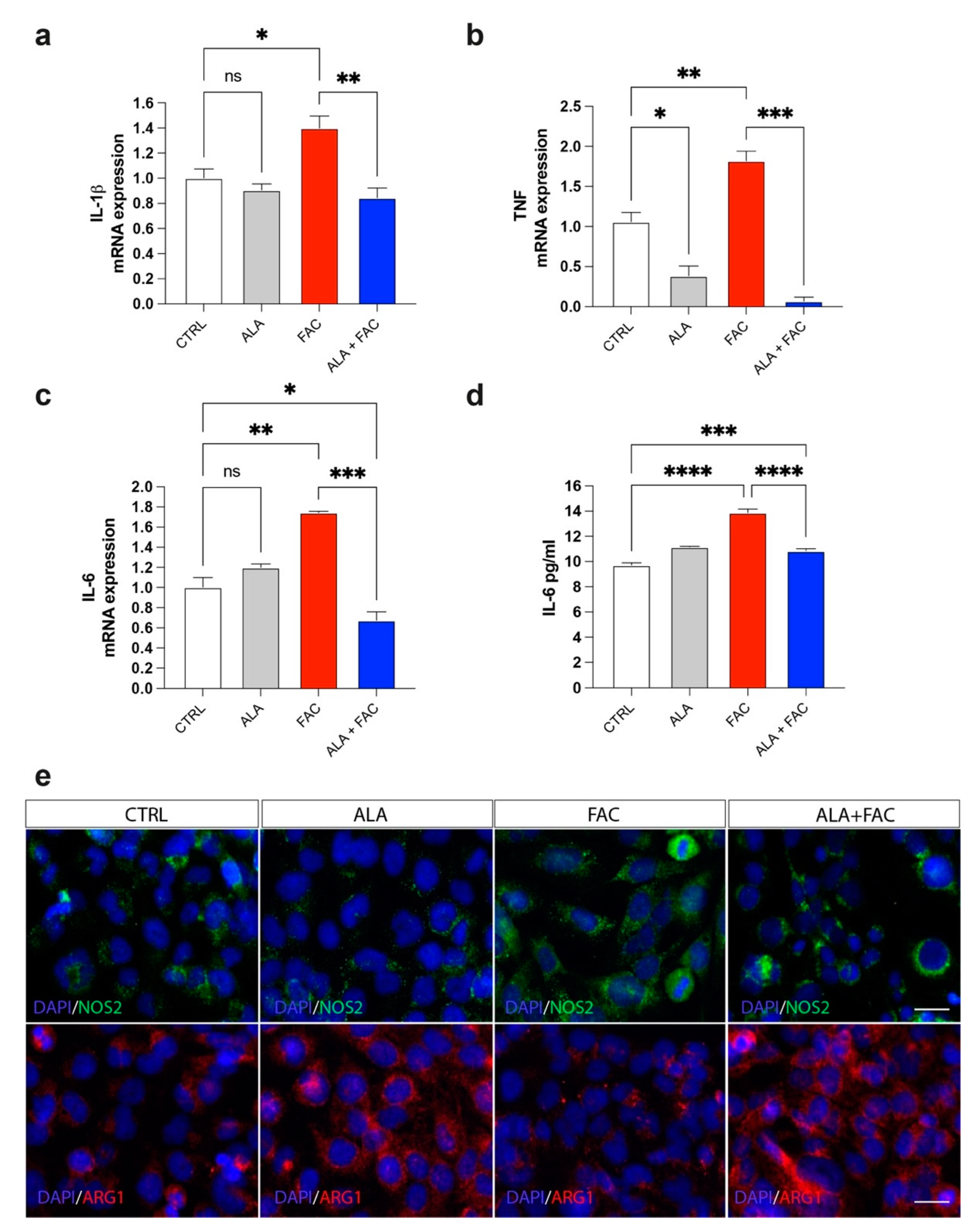
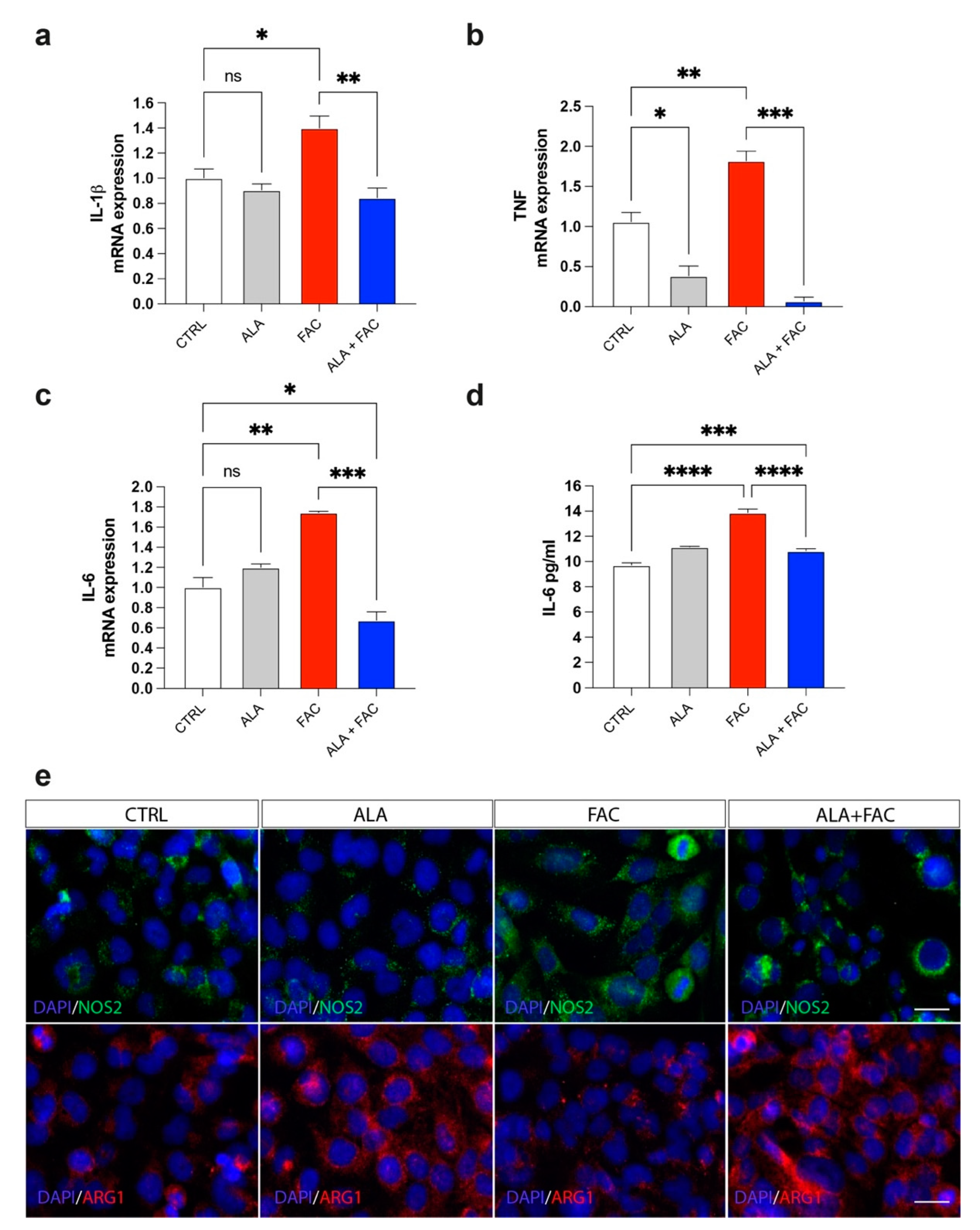
3.3. Iron Overload and Inflammation
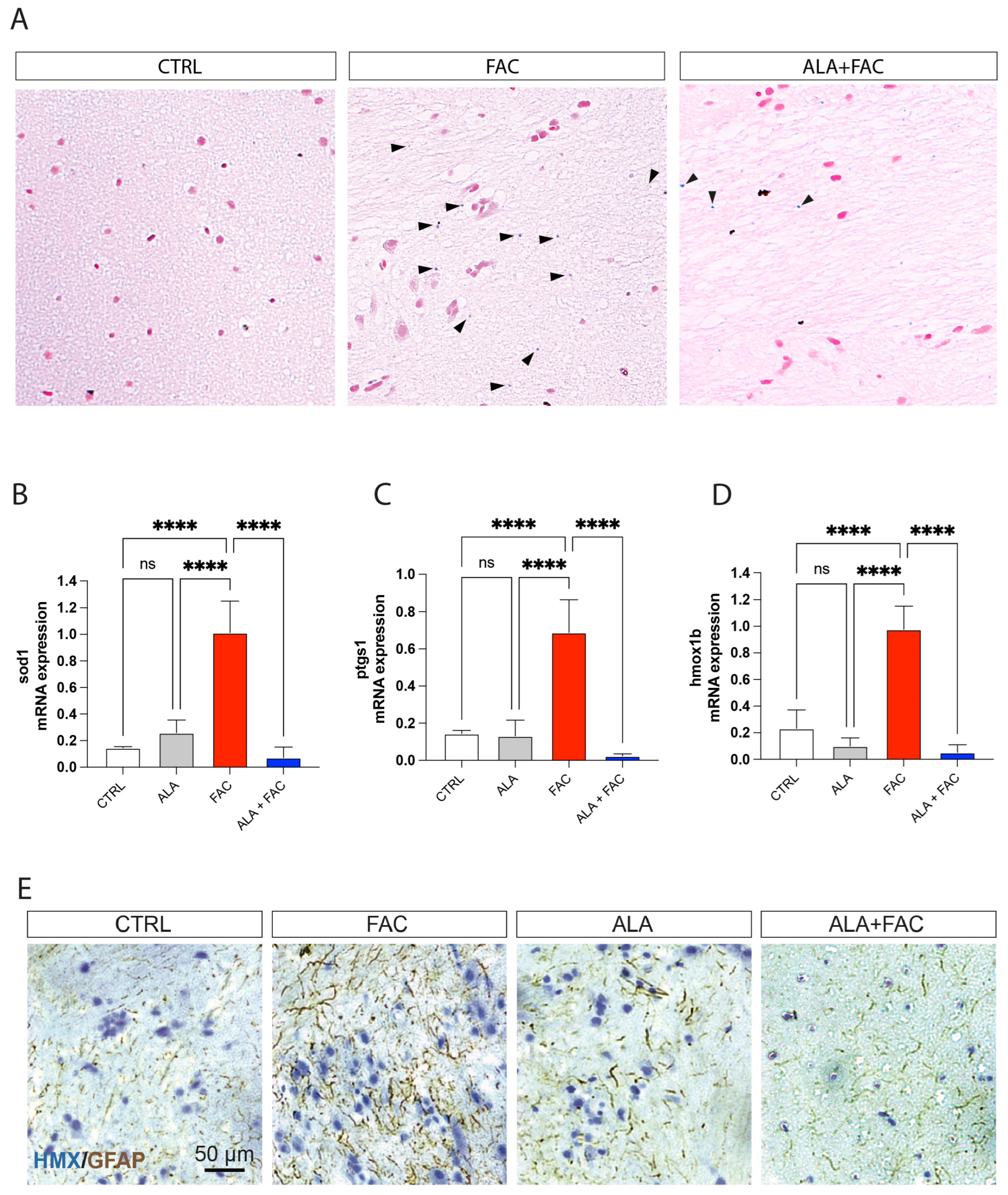
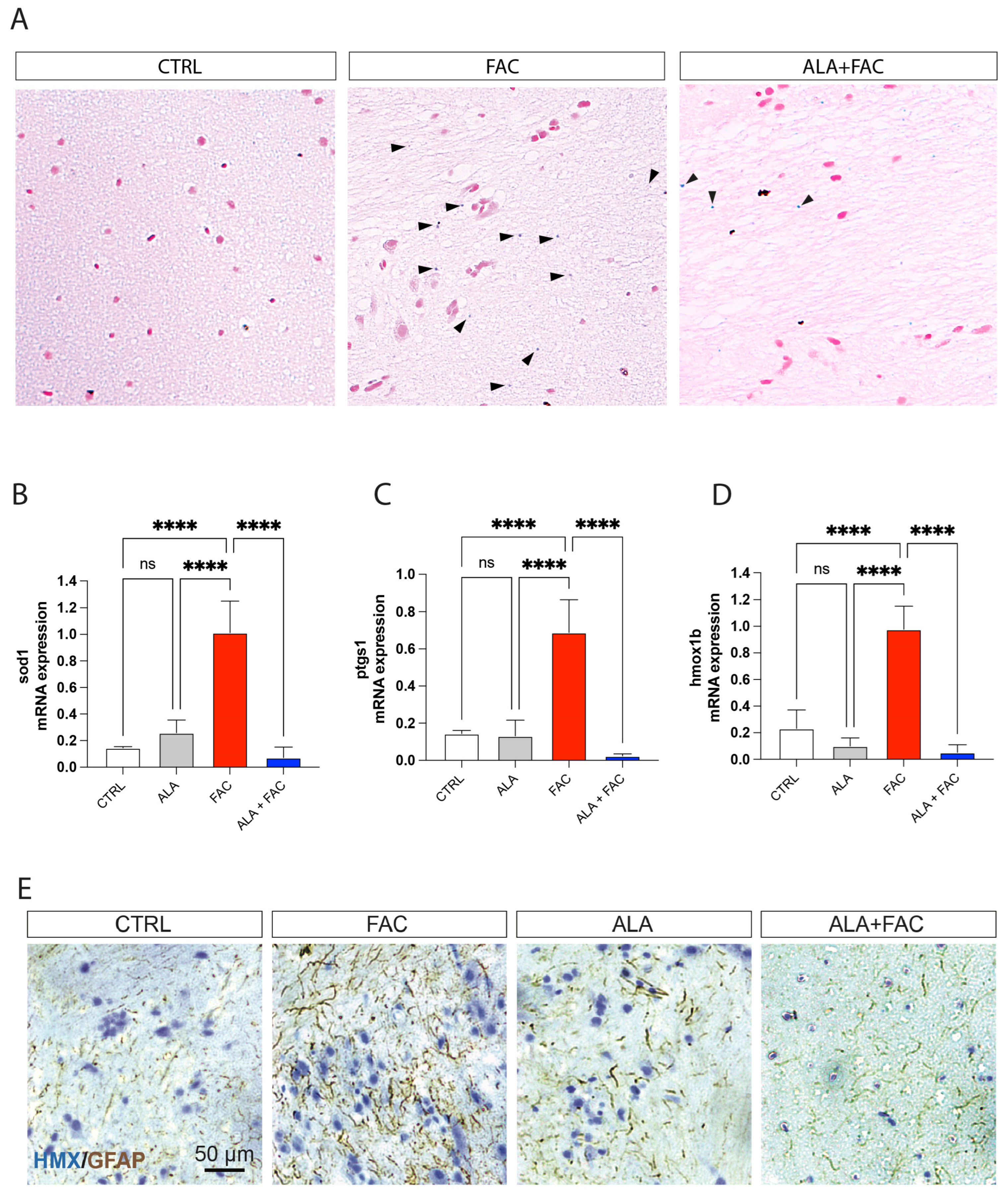
3.4. Iron Overload In Vivo Zebrafish Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Asch, C.J.; Luitse, M.J.; Rinkel, G.J.; van der Tweel, I.; Algra, A.; Klijn, C.J. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: A systematic review and meta-analysis. Lancet Neurol. 2010, 9, 167–176. [Google Scholar] [CrossRef]
- Bako, A.T.; Pan, A.; Potter, T.; Tannous, J.; Johnson, C.; Baig, E.; Meeks, J.; Woo, D.; Vahidy, F.S. Contemporary Trends in the Nationwide Incidence of Primary Intracerebral Hemorrhage. Stroke 2022, 53, e70–e74. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.I.; Freeman, W.D. Spontaneous intracerebral hemorrhage. Semin. Neurol. 2010, 30, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Al-Shahi Salman, R.; Warach, S.; Launer, L.J.; Van Buchem, M.A.; Breteler, M.M.; Microbleed Study, G. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef]
- Dye, J.A.; Rees, G.; Yang, I.; Vespa, P.M.; Martin, N.A.; Vinters, H.V. Neuropathologic analysis of hematomas evacuated from patients with spontaneous intracerebral hemorrhage. Neuropathology 2014, 34, 253–260. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappala, A.; Cammarata, F.P.; et al. The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression. Biomedicines 2022, 10, 806. [Google Scholar] [CrossRef]
- Wilkinson, D.A.; Pandey, A.S.; Thompson, B.G.; Keep, R.F.; Hua, Y.; Xi, G. Injury mechanisms in acute intracerebral hemorrhage. Neuropharmacology 2018, 134, 240–248. [Google Scholar] [CrossRef]
- Ziai, W.C.; Carhuapoma, J.R. Intracerebral Hemorrhage. Continuum 2018, 24, 1603–1622. [Google Scholar] [CrossRef]
- Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 2016, 1, 172–184. [Google Scholar] [CrossRef]
- Mendelow, A.D.; Gregson, B.A.; Fernandes, H.M.; Murray, G.D.; Teasdale, G.M.; Hope, D.T.; Karimi, A.; Shaw, M.D.; Barer, D.H.; Investigators, S. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): A randomised trial. Lancet 2005, 365, 387–397. [Google Scholar] [CrossRef]
- Morgenstern, L.B.; Demchuk, A.M.; Kim, D.H.; Frankowski, R.F.; Grotta, J.C. Rebleeding leads to poor outcome in ultra-early craniotomy for intracerebral hemorrhage. Neurology 2001, 56, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Imberti, R.; Pietrobono, L.; Klersy, C.; Gamba, G.; Iotti, G.A.; Cornara, G. Intraoperative intravenous administration of rFVIIa and hematoma volume after early surgery for spontaneous intracerebral hemorrhage: A randomized prospective phase II study. Minerva Anestesiol. 2012, 78, 168–175. [Google Scholar]
- Beez, T.; Steiger, H.J.; Etminan, N. Pharmacological targeting of secondary brain damage following ischemic or hemorrhagic stroke, traumatic brain injury, and bacterial meningitis—a systematic review and meta-analysis. BMC Neurol. 2017, 17, 209. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hua, Y.; Keep, R.F.; Nakamura, T.; Hoff, J.T.; Xi, G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke 2003, 34, 2964–2969. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. The possible role of iron in the etiopathology of Parkinson’s disease. Mov. Disord. 1993, 8, 1–12. [Google Scholar] [CrossRef]
- Zheng, W.; Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Excess iron: Considerations related to development and early growth. Am. J. Clin. Nutr. 2017, 106, 1600S–1605S. [Google Scholar] [CrossRef]
- Xiong, H.; Tuo, Q.Z.; Guo, Y.J.; Lei, P. Diagnostics and Treatments of Iron-Related CNS Diseases. Adv. Exp. Med. Biol. 2019, 1173, 179–194. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef]
- Zhao, F.; Hua, Y.; He, Y.; Keep, R.F.; Xi, G. Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke 2011, 42, 3587–3593. [Google Scholar] [CrossRef]
- Selim, M.; Yeatts, S.; Goldstein, J.N.; Gomes, J.; Greenberg, S.; Morgenstern, L.B.; Schlaug, G.; Torbey, M.; Waldman, B.; Xi, G.; et al. Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke 2011, 42, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Wang, J.; Qian, Z.M.; Yang, Q.W. Iron and intracerebral hemorrhage: From mechanism to translation. Transl. Stroke Res. 2014, 5, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.F.; Claus, C.P.; Vreman, H.J.; Wong, R.J.; Noble-Haeusslein, L.J. Heme regulation in traumatic brain injury: Relevance to the adult and developing brain. J. Cereb. Blood Flow Metab. 2005, 25, 1401–1417. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Xue, M.; Yong, V.W. Microglia and macrophage phenotypes in intracerebral haemorrhage injury: Therapeutic opportunities. Brain 2020, 143, 1297–1314. [Google Scholar] [CrossRef] [PubMed]
- DeGregorio-Rocasolano, N.; Marti-Sistac, O.; Gasull, T. Deciphering the Iron Side of Stroke: Neurodegeneration at the Crossroads Between Iron Dyshomeostasis, Excitotoxicity, and Ferroptosis. Front. Neurosci. 2019, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Bernstock, J.D.; Spitale, F.M.; Giallongo, C.; Giunta, M.A.S.; Li Volti, G.; Gulisano, M.; Leanza, G.; Tibullo, D.; Parenti, R.; et al. Clobetasol Modulates Adult Neural Stem Cell Growth via Canonical Hedgehog Pathway Activation. Int. J. Mol. Sci. 2019, 20, 1991. [Google Scholar] [CrossRef]
- Hui, Z.; Wang, S.; Li, J.; Wang, J.; Zhang, Z. Compound Tongluo Decoction inhibits endoplasmic reticulum stress-induced ferroptosis and promoted angiogenesis by activating the Sonic Hedgehog pathway in cerebral infarction. J. Ethnopharmacol. 2022, 283, 114634. [Google Scholar] [CrossRef]
- Vicario, N.; Calabrese, G.; Zappala, A.; Parenti, C.; Forte, S.; Graziano, A.C.E.; Vanella, L.; Pellitteri, R.; Cardile, V.; Parenti, R. Inhibition of Cx43 mediates protective effects on hypoxic/reoxygenated human neuroblastoma cells. J. Cell. Mol. Med. 2017, 21, 2563–2572. [Google Scholar] [CrossRef]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and indirect antioxidant properties of alpha-lipoic acid and therapeutic potential. Mol. Nutr. Food Res. 2013, 57, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Cadenas, E. Lipoic acid: Energy metabolism and redox regulation of transcription and cell signaling. J. Clin. Biochem. Nutr. 2011, 48, 26–32. [Google Scholar] [CrossRef]
- Camiolo, G.; Tibullo, D.; Giallongo, C.; Romano, A.; Parrinello, N.L.; Musumeci, G.; Di Rosa, M.; Vicario, N.; Brundo, M.V.; Amenta, F.; et al. alpha-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int. J. Mol. Sci. 2019, 20, 609. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, G.; Barbato, A.; Giallongo, C.; Vicario, N.; Romano, A.; Parrinello, N.L.; Parenti, R.; Sandoval, J.C.; Garcia-Moreno, D.; Lazzarino, G.; et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol. 2020, 36, 101611. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, M.; Sferrazzo, G.; Pittala, V.; Di Rosa, M.; Vanella, L.; Salerno, L.; Sorrenti, V.; Carota, G.; Parrinello, N.; Raffaele, M.; et al. Non-competitive heme oxygenase-1 activity inhibitor reduces non-small cell lung cancer glutathione content and regulates cell proliferation. Mol. Biol. Rep. 2020, 47, 1949–1964. [Google Scholar] [CrossRef]
- Giallongo, C.; Dulcamare, I.; Tibullo, D.; Del Fabro, V.; Vicario, N.; Parrinello, N.; Romano, A.; Scandura, G.; Lazzarino, G.; Conticello, C.; et al. CXCL12/CXCR4 axis supports mitochondrial trafficking in tumor myeloma microenvironment. Oncogenesis 2022, 11, 6. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Puglisi, F.; Barbato, A.; Vicario, N.; Cambria, D.; Parrinello, N.L.; Romano, A.; Conticello, C.; Forte, S.; et al. Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers 2020, 12, 1999. [Google Scholar] [CrossRef]
- Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Wissam Saab, M.; Tavazzi, B.; et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules 2020, 10, 696. [Google Scholar] [CrossRef]
- Torrisi, F.; Minafra, L.; Cammarata, F.P.; Savoca, G.; Calvaruso, M.; Vicario, N.; Maccari, L.; Peres, E.A.; Ozcelik, H.; Bernaudin, M.; et al. SRC Tyrosine Kinase Inhibitor and X-rays Combined Effect on Glioblastoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 3917. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; Lo Furno, D.; Zappala, A.; Valable, S.; Li Volti, G.; Tibullo, D.; Vicario, N.; Parenti, R. Connexin 43 and Sonic Hedgehog Pathway Interplay in Glioblastoma Cell Proliferation and Migration. Biology 2021, 10, 767. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Tibullo, D.; Vicario, N.; Giallongo, C.; La Spina, E.; Romano, A.; Lombardo, S.; Moretti, M.; Masia, F.; Coda, A.R.D.; et al. IGFBP-6/sonic hedgehog/TLR4 signalling axis drives bone marrow fibrotic transformation in primary myelofibrosis. Aging 2021, 13, 25055–25071. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Longo, A.; Vicario, N.; Romano, A.; Barbato, A.; Di Rosa, M.; Barbagallo, I.; Anfuso, C.D.; Lupo, G.; Gulino, R.; et al. Ixazomib Improves Bone Remodeling and Counteracts sonic Hedgehog signaling Inhibition Mediated by Myeloma Cells. Cancers 2020, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, M.; Kovacovicova, K.; Biagini, T.; Lo Re, O.; Frohlich, J.; Giallongo, S.; Nhan, J.D.; Giannone, A.G.; Cabibi, D.; Ivanov, M.; et al. Nociceptin/orphanin FQ opioid receptor (NOP) selective ligand MCOPPB links anxiolytic and senolytic effects. Geroscience 2022, 44, 463–483. [Google Scholar] [CrossRef]
- Vicario, N.; Spitale, F.M.; Tibullo, D.; Giallongo, C.; Amorini, A.M.; Scandura, G.; Spoto, G.; Saab, M.W.; D’Aprile, S.; Alberghina, C.; et al. Clobetasol promotes neuromuscular plasticity in mice after motoneuronal loss via sonic hedgehog signaling, immunomodulation and metabolic rebalancing. Cell Death Dis. 2021, 12, 625. [Google Scholar] [CrossRef]
- Gulino, R.; Vicario, N.; Giunta, M.A.S.; Spoto, G.; Calabrese, G.; Vecchio, M.; Gulisano, M.; Leanza, G.; Parenti, R. Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss. Int. J. Mol. Sci. 2019, 20, 1500. [Google Scholar] [CrossRef]
- Parenti, R.; Puzzo, L.; Vecchio, G.M.; Gravina, L.; Salvatorelli, L.; Musumeci, G.; Vasquez, E.; Magro, G. Immunolocalization of Wilms’ Tumor protein (WT1) in developing human peripheral sympathetic and gastroenteric nervous system. Acta Histochem. 2014, 116, 48–54. [Google Scholar] [CrossRef]
- Parenti, R.; Cicirata, F.; Panto, M.R.; Serapide, M.F. The projections of the lateral reticular nucleus to the deep cerebellar nuclei. An experimental analysis in the rat. Eur. J. Neurosci. 1996, 8, 2157–2167. [Google Scholar] [CrossRef]
- Longhitano, L.; Vicario, N.; Tibullo, D.; Giallongo, C.; Broggi, G.; Caltabiano, R.; Barbagallo, G.M.V.; Altieri, R.; Baghini, M.; Di Rosa, M.; et al. Lactate Induces the Expressions of MCT1 and HCAR1 to Promote Tumor Growth and Progression in Glioblastoma. Front. Oncol. 2022, 12, 871798. [Google Scholar] [CrossRef]
- Spitale, F.M.; Vicario, N.; Rosa, M.D.; Tibullo, D.; Vecchio, M.; Gulino, R.; Parenti, R. Increased expression of connexin 43 in a mouse model of spinal motoneuronal loss. Aging 2020, 12, 12598–12608. [Google Scholar] [CrossRef]
- Vicario, N.; Denaro, S.; Turnaturi, R.; Longhitano, L.; Spitale, F.M.; Spoto, S.; Marrazzo, A.; Zappala, A.; Tibullo, D.; Li Volti, G.; et al. Mu and Delta Opioid Receptor Targeting Reduces Connexin 43-Based Heterocellular Coupling during Neuropathic Pain. Int. J. Mol. Sci. 2022, 23, 5864. [Google Scholar] [CrossRef]
- Fidilio, A.; Grasso, M.; Turnaturi, R.; Caruso, G.; Spitale, F.M.; Vicario, N.; Parenti, R.; Spoto, S.; Musso, N.; Marrazzo, A.; et al. The Multimodal MOPr/DOPr Agonist LP2 Reduces Allodynia in Chronic Constriction Injured Rats by Rescue of TGF-beta1 Signalling. Front. Pharmacol. 2021, 12, 749365. [Google Scholar] [CrossRef]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Castrogiovanni, P.; Imbesi, R.; Tibullo, D.; Li Volti, G.; Barbagallo, I.; Vicario, N.; Musumeci, G.; Di Rosa, M. Middle-aged healthy women and Alzheimer’s disease patients present an overlapping of brain cell transcriptional profile. Neuroscience 2019, 406, 333–344. [Google Scholar] [CrossRef]
- Vicario, N.; Turnaturi, R.; Spitale, F.M.; Torrisi, F.; Zappala, A.; Gulino, R.; Pasquinucci, L.; Chiechio, S.; Parenti, C.; Parenti, R. Intercellular communication and ion channels in neuropathic pain chronicization. Inflamm. Res. 2020, 69, 841–850. [Google Scholar] [CrossRef]
- Selim, M. Deferoxamine mesylate: A new hope for intracerebral hemorrhage: From bench to clinical trials. Stroke 2009, 40, S90–S91. [Google Scholar] [CrossRef]
- Yeatts, S.D.; Palesch, Y.Y.; Moy, C.S.; Selim, M. High dose deferoxamine in intracerebral hemorrhage (HI-DEF) trial: Rationale, design, and methods. Neurocritical Care 2013, 19, 257–266. [Google Scholar] [CrossRef]
- Okauchi, M.; Hua, Y.; Keep, R.F.; Morgenstern, L.B.; Schallert, T.; Xi, G. Deferoxamine treatment for intracerebral hemorrhage in aged rats: Therapeutic time window and optimal duration. Stroke 2010, 41, 375–382. [Google Scholar] [CrossRef]
- Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Deferoxamine therapy for intracerebral hemorrhage. Acta Neurochir. Suppl. 2008, 105, 3–6. [Google Scholar] [CrossRef]
- Zeng, L.; Tan, L.; Li, H.; Zhang, Q.; Li, Y.; Guo, J. Deferoxamine therapy for intracerebral hemorrhage: A systematic review. PLoS ONE 2018, 13, e0193615. [Google Scholar] [CrossRef]
- Freedman, M.H.; Boyden, M.; Taylor, M.; Skarf, B. Neurotoxicity associated with deferoxamine therapy. Toxicology 1988, 49, 283–290. [Google Scholar] [CrossRef]
- Levine, J.E.; Cohen, A.; MacQueen, M.; Martin, M.; Giardina, P.J. Sensorimotor neurotoxicity associated with high-dose deferoxamine treatment. J. Pediatr. Hematol. Oncol. 1997, 19, 139–141. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E.A. Iron overload in hematological disorders. Presse Med. 2017, 46, e296–e305. [Google Scholar] [CrossRef]
- Rostoker, G.; Vaziri, N.D. Iatrogenic iron overload and its potential consequences in patients on hemodialysis. Presse Med 2017, 46, e312–e328. [Google Scholar] [CrossRef]
- Kawabata, H. The mechanisms of systemic iron homeostasis and etiology, diagnosis, and treatment of hereditary hemochromatosis. Int. J. Hematol. 2018, 107, 31–43. [Google Scholar] [CrossRef]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1779. [Google Scholar] [CrossRef]
- Zhang, X.; Surguladze, N.; Slagle-Webb, B.; Cozzi, A.; Connor, J.R. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia 2006, 54, 795–804. [Google Scholar] [CrossRef]
- Kenkhuis, B.; van Eekeren, M.; Parfitt, D.A.; Ariyurek, Y.; Banerjee, P.; Priller, J.; van der Weerd, L.; van Roon-Mom, W.M.C. Iron accumulation induces oxidative stress, while depressing inflammatory polarization in human iPSC-derived microglia. Stem Cell Rep. 2022, 17, 1351–1365. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Borquez, D.A.; Nunez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef]
- Gurer, H.; Ozgunes, H.; Oztezcan, S.; Ercal, N. Antioxidant role of alpha-lipoic acid in lead toxicity. Free Radic. Biol. Med. 1999, 27, 75–81. [Google Scholar] [CrossRef]
- Ou, P.; Tritschler, H.J.; Wolff, S.P. Thioctic (lipoic) acid: A therapeutic metal-chelating antioxidant? Biochem. Pharm. 1995, 50, 123–126. [Google Scholar] [CrossRef]
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 1997, 29, 315–331. [Google Scholar] [CrossRef]
- Stepien, K.M.; Heaton, R.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.; Hargreaves, I.P. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Li Volti, G.; Tibullo, D.; Vanella, L.; Giallongo, C.; Di Raimondo, F.; Forte, S.; Di Rosa, M.; Signorelli, S.S.; Barbagallo, I. The Heme Oxygenase System in Hematological Malignancies. Antioxid Redox Signal. 2017, 27, 363–377. [Google Scholar] [CrossRef]
- Barbagallo, I.; Giallongo, C.; Volti, G.L.; Distefano, A.; Camiolo, G.; Raffaele, M.; Salerno, L.; Pittala, V.; Sorrenti, V.; Avola, R.; et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol. Neurobiol. 2019, 56, 1451–1460. [Google Scholar] [CrossRef]
- Scandura, G.; Giallongo, C.; Puglisi, F.; Romano, A.; Parrinello, N.L.; Zuppelli, T.; Longhitano, L.; Giallongo, S.; Di Rosa, M.; Musumeci, G.; et al. TLR4 Signaling and Heme Oxygenase-1/Carbon Monoxide Pathway Crosstalk Induces Resiliency of Myeloma Plasma Cells to Bortezomib Treatment. Antioxidants 2022, 11, 767. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Hoeft, K.; Bloch, D.B.; Graw, J.A.; Malhotra, R.; Ichinose, F.; Bagchi, A. Iron Loading Exaggerates the Inflammatory Response to the Toll-like Receptor 4 Ligand Lipopolysaccharide by Altering Mitochondrial Homeostasis. Anesthesiology 2017, 127, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Lopez-Alonso, I.; Gonzalez-Lopez, A.; Amado-Rodriguez, L.; Batalla-Solis, E.; Astudillo, A.; Blazquez-Prieto, J.; Fernandez, A.F.; Galvan, J.A.; dos Santos, C.C.; et al. Defective autophagy impairs ATF3 activity and worsens lung injury during endotoxemia. J. Mol. Med. 2014, 92, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef]
- Dairaghi, D.J.; Shadel, G.S.; Clayton, D.A. Human mitochondrial transcription factor A and promoter spacing integrity are required for transcription initiation. Biochim. Biophys. Acta 1995, 1271, 127–134. [Google Scholar] [CrossRef]
- Fayez, A.M.; Zakaria, S.; Moustafa, D. Alpha lipoic acid exerts antioxidant effect via Nrf2/HO-1 pathway activation and suppresses hepatic stellate cells activation induced by methotrexate in rats. Biomed. Pharmacother. 2018, 105, 428–433. [Google Scholar] [CrossRef]
- Kim, S.M.; Ha, J.S.; Han, A.R.; Cho, S.W.; Yang, S.J. Effects of alpha-lipoic acid on LPS-induced neuroinflammation and NLRP3 inflammasome activation through the regulation of BV-2 microglial cells activation. BMB Rep. 2019, 52, 613–618. [Google Scholar] [CrossRef]
- Hamilton, J.L.; Hatef, A.; Imran ul-Haq, M.; Nair, N.; Unniappan, S.; Kizhakkedathu, J.N. Clinically approved iron chelators influence zebrafish mortality, hatching morphology and cardiac function. PLoS ONE 2014, 9, e109880. [Google Scholar] [CrossRef]
- Nasrallah, G.K.; Younes, N.N.; Baji, M.H.; Shraim, A.M.; Mustafa, I. Zebrafish larvae as a model to demonstrate secondary iron overload. Eur. J. Haematol. 2018, 100, 536–543. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Human) | Forward Primer (5′ → 3′) | Reverse Primer (5′ → 3′) |
|---|---|---|
| HO-1 | GTTGGGGTGGTTTTTGAGCC | TTAGACCAAGGCCACAGTGC |
| DMT1 | CGGAATAGGAAGTGCCATCCA | GGGAGCAAGGAAAAAGAACTACA |
| FPN1 | CTCCCAAACCGCTTCCATAAG | TCTTCTGCGGCTGCTATCG |
| IL-6 | CCACCGGGAACGAAAGAGAA | GAGAAGGCAACTGGACCGAA |
| IL-1β | AGCTCGCCAGTGAAATGATG | GTCGGAGATTCGTAGCTGGA |
| TNF | GCAACAAGACCACCACTTCG | GATCAAAGCTGTAGGCCCCA |
| GAPDH | TTCTTTTGCGTCGCCAGCC | CTTCCCGTTCTCAGCCTTGAC |
| Gene (Zebrafish) | Forward Primer (5′ → 3′) | Reverse Primer (5′ → 3′) |
|---|---|---|
| hmox1b | CTCTCCAGCCCTTCAGTTCG | AAGCGTAAACTCCCATGCCA |
| sod1 | GTGACAACACAAACGGCTGC | GGCATCAGCGGTCACATTAC |
| ptgs1 | ACTTTACCACTGGCACCCAC | ACGATGACCCTCTCAGCAAC |
| gapdh | CATCTTTGACGCTGGTGCTG | TGGGAGAATGGTCGCGTATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carota, G.; Distefano, A.; Spampinato, M.; Giallongo, C.; Broggi, G.; Longhitano, L.; Palumbo, G.A.; Parenti, R.; Caltabiano, R.; Giallongo, S.; et al. Neuroprotective Role of α-Lipoic Acid in Iron-Overload-Mediated Toxicity and Inflammation in In Vitro and In Vivo Models. Antioxidants 2022, 11, 1596. https://doi.org/10.3390/antiox11081596
Carota G, Distefano A, Spampinato M, Giallongo C, Broggi G, Longhitano L, Palumbo GA, Parenti R, Caltabiano R, Giallongo S, et al. Neuroprotective Role of α-Lipoic Acid in Iron-Overload-Mediated Toxicity and Inflammation in In Vitro and In Vivo Models. Antioxidants. 2022; 11(8):1596. https://doi.org/10.3390/antiox11081596
Chicago/Turabian StyleCarota, Giuseppe, Alfio Distefano, Mariarita Spampinato, Cesarina Giallongo, Giuseppe Broggi, Lucia Longhitano, Giuseppe A. Palumbo, Rosalba Parenti, Rosario Caltabiano, Sebastiano Giallongo, and et al. 2022. "Neuroprotective Role of α-Lipoic Acid in Iron-Overload-Mediated Toxicity and Inflammation in In Vitro and In Vivo Models" Antioxidants 11, no. 8: 1596. https://doi.org/10.3390/antiox11081596
APA StyleCarota, G., Distefano, A., Spampinato, M., Giallongo, C., Broggi, G., Longhitano, L., Palumbo, G. A., Parenti, R., Caltabiano, R., Giallongo, S., Di Rosa, M., Polosa, R., Bramanti, V., Vicario, N., Li Volti, G., & Tibullo, D. (2022). Neuroprotective Role of α-Lipoic Acid in Iron-Overload-Mediated Toxicity and Inflammation in In Vitro and In Vivo Models. Antioxidants, 11(8), 1596. https://doi.org/10.3390/antiox11081596