
Gq Signaling in Autophagy Control: Between Chemical and Mechanical Cues


Abstract
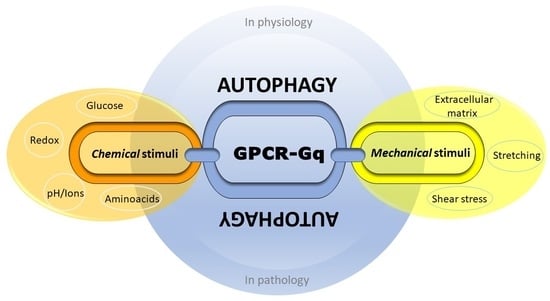
1. Introduction
2. GPCRs Regulation and Functions, beyond the Classical Modulation
3. New Avenues in Gαq/11 Signaling Complexes
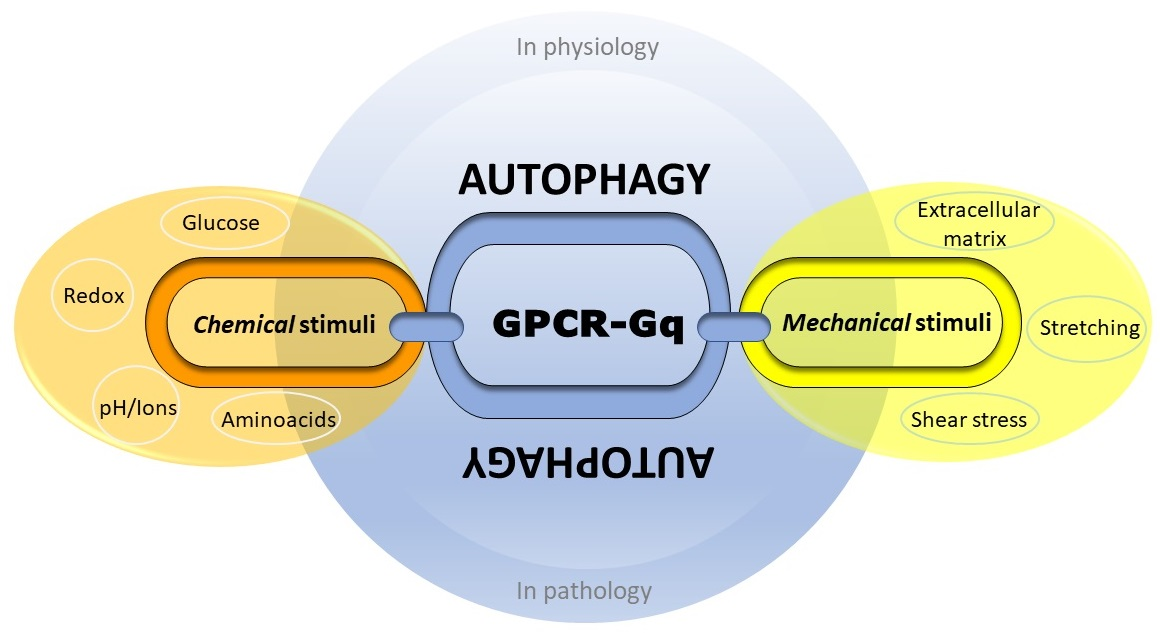
4. Autophagy between Nutrient, Mechanical and Oxidative Stress: An Emerging Role of Gαq
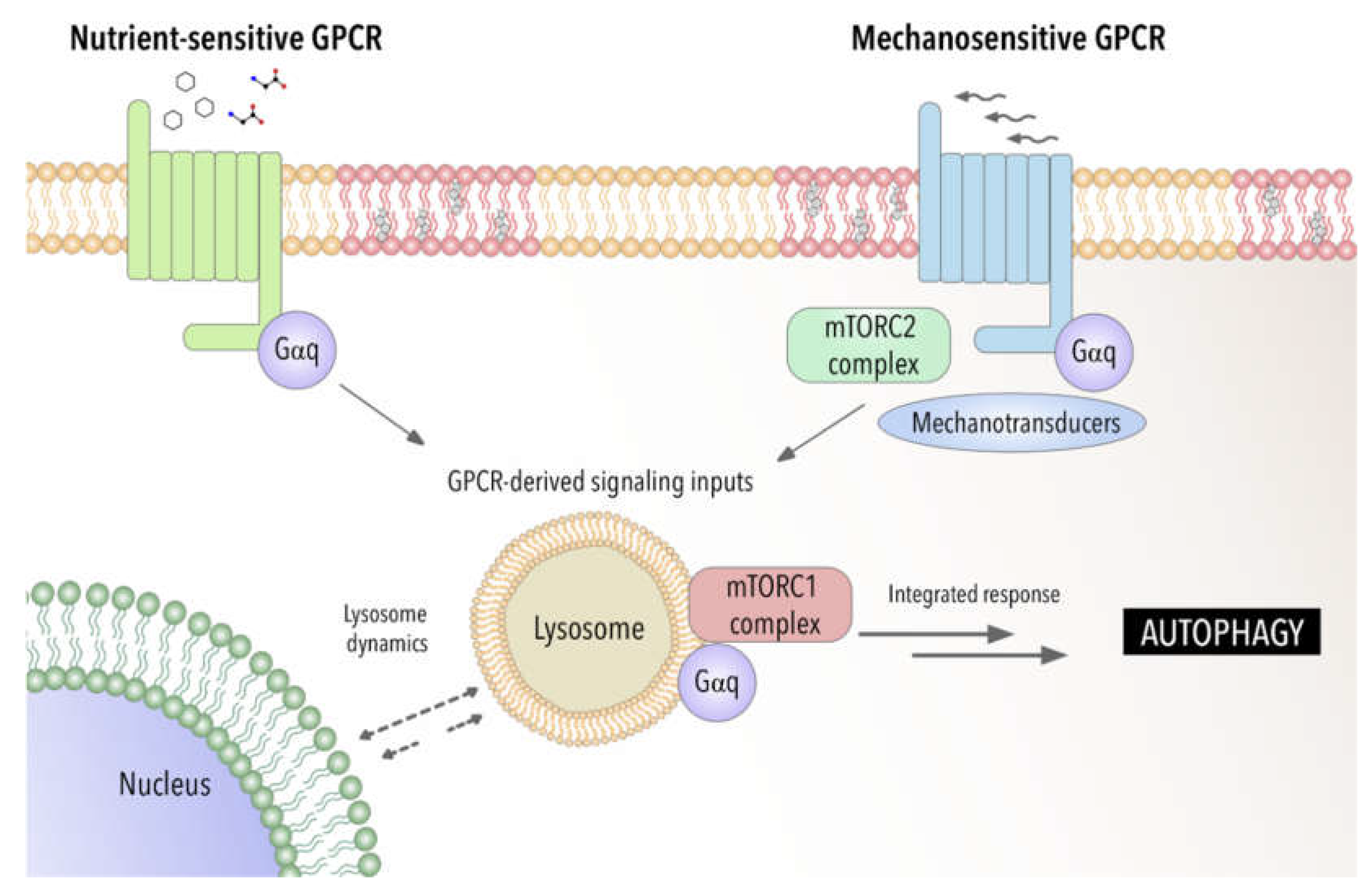
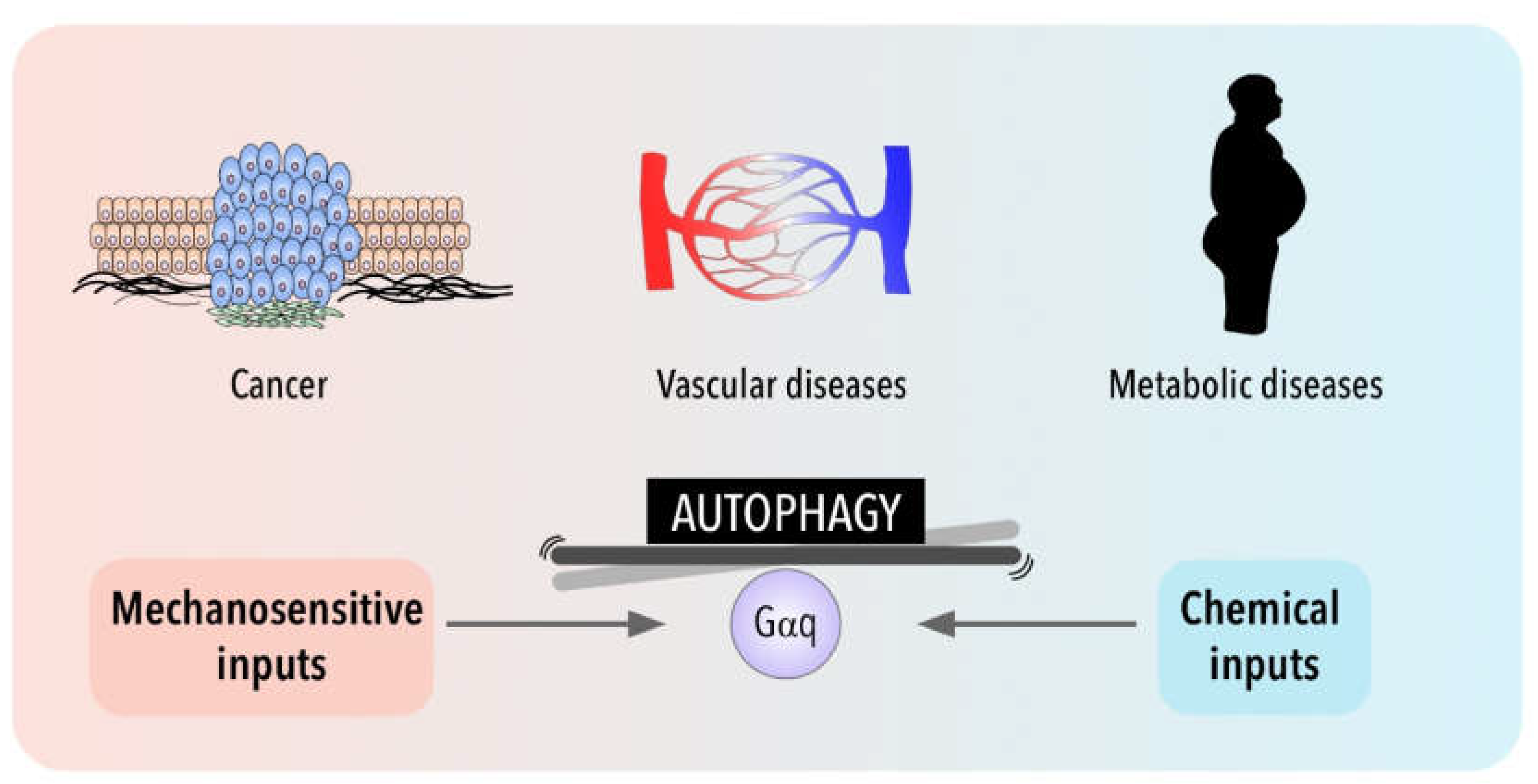
5. Autophagy in Disease for Good and for Bad: Gαq Involvement
6. Conclusions
Funding
Conflicts of Interest
References
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Lu, B.; Evans, R.; Gutkind, J.S. Signals and Receptors. Cold Spring Harb. Perspect. Biol. 2016, 8, a005900. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and Dynamics of GPCR Signaling Complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Gutkind, J.S. G-Protein-Coupled Receptors and Signaling Networks: Emerging Paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and Homeostasis of the Extracellular Matrix: Implications for Fibrotic Diseases and Cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef]
- Mahoney, J.P.; Sunahara, R.K. Mechanistic Insights into GPCR–G Protein Interactions. Curr. Opin. Struct. Biol. 2016, 41, 247–254. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-Transmembrane Receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Chen, C.G.; Iozzo, R.V. Extracellular Matrix Guidance of Autophagy: A Mechanism Regulating Cancer Growth. Open Biol. 2022, 12, 210304. [Google Scholar] [CrossRef]
- Hupfer, A.; Brichkina, A.; Koeniger, A.; Keber, C.; Denkert, C.; Pfefferle, P.; Helmprobst, F.; Pagenstecher, A.; Visekruna, A.; Lauth, M. Matrix Stiffness Drives Stromal Autophagy and Promotes Formation of a Protumorigenic Niche. Proc. Natl. Acad. Sci. USA 2021, 118, e2105367118. [Google Scholar] [CrossRef]
- Ge, H.; Tian, M.; Pei, Q.; Tan, F.; Pei, H. Extracellular Matrix Stiffness: New Areas Affecting Cell Metabolism. Front. Oncol. 2021, 11, 631991. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Seyedabadi, M.; Gharghabi, M.; Gurevich, E.V.; Gurevich, V.V. Structural Basis of GPCR Coupling to Distinct Signal Transducers: Implications for Biased Signaling. Trends Biochem. Sci. 2022, 47, 570–581. [Google Scholar] [CrossRef]
- Vizurraga, A.; Adhikari, R.; Yeung, J.; Yu, M.; Tall, G.G. Mechanisms of Adhesion G Protein–Coupled Receptor Activation. J. Biol. Chem. 2020, 295, 14065–14083. [Google Scholar] [CrossRef] [PubMed]
- Wauson, E.M.; Dbouk, H.A.; Ghosh, A.B.; Cobb, M.H. G Protein-Coupled Receptors and the Regulation of Autophagy. Trends Endocrinol. Metab. 2014, 25, 274–282. [Google Scholar] [CrossRef]
- Wauson, E.M.; Zaganjor, E.; Lee, A.-Y.; Guerra, M.L.; Ghosh, A.B.; Bookout, A.L.; Chambers, C.P.; Jivan, A.; McGlynn, K.; Hutchison, M.R.; et al. The G Protein-Coupled Taste Receptor T1R1/T1R3 Regulates MTORC1 and Autophagy. Mol. Cell 2012, 47, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.-A.; Yan, Y.; Kee, T.R.; Cazzaro, S.; McGill Percy, K.C.; Wang, X.; Liu, T.; Liggett, S.B.; Kang, D.E. β-Arrestin1 Promotes Tauopathy by Transducing GPCR Signaling, Disrupting Microtubules and Autophagy. Life Sci. Alliance 2022, 5, e202101183. [Google Scholar] [CrossRef]
- Cabezudo, S.; Sanz-Flores, M.; Caballero, A.; Tasset, I.; Rebollo, E.; Diaz, A.; Aragay, A.M.; Cuervo, A.M.; Mayor, F.; Ribas, C. Gαq Activation Modulates Autophagy by Promoting MTORC1 Signaling. Nat. Commun. 2021, 12, 4540. [Google Scholar] [CrossRef] [PubMed]
- Attwood, T.K.; Findlay, J.B.C. Fingerprinting G-Protein-Coupled Receptors. Protein Eng. Des. Sel. 1994, 7, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Culhane, K.J.; Gupte, T.M.; Madhugiri, I.; Gadgil, C.J.; Sivaramakrishnan, S. Kinetic Model of GPCR-G Protein Interactions Reveals Allokairic Modulation of Signaling. Nat. Commun. 2022, 13, 1202. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. β-Arrestin-Mediated Receptor Trafficking and Signal Transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Dwivedi, H.; Baidya, M.; Kumar, M.; Shukla, A.K. Novel Structural Insights into GPCR–β-Arrestin Interaction and Signaling. Trends Cell Biol. 2017, 27, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Tanowitz, M.; von Zastrow, M. A Novel Endocytic Recycling Signal That Distinguishes the Membrane Trafficking of Naturally Occurring Opioid Receptors. J. Biol. Chem. 2003, 278, 45978–45986. [Google Scholar] [CrossRef]
- Pavlos, N.J.; Friedman, P.A. GPCR Signaling and Trafficking: The Long and Short of It. Trends Endocrinol. Metab. 2017, 28, 213–226. [Google Scholar] [CrossRef]
- Sadoshima, J.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine Release of Angiotensin II Mediates Stretch-Induced Hypertrophy of Cardiac Myocytes in Vitro. Cell 1993, 75, 977–984. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Laher, I.; Matrougui, K.; Guerineau, N.C.; Henrion, D. Emerging Role of G Protein-Coupled Receptors in Microvascular Myogenic Tone. Cardiovasc. Res. 2012, 95, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Iring, A.; Strilic, B.; Albarrán Juárez, J.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 Control Blood Pressure by Mediating Endothelial Mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086. [Google Scholar] [CrossRef]
- Schnitzler, M.M.Y.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-Coupled Receptors as Mechanosensors Mediating Myogenic Vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef] [PubMed]
- Iliff, A.J.; Xu, X.Z.S. A Mechanosensitive GPCR That Detects the Bloody Force. Cell 2018, 173, 542–544. [Google Scholar] [CrossRef]
- Marullo, S.; Doly, S.; Saha, K.; Enslen, H.; Scott, M.G.H.; Coureuil, M. Mechanical GPCR Activation by Traction Forces Exerted on Receptor N-Glycans. ACS Pharmacol. Transl. Sci. 2020, 3, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Langenhan, T.; Piao, X.; Monk, K.R. Adhesion G Protein-Coupled Receptors in Nervous System Development and Disease. Nat. Rev. Neurosci. 2016, 17, 550–561. [Google Scholar] [CrossRef]
- Folts, C.J.; Giera, S.; Li, T.; Piao, X. Adhesion G Protein-Coupled Receptors as Drug Targets for Neurological Diseases. Trends Pharmacol. Sci. 2019, 40, 278–293. [Google Scholar] [CrossRef]
- Makino, A.; Prossnitz, E.R.; Bünemann, M.; Wang, J.M.; Yao, W.; Schmid-Schönbein, G.W. G Protein-Coupled Receptors Serve as Mechanosensors for Fluid Shear Stress in Neutrophils. Am. J. Physiol.-Cell Physiol. 2006, 290, C1633–C1639. [Google Scholar] [CrossRef]
- White, J.P.; Wrann, C.D.; Rao, R.R.; Nair, S.K.; Jedrychowski, M.P.; You, J.-S.; Martínez-Redondo, V.; Gygi, S.P.; Ruas, J.L.; Hornberger, T.A.; et al. G Protein-Coupled Receptor 56 Regulates Mechanical Overload-Induced Muscle Hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 15756–15761. [Google Scholar] [CrossRef]
- Ozkan, A.D.; Gettas, T.; Sogata, A.; Phaychanpheng, W.; Zhou, M.; Lacroix, J.J. Mechanical and Chemical Activation of GPR68 Probed with a Genetically Encoded Fluorescent Reporter. J. Cell Sci. 2021, 134, jcs255455. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Kornmann, B. Mechanical Forces on Cellular Organelles. J. Cell Sci. 2018, 131, jcs218479. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lai, L.; Wang, B.; Juang, S.; Chang, C.; Leu, J.; Shyu, K. Mechanical Stress Enhances Serotonin 2B Receptor Modulating Brain Natriuretic Peptide through Nuclear Factor-ΚB in Cardiomyocytes. Cardiovasc. Res. 2006, 72, 303–312. [Google Scholar] [CrossRef]
- Myagmar, B.-E.; Ismaili, T.; Swigart, P.M.; Raghunathan, A.; Baker, A.J.; Sahdeo, S.; Blevitt, J.M.; Milla, M.E.; Simpson, P.C. Coupling to Gq Signaling Is Required for Cardioprotection by an Alpha-1A-Adrenergic Receptor Agonist. Circ. Res. 2019, 125, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Sainz, E.; Clark, W.A.; Jensen, R.T.; Battey, J.F.; Northup, J.K. The Bombesin Receptor Subtypes Have Distinct G Protein Specificities. J. Biol. Chem. 1999, 274, 11573–11581. [Google Scholar] [CrossRef]
- Wang, N.; He, X.; Zhao, J.; Jiang, H.; Cheng, X.; Xia, Y.; Eric Xu, H.; He, Y. Structural Basis of Leukotriene B4 Receptor 1 Activation. Nat. Commun. 2022, 13, 1156. [Google Scholar] [CrossRef]
- Mobbs, J.I.; Belousoff, M.J.; Harikumar, K.G.; Piper, S.J.; Xu, X.; Furness, S.G.B.; Venugopal, H.; Christopoulos, A.; Danev, R.; Wootten, D.; et al. Structures of the Human Cholecystokinin 1 (CCK1) Receptor Bound to Gs and Gq Mimetic Proteins Provide Insight into Mechanisms of G Protein Selectivity. PLoS Biol. 2021, 19, e3001295. [Google Scholar] [CrossRef] [PubMed]
- Capra, V. Molecular and Functional Aspects of Human Cysteinyl Leukotriene Receptors. Pharmacol. Res. 2004, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Storch, U.; Blodow, S.; Gudermann, T.; Mederos y Schnitzler, M. Cysteinyl Leukotriene 1 Receptors as Novel Mechanosensors Mediating Myogenic Tone Together with Angiotensin II Type 1 Receptors—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 121–126. [Google Scholar] [CrossRef]
- Legler, D.F.; Bruckner, M.; Uetz-von Allmen, E.; Krause, P. Prostaglandin E2 at New Glance: Novel Insights in Functional Diversity Offer Therapeutic Chances. Int. J. Biochem. Cell Biol. 2010, 42, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.P.; Cullen, D.M.; Gong, G.; Recker, R.R. Bone Biomechanical Properties in Prostaglandin EP1 and EP2 Knockout Mice. Bone 2001, 29, 121–125. [Google Scholar] [CrossRef]
- Eguchi, S.; Hirata, Y.; Imai, T.; Marumo, F. Endothelin Receptor Subtypes Are Coupled to Adenylate Cyclase via Different Guanyl Nucleotide-Binding Proteins in Vasculature. Endocrinology 1993, 132, 524–529. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.L.; Gyoneva, S.; Vellano, C.P.; Smrcka, A.V.; Traynelis, S.F.; Hepler, J.R. Protease-Activated Receptor 1 (PAR1) Coupling to Gq/11 but Not to Gi/o or G12/13 Is Mediated by Discrete Amino Acids within the Receptor Second Intracellular Loop. Cell. Signal. 2012, 24, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, J.-H.; Nam, D.-H.; Kim, G.-Y.; Lim, J.H.; Kim, J.-R.; Woo, C.-H. PAR-1 Is a Novel Mechano-Sensor Transducing Laminar Flow-Mediated Endothelial Signaling. Sci. Rep. 2018, 8, 15172. [Google Scholar] [CrossRef]
- Liu, H.-X.; Hökfelt, T. The Participation of Galanin in Pain Processing at the Spinal Level. Trends Pharmacol. Sci. 2002, 23, 468–474. [Google Scholar] [CrossRef]
- Abizaid, A.; Hougland, J.L. Ghrelin Signaling: GOAT and GHS-R1a Take a LEAP in Complexity. Trends Endocrinol. Metab. 2020, 31, 107–117. [Google Scholar] [CrossRef]
- Liu, F.; Usui, I.; Evans, L.G.; Austin, D.A.; Mellon, P.L.; Olefsky, J.M.; Webster, N.J.G. Involvement of Both Gq/11 and Gs Proteins in Gonadotropin-Releasing Hormone Receptor-Mediated Signaling in LβT2 Cells. J. Biol. Chem. 2002, 277, 32099–32108. [Google Scholar] [CrossRef] [PubMed]
- Erdogmus, S.; Storch, U.; Danner, L.; Becker, J.; Winter, M.; Ziegler, N.; Wirth, A.; Offermanns, S.; Hoffmann, C.; Gudermann, T.; et al. Helix 8 Is the Essential Structural Motif of Mechanosensitive GPCRs. Nat. Commun. 2019, 10, 5784. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, T.; Wu, Y.; Xu, H.; Xie, C.; Dong, Y.; Zhong, L.; Wang, Z.; Zhao, H.; Zhou, Y.; et al. GPR39 Overexpression in OSCC Promotes YAP-Sustained Malignant Progression. J. Dent. Res. 2020, 99, 949–958. [Google Scholar] [CrossRef]
- Xu, J.; Mathur, J.; Vessières, E.; Hammack, S.; Nonomura, K.; Favre, J.; Grimaud, L.; Petrus, M.; Francisco, A.; Li, J.; et al. GPR68 Senses Flow and Is Essential for Vascular Physiology. Cell 2018, 173, 762–775.e16. [Google Scholar] [CrossRef]
- Adjobo-Hermans, M.J.; Goedhart, J.; van Weeren, L.; Nijmeijer, S.; Manders, E.M.; Offermanns, S.; Gadella, T.W. Real-Time Visualization of Heterotrimeric G Protein Gq Activation in Living Cells. BMC Biol. 2011, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Qu, Q.; Robertson, M.J.; Skiniotis, G.; Kobilka, B.K. Structures of the M1 and M2 Muscarinic Acetylcholine Receptor/G-Protein Complexes. Science 2019, 364, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Kassmann, M.; Nickel, S.; Zhang, C.; Alenina, N.; Anistan, Y.M.; Schleifenbaum, J.; Bader, M.; Welsh, D.G.; Huang, Y.; et al. Myogenic Vasoconstriction Requires Canonical Gq/11 Signaling of the Angiotensin II Type 1 Receptor. J. Am. Heart Assoc. 2022, 11, e022070. [Google Scholar] [CrossRef] [PubMed]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological and Pathogenic Actions of Angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Han, G.W.; Batyuk, A.; Ishchenko, A.; White, K.L.; Patel, N.; Sadybekov, A.; Zamlynny, B.; Rudd, M.T.; Hollenstein, K.; et al. Structural Basis for Selectivity and Diversity in Angiotensin II Receptors. Nature 2017, 544, 327–332. [Google Scholar] [CrossRef]
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical Stress Activates Angiotensin II Type 1 Receptor without the Involvement of Angiotensin II. Nat. Cell Biol. 2004, 6, 499–506. [Google Scholar] [CrossRef]
- Hawes, B.E.; Kil, E.; Green, B.; O’Neill, K.; Fried, S.; Graziano, M.P. The Melanin-Concentrating Hormone Receptor Couples to Multiple G Proteins to Activate Diverse Intracellular Signaling Pathways. Endocrinology 2000, 141, 4524–4532. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Zhang, Y.-L.; Frangos, J.A. G Protein-Coupled Receptors Sense Fluid Shear Stress in Endothelial Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef]
- Sun, Y.; Leng, P.; Guo, P.; Gao, H.; Liu, Y.; Li, C.; Li, Z.; Zhang, H. G Protein Coupled Estrogen Receptor Attenuates Mechanical Stress-Mediated Apoptosis of Chondrocyte in Osteoarthritis via Suppression of Piezo1. Mol. Med. 2021, 27, 96. [Google Scholar] [CrossRef]
- Gao, H.; Tian, K.; Feng, X.; Yan, M.; Gao, C.; Jiang, Y.; Zhu, C.; Zhu, H.; Liu, X.; Peng, Y. Free Fatty Acid Receptor 2 Promotes Cardiomyocyte Hypertrophy by Activating STAT3 and GATA4. Food Sci. Hum. Wellness 2022, 11, 405–417. [Google Scholar] [CrossRef]
- Mahon, M.J. The Parathyroid Hormone Receptorsome and the Potential for Therapeutic Intervention. Curr. Drug Targets 2012, 13, 116–128. [Google Scholar] [CrossRef]
- Ascolani, G.; Skerry, T.M.; Lacroix, D.; Dall’Ara, E.; Shuaib, A. Analysis of Mechanotransduction Dynamics during Combined Mechanical Stimulation and Modulation of the Extracellular-Regulated Kinase Cascade Uncovers Hidden Information within the Signalling Noise. Interface Focus 2021, 11, 20190136. [Google Scholar] [CrossRef]
- Terrillon, S.; Barberis, C.; Bouvier, M. Heterodimerization of V1a and V2 Vasopressin Receptors Determines the Interaction with β-Arrestin and Their Trafficking Patterns. Proc. Natl. Acad. Sci. USA 2004, 101, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Sun, Y.-J.; Ma, M.-L.; Wang, Y.; Lin, H.; Li, R.-R.; Liang, Z.-L.; Gao, Y.; Yang, Z.; He, D.-F.; et al. Gq Activity- and β-Arrestin-1 Scaffolding-Mediated ADGRG2/CFTR Coupling Are Required for Male Fertility. eLife 2018, 7, e33432. [Google Scholar] [CrossRef] [PubMed]
- Erb, L.; Weisman, G.A. Coupling of P2Y Receptors to G Proteins and Other Signaling Pathways. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 789–803. [Google Scholar] [CrossRef]
- Sathanoori, R.; Bryl-Gorecka, P.; Müller, C.E.; Erb, L.; Weisman, G.A.; Olde, B.; Erlinge, D. P2Y2 Receptor Modulates Shear Stress-Induced Cell Alignment and Actin Stress Fibers in Human Umbilical Vein Endothelial Cells. Cell. Mol. Life Sci. 2017, 74, 731–746. [Google Scholar] [CrossRef]
- Jelinek, V.; Mösslein, N.; Bünemann, M. Structures in G Proteins Important for Subtype Selective Receptor Binding and Subsequent Activation. Commun. Biol. 2021, 4, 635. [Google Scholar] [CrossRef]
- Campbell, A.P.; Smrcka, A.V. Targeting G Protein-Coupled Receptor Signalling by Blocking G Proteins. Nat. Rev. Drug Discov. 2018, 17, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, G.; Cabezudo, S.; García-Hoz, C.; Benincá, C.; Aragay, A.M.; Mayor, F.; Ribas, C. Gαq Signalling: The New and the Old. Cell. Signal. 2014, 26, 833–848. [Google Scholar] [CrossRef]
- Kang, Y.; Kuybeda, O.; de Waal, P.W.; Mukherjee, S.; van Eps, N.; Dutka, P.; Zhou, X.E.; Bartesaghi, A.; Erramilli, S.; Morizumi, T.; et al. Cryo-EM Structure of Human Rhodopsin Bound to an Inhibitory G Protein. Nature 2018, 558, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the Μ-Opioid Receptor–Gi Protein Complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef]
- Xia, R.; Wang, N.; Xu, Z.; Lu, Y.; Song, J.; Zhang, A.; Guo, C.; He, Y. Cryo-EM Structure of the Human Histamine H1 Receptor/Gq Complex. Nat. Commun. 2021, 12, 2086. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, D.; Fu, Y.; Chen, A.; Yang, X.; Zhang, H. Cryo-EM Structures of Human Bradykinin Receptor-Gq Proteins Complexes. Nat. Commun. 2022, 13, 714. [Google Scholar] [CrossRef]
- Okashah, N.; Wan, Q.; Ghosh, S.; Sandhu, M.; Inoue, A.; Vaidehi, N.; Lambert, N.A. Variable G Protein Determinants of GPCR Coupling Selectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 12054–12059. [Google Scholar] [CrossRef]
- Hubbard, K.B.; Hepler, J.R. Cell Signalling Diversity of the Gqα Family of Heterotrimeric G Proteins. Cell. Signal. 2006, 18, 135–150. [Google Scholar] [CrossRef]
- Davignon, I.; Barnard, M.; Gavrilova, O.; Sweet, K.; Wilkie, T.M. Gene Structure of MurineGna11andGna15:Tandemly Duplicated Gq Class G Protein α Subunit Genes. Genomics 1996, 31, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, T.M.; Scherle, P.A.; Strathmann, M.P.; Slepak, V.Z.; Simon, M.I. Characterization of G-Protein Alpha Subunits in the Gq Class: Expression in Murine Tissues and in Stromal and Hematopoietic Cell Lines. Proc. Natl. Acad. Sci. USA 1991, 88, 10049–10053. [Google Scholar] [CrossRef]
- Sánchez-Fernández, G.; Cabezudo, S.; Caballero, Á.; García-Hoz, C.; Tall, G.G.; Klett, J.; Michnick, S.W.; Mayor, F.; Ribas, C. Protein Kinase C ζ Interacts with a Novel Binding Region of Gαq to Act as a Functional Effector. J. Biol. Chem. 2016, 291, 9513–9525. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.I.; Strathmann, M.P.; Gautam, N. Diversity of G Proteins in Signal Transduction. Science 1991, 252, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Wedegaertner, P.B. G Protein Trafficking. In GPCR Signalling Complexes—Synthesis, Assembly, Trafficking and Specificity; Subcellular Biochemistry; Dupré, D., Hébert, T., Jockers, R., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 63, pp. 193–223. [Google Scholar] [CrossRef]
- Siderovski, D.P.; Willard, F.S. The GAPs, GEFs, and GDIs of Heterotrimeric G-Protein Alpha Subunits. Int. J. Biol. Sci. 2005, 1, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, S. Cellular Regulation of RGS Proteins: Modulators and Integrators of G Protein Signaling. Pharmacol. Rev. 2002, 54, 527–559. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zeng, W.; Popov, S.; Berman, D.M.; Davignon, I.; Yu, K.; Yowe, D.; Offermanns, S.; Muallem, S.; Wilkie, T.M. RGS Proteins Determine Signaling Specificity of Gq-Coupled Receptors. J. Biol. Chem. 1999, 274, 3549–3556. [Google Scholar] [CrossRef] [PubMed]
- Masuho, I.; Balaji, S.; Muntean, B.S.; Skamangas, N.K.; Chavali, S.; Tesmer, J.J.G.; Babu, M.M.; Martemyanov, K.A. A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell 2020, 183, 503–521.e19. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Okamoto, M.; Sugawara, Y.; Mizuno, N.; Yamauchi, J.; Itoh, H. Ric-8A Potentiates Gq-Mediated Signal Transduction by Acting Downstream of G Protein-Coupled Receptor in Intact Cells. Genes Cells 2006, 11, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Hampoelz, B.; Hoeller, O.; Bowman, S.K.; Dunican, D.; Knoblich, J.A. Drosophila Ric-8 Is Essential for Plasma-Membrane Localization of Heterotrimeric G Proteins. Nat. Cell Biol. 2005, 7, 1099–1105. [Google Scholar] [CrossRef]
- Tall, G.G. Ric-8 Regulation of Heterotrimeric G Proteins. J. Recept. Signal Transduct. 2013, 33, 139–143. [Google Scholar] [CrossRef]
- Gabay, M.; Pinter, M.E.; Wright, F.A.; Chan, P.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Tall, G.G. Ric-8 Proteins Are Molecular Chaperones That Direct Nascent G Protein α Subunit Membrane Association. Sci. Signal. 2011, 4, ra79. [Google Scholar] [CrossRef] [PubMed]
- Rojas, R.J.; Yohe, M.E.; Gershburg, S.; Kawano, T.; Kozasa, T.; Sondek, J. Gαq Directly Activates P63RhoGEF and Trio via a Conserved Extension of the Dbl Homology-Associated Pleckstrin Homology Domain. J. Biol. Chem. 2007, 282, 29201–29210. [Google Scholar] [CrossRef]
- Fukuhara, S.; Murga, C.; Zohar, M.; Igishi, T.; Gutkind, J.S. A Novel PDZ Domain Containing Guanine Nucleotide Exchange Factor Links Heterotrimeric G Proteins to Rho. J. Biol. Chem. 1999, 274, 5868–5879. [Google Scholar] [CrossRef]
- Lutz, S.; Freichel-Blomquist, A.; Yang, Y.; Rümenapp, U.; Jakobs, K.H.; Schmidt, M.; Wieland, T. The Guanine Nucleotide Exchange Factor P63RhoGEF, a Specific Link between Gq/11-Coupled Receptor Signaling and RhoA. J. Biol. Chem. 2005, 280, 11134–11139. [Google Scholar] [CrossRef] [PubMed]
- Vaqué, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A Genome-Wide RNAi Screen Reveals a Trio-Regulated Rho GTPase Circuitry Transducing Mitogenic Signals Initiated by G Protein-Coupled Receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef]
- Popova, J.S.; Rasenick, M.M. Gβγ Mediates the Interplay between Tubulin Dimers and Microtubules in the Modulation of Gq Signaling. J. Biol. Chem. 2003, 278, 34299–34308. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ciruela, F.; McIlhinney, R.A.J. Metabotropic Glutamate Receptor Type 1α and Tubulin Assemble into Dynamic Interacting Complexes. J. Neurochem. 2008, 76, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R. Plasma Membrane Microdomains. Curr. Opin. Cell Biol. 2002, 14, 483–487. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Kurzchalia, T.V.; Partan, R.G. Membrane Microdomains and Caveolae. Curr. Opin. Cell Biol. 1999, 11, 424–431. [Google Scholar] [CrossRef]
- Parton, R.G.; del Pozo, M.A. Caveolae as Plasma Membrane Sensors, Protectors and Organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Le Roux, A.-L.; Quiroga, X.; Walani, N.; Arroyo, M.; Roca-Cusachs, P. The Plasma Membrane as a Mechanochemical Transducer. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180221. [Google Scholar] [CrossRef]
- Ostrom, R.S.; Insel, P.A. The Evolving Role of Lipid Rafts and Caveolae in G Protein-Coupled Receptor Signaling: Implications for Molecular Pharmacology. Br. J. Pharmacol. 2004, 143, 235–245. [Google Scholar] [CrossRef]
- Mizuno, N.; Itoh, H. Functions and Regulatory Mechanisms of Gq-Signaling Pathways. Neurosignals 2009, 17, 42–54. [Google Scholar] [CrossRef]
- Oh, P.; Schnitzer, J.E. Segregation of Heterotrimeric G Proteins in Cell Surface Microdomains. Mol. Biol. Cell 2001, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vicente, R.; Pavón, D.M.; Martín-Padura, I.; Català-Montoro, M.; Díez-Sánchez, A.; Quílez-Álvarez, A.; López, J.A.; Sánchez-Álvarez, M.; Vázquez, J.; Strippoli, R.; et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep. 2018, 25, 1622–1635.e6. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef]
- Calizo, R.C.; Scarlata, S. A Role for G-Proteins in Directing G-Protein-Coupled Receptor–Caveolae Localization. Biochemistry 2012, 51, 9513–9523. [Google Scholar] [CrossRef]
- Qifti, A.; Garwain, O.; Scarlata, S. Mechanical Stretch Redefines Membrane Gαq–Calcium Signaling Complexes. J. Membr. Biol. 2019, 252, 307–315. [Google Scholar] [CrossRef]
- Rizzo, V.; McIntosh, D.P.; Oh, P.; Schnitzer, J.E. In Situ Flow Activates Endothelial Nitric Oxide Synthase in Luminal Caveolae of Endothelium with Rapid Caveolin Dissociation and Calmodulin Association. J. Biol. Chem. 1998, 273, 34724–34729. [Google Scholar] [CrossRef]
- Sugawara, Y.; Nishii, H.; Takahashi, T.; Yamauchi, J.; Mizuno, N.; Tago, K.; Itoh, H. The Lipid Raft Proteins Flotillins/Reggies Interact with Gαq and Are Involved in Gq-Mediated P38 Mitogen-Activated Protein Kinase Activation through Tyrosine Kinase. Cell. Signal. 2007, 19, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular Definitions of Autophagy and Related Processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Tee, A.R. MTOR and Autophagy: A Dynamic Relationship Governed by Nutrients and Energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.-M.; Starker, C.; Nho, R.S.; et al. The ULK1 Complex Mediates MTORC1 Signaling to the Autophagy Initiation Machinery via Binding and Phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-Dependent MTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Case, N.; Thomas, J.; Sen, B.; Styner, M.; Xie, Z.; Galior, K.; Rubin, J. Mechanical Regulation of Glycogen Synthase Kinase 3β (GSK3β) in Mesenchymal Stem Cells Is Dependent on Akt Protein Serine 473 Phosphorylation via MTORC2 Protein. J. Biol. Chem. 2011, 286, 39450–39456. [Google Scholar] [CrossRef]
- Hernández-Cáceres, M.P.; Munoz, L.; Pradenas, J.M.; Pena, F.; Lagos, P.; Aceiton, P.; Owen, G.I.; Morselli, E.; Criollo, A.; Ravasio, A.; et al. Mechanobiology of Autophagy: The Unexplored Side of Cancer. Front. Oncol. 2021, 11, 632956. [Google Scholar] [CrossRef]
- Dupont, N.; Codogno, P. Autophagy Transduces Physical Constraints into Biological Responses. Int. J. Biochem. Cell Biol. 2016, 79, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Martinac, B.; Poole, K. Mechanically Activated Ion Channels. Int. J. Biochem. Cell Biol. 2018, 97, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Saotome, K.; Murthy, S.E.; Kefauver, J.M.; Whitwam, T.; Patapoutian, A.; Ward, A.B. Structure of the Mechanically Activated Ion Channel Piezo1. Nature 2018, 554, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H.A. The Primary Cilium as Sensor of Fluid Flow: New Building Blocks to the Model. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am. J. Physiol. -Cell Physiol. 2015, 308, C198–C208. [Google Scholar] [CrossRef] [PubMed]
- Baschieri, F.; Dayot, S.; Elkhatib, N.; Ly, N.; Capmany, A.; Schauer, K.; Betz, T.; Vignjevic, D.M.; Poincloux, R.; Montagnac, G. Frustrated Endocytosis Controls Contractility-Independent Mechanotransduction at Clathrin-Coated Structures. Nat. Commun. 2018, 9, 3825. [Google Scholar] [CrossRef]
- Kirby, T.J.; Lammerding, J. Emerging Views of the Nucleus as a Cellular Mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Helle, S.C.J.; Feng, Q.; Aebersold, M.J.; Hirt, L.; Grüter, R.R.; Vahid, A.; Sirianni, A.; Mostowy, S.; Snedeker, J.G.; Šarić, A.; et al. Mechanical Force Induces Mitochondrial Fission. eLife 2017, 6, e30292. [Google Scholar] [CrossRef]
- Guet, D.; Mandal, K.; Pinot, M.; Hoffmann, J.; Abidine, Y.; Sigaut, W.; Bardin, S.; Schauer, K.; Goud, B.; Manneville, J.-B. Mechanical Role of Actin Dynamics in the Rheology of the Golgi Complex and in Golgi-Associated Trafficking Events. Curr. Biol. 2014, 24, 1700–1711. [Google Scholar] [CrossRef]
- Aguilera, M.O.; Berón, W.; Colombo, M.I. The Actin Cytoskeleton Participates in the Early Events of Autophagosome Formation upon Starvation Induced Autophagy. Autophagy 2012, 8, 1590–1603. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The Cytoskeleton–Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Hsu, P.; Shi, Y. Regulation of Autophagy by Mitochondrial Phospholipids in Health and Diseases. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2017, 1862, 114–129. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Alan Diehl, J.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of Autophagy during ECM Detachment Is Linked to a Selective Inhibition of MTORC1 by PERK. Oncogene 2013, 32, 4932–4940. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, D.; Sun, Y.; Gao, D.; Li, X.; Yang, J.; Ma, Z. Detachment-Based Equilibrium of Anoikic Cell Death and Autophagic Cell Survival through Adaptor Protein P66Shc. Anat. Rec. 2016, 299, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, A.; Debnath, J. The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J. Mol. Biol. 2017, 429, 515–530. [Google Scholar] [CrossRef]
- Anlaş, A.A.; Nelson, C.M. Soft Microenvironments Induce Chemoresistance by Increasing Autophagy Downstream of Integrin-Linked Kinase. Cancer Res. 2020, 80, 4103–4113. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Instructive Roles of Extracellular Matrix on Autophagy. Am. J. Pathol. 2014, 184, 2146–2153. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin Causes Autophagy in Endothelial Cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef]
- Nguyen, T.M.B.; Subramanian, I.V.; Xiao, X.; Ghosh, G.; Nguyen, P.; Kelekar, A.; Ramakrishnan, S. Endostatin Induces Autophagy in Endothelial Cells by Modulating Beclin 1 and β-Catenin Levels. J. Cell. Mol. Med. 2009, 13, 3687–3698. [Google Scholar] [CrossRef] [PubMed]
- Castagnaro, S.; Gambarotto, L.; Cescon, M.; Bonaldo, P. Autophagy in the Mesh of Collagen VI. Matrix Biol. 2021, 100–101, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Carmignac, V.; Svensson, M.; Körner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy Is Increased in Laminin A2 Chain-Deficient Muscle and Its Inhibition Improves Muscle Morphology in a Mouse Model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902. [Google Scholar] [CrossRef]
- García-Hoz, C.; Sánchez-Fernández, G.; Díaz-Meco, M.T.; Moscat, J.; Mayor, F.; Ribas, C. Gαq Acts as an Adaptor Protein in Protein Kinase Cζ (PKCζ)-Mediated ERK5 Activation by G Protein-Coupled Receptors (GPCR). J. Biol. Chem. 2010, 285, 13480–13489. [Google Scholar] [CrossRef] [PubMed]
- Nigro, P.; Abe, J.; Berk, B.C. Flow Shear Stress and Atherosclerosis: A Matter of Site Specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Scholz, N. Cancer Cell Mechanics: Adhesion G Protein-Coupled Receptors in Action? Front. Oncol. 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A. Cytoskeletal Elements Are Required for the Formation and Maturation of Autophagic Vacuoles. J. Cell. Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Lim, C.-Y.; Zoncu, R. The Lysosome as a Command-and-Control Center for Cellular Metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Cabukusta, B.; Neefjes, J. Mechanisms of Lysosomal Positioning and Movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef]
- Kesisova, I.A.; Robinson, B.P.; Spiliotis, E.T. A Septin GTPase Scaffold of Dynein–Dynactin Motors Triggers Retrograde Lysosome Transport. J. Cell Biol. 2021, 220, e202005219. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal Positioning Coordinates Cellular Nutrient Responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 Effector Protein RILP Controls Lysosomal Transport by Inducing the Recruitment of Dynein-Dynactin Motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A Key to Lysosome Biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Zhou, H.; Jian, Y.; Li, L.; Wang, M.; Liu, N.; Yin, Q.; Liang, Z.; Guo, W.; Yang, C. The Rab7 Effector WDR91 Promotes Autophagy-Lysosome Degradation in Neurons by Regulating Lysosome Fusion. J. Cell Biol. 2021, 220, e202007061. [Google Scholar] [CrossRef] [PubMed]
- Willett, R.; Martina, J.A.; Zewe, J.P.; Wills, R.; Hammond, G.R.V.; Puertollano, R. TFEB Regulates Lysosomal Positioning by Modulating TMEM55B Expression and JIP4 Recruitment to Lysosomes. Nat. Commun. 2017, 8, 1580. [Google Scholar] [CrossRef]
- Settembre, C.; di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Wright, S.C.; Lukasheva, V.; le Gouill, C.; Kobayashi, H.; Breton, B.; Mailhot-Larouche, S.; Blondel-Tepaz, É.; Antunes Vieira, N.; Costa-Neto, C.; Héroux, M.; et al. BRET-Based Effector Membrane Translocation Assay Monitors GPCR-Promoted and Endocytosis-Mediated Gq Activation at Early Endosomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2025846118. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-C.; Liu, P.-F.; Chang, C.-H.; Lin, Y.-C.; Chen, Y.-J.; Shu, C.-W. The Interplay of Autophagy and Oxidative Stress in the Pathogenesis and Therapy of Retinal Degenerative Diseases. Cell Biosci. 2022, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS: Thiol Network Is the Principal Suspect for Autophagy Commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Gu, L.; Smerin, D.; Mao, S.; Xiong, X. The Interrelation between Reactive Oxygen Species and Autophagy in Neurological Disorders. Oxidative Med. Cell. Longev. 2017, 2017, 8495160. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of Reactive Oxygen Species by Mitochondria. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive Oxygen Species Are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 Inhibits Tumorigenesis via Regulating PTEN/AKT Activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef]
- Mahalingaiah, P.K.S.; Singh, K.P. Chronic Oxidative Stress Increases Growth and Tumorigenic Potential of MCF-7 Breast Cancer Cells. PLoS ONE 2014, 9, e87371. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zheng, B.; Wang, C.; Yang, Y.; Luo, W.; Ma, S.; Zhang, X.; Ma, D.; Sun, Y.; Yang, Z.; et al. Oxidative Stress Induces Neuronal Apoptosis through Suppressing Transcription Factor EB Phosphorylation at Ser467. Cell. Physiol. Biochem. 2018, 46, 1536–1554. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and Decoding Ubiquitin Chains for Mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs Govern Autophagosome Biogenesis during Mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Goh, J.-Y.; Xiao, L.; Xian, H.; Lim, K.-L.; Liou, Y.-C. Reactive Oxygen Species Trigger Parkin/PINK1 Pathway–Dependent Mitophagy by Inducing Mitochondrial Recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef]
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.-L.; Tan, E.-K. Superoxide Drives Progression of Parkin/PINK1-Dependent Mitophagy Following Translocation of Parkin to Mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.; Chen, Y.; Liu, H.; Zhou, H.; Bai, Z.; Hu, Z.; Guo, X. PINK1 Protects against Oxidative Stress Induced Senescence of Human Nucleus Pulposus Cells via Regulating Mitophagy. Biochem. Biophys. Res. Commun. 2018, 504, 406–414. [Google Scholar] [CrossRef]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.-H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-Dependent Regulation of Parkin and DJ-1 Localization during Oxidative Stress in Neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial Outer-Membrane Protein FUNDC1 Mediates Hypoxia-Induced Mitophagy in Mammalian Cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates Both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [PubMed]
- Benincá, C.; Planagumà, J.; de Freitas Shuck, A.; Acín-Perez, R.; Muñoz, J.P.; de Almeida, M.M.; Brown, J.H.; Murphy, A.N.; Zorzano, A.; Enríquez, J.A.; et al. A New Non-Canonical Pathway of Gαq Protein Regulating Mitochondrial Dynamics and Bioenergetics. Cell. Signal. 2014, 26, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Qin, F.; Lennon, S.L.; Zhang, J.; Tong, X.; Mazzini, M.J.; Kang, Y.J.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Short Communication: Oxidative Posttranslational Modifications Mediate Decreased SERCA Activity and Myocyte Dysfunction in Gαq-Overexpressing Mice. Circ. Res. 2010, 107, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Westenbrink, B.D.; Ling, H.; Divakaruni, A.S.; Gray, C.B.B.; Zambon, A.C.; Dalton, N.D.; Peterson, K.L.; Gu, Y.; Matkovich, S.J.; Murphy, A.N.; et al. Mitochondrial Reprogramming Induced by CaMKIIδ Mediates Hypertrophy Decompensation. Circ. Res. 2015, 116, e28–e39. [Google Scholar] [CrossRef]
- Jhun, B.S.; O-Uchi, J.; Adaniya, S.M.; Mancini, T.J.; Cao, J.L.; King, M.E.; Landi, A.K.; Ma, H.; Shin, M.; Yang, D.; et al. Protein Kinase D Activation Induces Mitochondrial Fragmentation and Dysfunction in Cardiomyocytes. J. Physiol. 2018, 596, 827–855. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Yang, C.-C.; Cho, R.-L.; Wang, C.-Y.; Hsiao, L.-D.; Yang, C.-M. Sphingosine 1-Phosphate-Induced ICAM-1 Expression via NADPH Oxidase/ROS-Dependent NF-ΚB Cascade on Human Pulmonary Alveolar Epithelial Cells. Front. Pharmacol. 2016, 7, 80. [Google Scholar] [CrossRef]
- Kenific, C.M.; Wittmann, T.; Debnath, J. Autophagy in Adhesion and Migration. J. Cell Sci. 2016, 129, 3685–3693. [Google Scholar] [CrossRef] [PubMed]
- King, J.S. Mechanical Stress Meets Autophagy: Potential Implications for Physiology and Pathology. Trends Mol. Med. 2012, 18, 583–588. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Komatsu, M. Pathophysiological Role of Autophagy: Lesson from Autophagy-Deficient Mouse Models. Exp. Anim. 2011, 60, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Koya, D. Autophagy in Metabolic Disease and Ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Van Beek, N.; Klionsky, D.J.; Reggiori, F. Genetic Aberrations in Macroautophagy Genes Leading to Diseases. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, D.; Wang, C.; Luo, Y.; Ding, X.; Yang, X.; Silva, F.; Arenas, S.; Weaver, J.M.; Mandell, M.; et al. Sustained Activation of Autophagy Suppresses Adipocyte Maturation via a Lipolysis-Dependent Mechanism. Autophagy 2020, 16, 1668–1682. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy: A Potential Link between Obesity and Insulin Resistance. Cell Metab. 2010, 11, 449–451. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef]
- Park, E.-Y.; Park, J.-B. High Glucose-Induced Oxidative Stress Promotes Autophagy through Mitochondrial Damage in Rat Notochordal Cells. Int. Orthop. 2013, 37, 2507–2514. [Google Scholar] [CrossRef]
- Oliveira de Souza, C.; Sun, X.; Oh, D. Metabolic Functions of G Protein-Coupled Receptors and β-Arrestin-Mediated Signaling Pathways in the Pathophysiology of Type 2 Diabetes and Obesity. Front. Endocrinol. 2021, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- Blad, C.C.; Tang, C.; Offermanns, S. G Protein-Coupled Receptors for Energy Metabolites as New Therapeutic Targets. Nat. Rev. Drug Discov. 2012, 11, 603–619. [Google Scholar] [CrossRef]
- Offermanns, S. Free Fatty Acid (FFA) and Hydroxy Carboxylic Acid (HCA) Receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 407–434. [Google Scholar] [CrossRef] [PubMed]
- Wauson, E.M.; Zaganjor, E.; Cobb, M.H. Amino Acid Regulation of Autophagy through the GPCR TAS1R1-TAS1R3. Autophagy 2013, 9, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Wauson, E.M.; Lorente-Rodríguez, A.; Cobb, M.H. Minireview: Nutrient Sensing by G Protein-Coupled Receptors. Mol. Endocrinol. 2013, 27, 1188–1197. [Google Scholar] [CrossRef]
- Lizaso, A.; Tan, K.-T.; Lee, Y.-H. β-Adrenergic Receptor-Stimulated Lipolysis Requires the RAB7-Mediated Autolysosomal Lipid Degradation. Autophagy 2013, 9, 1228–1243. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.-H.; Kim, J.-W.; et al. Loss of Autophagy Diminishes Pancreatic β Cell Mass and Function with Resultant Hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. Feedback on Fat: P62-MTORC1-Autophagy Connections. Cell 2011, 147, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Noda, S.E.; Kaltenbronn, K.M.; Frankfater, C.; Makepeace, C.M.; Fettig, N.; Piggott, K.D.; Custer, P.L.; Ippolito, J.E.; Blumer, K.J. Oncogenic Gq/11 Signaling Acutely Drives and Chronically Sustains Metabolic Reprogramming in Uveal Melanoma. J. Biol. Chem. 2022, 298, 101495. [Google Scholar] [CrossRef]
- Kimura, T.; Pydi, S.P.; Wang, L.; Haspula, D.; Cui, Y.; Lu, H.; König, G.M.; Kostenis, E.; Steinberg, G.R.; Gavrilova, O.; et al. Adipocyte Gq Signaling Is a Regulator of Glucose and Lipid Homeostasis in Mice. Nat. Commun. 2022, 13, 1652. [Google Scholar] [CrossRef] [PubMed]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between Mechanotransduction and Metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 22–38. [Google Scholar] [CrossRef]
- Kim, S.W.; Ehrman, J.; Ahn, M.-R.; Kondo, J.; Lopez, A.A.M.; Oh, Y.S.; Kim, X.H.; Crawley, S.W.; Goldenring, J.R.; Tyska, M.J.; et al. Shear Stress Induces Noncanonical Autophagy in Intestinal Epithelial Monolayers. Mol. Biol. Cell 2017, 28, 3043–3056. [Google Scholar] [CrossRef] [PubMed]
- Miceli, C.; Roccio, F.; Penalva-Mousset, L.; Burtin, M.; Leroy, C.; Nemazanyy, I.; Kuperwasser, N.; Pontoglio, M.; Friedlander, G.; Morel, E.; et al. The Primary Cilium and Lipophagy Translate Mechanical Forces to Direct Metabolic Adaptation of Kidney Epithelial Cells. Nat. Cell Biol. 2020, 22, 1091–1102. [Google Scholar] [CrossRef]
- He, C.; Sumpter, R., Jr.; Levine, B. Exercise Induces Autophagy in Peripheral Tissues and in the Brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Claude-Taupin, A.; Codogno, P.; Dupont, N. Links between Autophagy and Tissue Mechanics. J. Cell Sci. 2021, 134, jcs258589. [Google Scholar] [CrossRef]
- Mameli, E.; Martello, A.; Caporali, A. Autophagy at the Interface of Endothelial Cell Homeostasis and Vascular Disease. FEBS J. 2021, 289, 2976–2991. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; de Bruijn, J.; van Kuijk, K.; Riascos-Bernal, D.F.; Diaz, A.; Tasset, I.; Martín-Segura, A.; Gijbels, M.J.J.; Sander, B.; Kaushik, S.; et al. Protective Role of Chaperone-Mediated Autophagy against Atherosclerosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121133119. [Google Scholar] [CrossRef]
- Doronzo, G.; Astanina, E.; Corà, D.; Chiabotto, G.; Comunanza, V.; Noghero, A.; Neri, F.; Puliafito, A.; Primo, L.; Spampanato, C.; et al. TFEB Controls Vascular Development by Regulating the Proliferation of Endothelial Cells. EMBO J. 2019, 38, e98250. [Google Scholar] [CrossRef]
- Shadab, M.; Millar, M.W.; Slavin, S.A.; Leonard, A.; Fazal, F.; Rahman, A. Autophagy Protein ATG7 Is a Critical Regulator of Endothelial Cell Inflammation and Permeability. Sci. Rep. 2020, 10, 13708. [Google Scholar] [CrossRef]
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy Modulates Endothelial Junctions to Restrain Neutrophil Diapedesis during Inflammation. Immunity 2021, 54, 1989–2004.e9. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Cho, J.M.; Park, S.-K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress–Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Anandh Babu, P.V.; et al. Impairment of Autophagy in Endothelial Cells Prevents Shear-Stress-Induced Increases in Nitric Oxide Bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-X.; Qu, X.-L.; Chu, P.; Xie, D.-J.; Zhu, L.-L.; Chao, Y.-L.; Li, L.; Zhang, J.-J.; Chen, S.-L. Low Shear Stress Induces Vascular ENOS Uncoupling via Autophagy-Mediated ENOS Phosphorylation. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Dela Paz, N.G.; Melchior, B.; Frangos, J.A. Shear Stress Induces Gαq/11 Activation Independently of G Protein-Coupled Receptor Activation in Endothelial Cells. Am. J. Physiol. -Cell Physiol. 2017, 312, C428–C437. [Google Scholar] [CrossRef] [PubMed]
- Albarrán-Juárez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 Promote Endothelial Inflammation Depending on Flow Pattern and Integrin Activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Fujiwara, T.; et al. The Role of Autophagy Emerging in Postinfarction Cardiac Remodelling. Cardiovasc. Res. 2011, 91, 330–339. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Yan, L.; Vatner, D.E.; Kim, S.-J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in Chronically Ischemic Myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef]
- García-Hoz, C.; Sánchez-Fernández, G.; García-Escudero, R.; Fernández-Velasco, M.; Palacios-García, J.; Ruiz-Meana, M.; Díaz-Meco, M.T.; Leitges, M.; Moscat, J.; García-Dorado, D.; et al. Protein Kinase C (PKC) ζ-Mediated Gαq Stimulation of ERK5 Protein Pathway in Cardiomyocytes and Cardiac Fibroblasts. J. Biol. Chem. 2012, 287, 7792–7802. [Google Scholar] [CrossRef]
- Adams, J.W.; Pagel, A.L.; Means, C.K.; Oksenberg, D.; Armstrong, R.C.; Brown, J.H. Cardiomyocyte Apoptosis Induced by Gαq Signaling Is Mediated by Permeability Transition Pore Formation and Activation of the Mitochondrial Death Pathway. Circ. Res. 2000, 87, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Meleka, M.M.; Edwards, A.J.; Xia, J.; Dahlen, S.A.; Mohanty, I.; Medcalf, M.; Aggarwal, S.; Moeller, K.D.; Mortensen, O.V.; Osei-Owusu, P. Anti-Hypertensive Mechanisms of Cyclic Depsipeptide Inhibitor Ligands for Gq/11 Class G Proteins. Pharmacol. Res. 2019, 141, 264–275. [Google Scholar] [CrossRef]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef]
- Chude, C.; Amaravadi, R. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, D.-D.; Wang, L.-L.; Deng, R.; Zhu, X.-F. Decreased Expression of Autophagy-Related Proteins in Malignant Epithelial Ovarian Cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Hui, B.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy Inhibition Suppresses Pulmonary Metastasis of HCC in Mice via Impairing Anoikis Resistance and Colonization of HCC Cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Chmurska, A.; Matczak, K.; Marczak, A. Two Faces of Autophagy in the Struggle against Cancer. Int. J. Mol. Sci. 2021, 22, 2981. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-J.; Chang, W.-A.; Huang, M.-S.; Kuo, P.-L. Tumor Microenvironment: A New Treatment Target for Cancer. ISRN Biochem. 2014, 2014, 351959. [Google Scholar] [CrossRef]
- Das, J.; Maji, S.; Agarwal, T.; Chakraborty, S.; Maiti, T.K. Hemodynamic Shear Stress Induces Protective Autophagy in HeLa Cells through Lipid Raft-Mediated Mechanotransduction. Clin. Exp. Metastasis 2018, 35, 135–148. [Google Scholar] [CrossRef]
- Lien, S.-C.; Chang, S.-F.; Lee, P.-L.; Wei, S.-Y.; Chang, M.D.-T.; Chang, J.-Y.; Chiu, J.-J. Mechanical Regulation of Cancer Cell Apoptosis and Autophagy: Roles of Bone Morphogenetic Protein Receptor, Smad1/5, and P38 MAPK. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, Y.; Liu, W.; Liu, L.; Guo, Z.; Fan, C.; Wang, L.; Hu, J.; Li, B. Mechanical Stress-Dependent Autophagy Component Release via Extracellular Nanovesicles in Tumor Cells. ACS Nano 2019, 13, 4589–4602. [Google Scholar] [CrossRef]
- Yan, Z.; Su, G.; Gao, W.; He, J.; Shen, Y.; Zeng, Y.; Liu, X. Fluid Shear Stress Induces Cell Migration and Invasion via Activating Autophagy in HepG2 Cells. Cell Adhes. Migr. 2019, 13, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Follain, G.; Herrmann, D.; Harlepp, S.; Hyenne, V.; Osmani, N.; Warren, S.C.; Timpson, P.; Goetz, J.G. Fluids and Their Mechanics in Tumour Transit: Shaping Metastasis. Nat. Rev. Cancer 2020, 20, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Lund, A.W. Lymphatic and Interstitial Flow in the Tumour Microenvironment: Linking Mechanobiology with Immunity. Nat. Rev. Cancer 2012, 12, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Clocchiatti, A.; Dotto, G.P. A Role for Stromal Autophagy in Cancer-Associated Fibroblast Activation. Autophagy 2019, 15, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.I.; Kim, D.H.; Sung, K.W.; Shim, S.M.; Cha-Molstad, H.; Soung, N.K.; Lee, K.H.; Hwang, J.; Lee, H.G.; Kwon, Y.T.; et al. P62-Induced Cancer-Associated Fibroblast Activation via the Nrf2-ATF6 Pathway Promotes Lung Tumorigenesis. Cancers 2021, 13, 864. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordoñez, A.; Reina-Campos, M.; Kuo, H.-H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The Lactate-NAD+ Axis Activates Cancer-Associated Fibroblasts by Downregulating P62. Cell Rep. 2022, 39, 110792. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| GPCRs (Coupled to Gq Protein) | Chemical Class of Natural Ligand | Mechanical Stimulation | References |
|---|---|---|---|
| 5HT2A 5HT2B 5HT2C | Serotonin | Mechanical stretch | [43] |
| ADRA1A | Adrenaline/Noradrenaline | Shear stress | [44] |
| BB1 | Bombesin | No reported | [45] |
| BLT1 | Leukotrienes | No reported | [46] |
| CCK1 | Cholecystokinin gastrin | No reported | [47] |
| CysLT1 | Leukotrienes | Hypotonicity/ Increased intravascular pressure | [48,49] |
| EP1 | ProstaglandinE2 | No reported | [50,51] |
| ET1AR | Endothelin | Stretch | [34,52] |
| PAR1 | Thrombin | Laminar flow | [53,54] |
| Gal2 | Galanin | No reported | [55] |
| GHSR1a | Ghrelin | No reported | [56] |
| GnRH1 | Gonadotropin | Insensitive | [57,58] |
| GRP39 | Obestatin/Zinc | No reported | [59] |
| GPR68 GPR4 | Protons | Shear stress | [41,60] |
| H1R | Histamine | Hypotonicity, direct membrane stretches, shear stress, intravascular flow | [34,58,61] |
| M5R M1R M3R | Acetylcholine Acetylcholine | Hypotoniticy and membrane stretch No reported | [62,63] |
| AT1R | Angiotensin | Hypotonicity, direct membrane stretch, pressure overload, increased intravascular pressure Pressure overload | [64,65,66] |
| MCHR | Melanin | No reported | [67] |
| B2R | Bradykinin | Shear stress, hipotonicity, Increase in plasma membrane fluidity | [68] |
| GPER | Estrogen | Mechanical stress | [69] |
| FFAR1 | Fatty acids | No reported | [70] |
| PTH1R | PTH | Fluid shear stress | [71,72] |
| V1AR | Oxytocin Vasopressin | Stretch, shear stress | [34,73] |
| ADGRG2 | No identified | Luminal fluid | [74] |
| P2YR | nucleotides | Fluid shear stress/Mechanical stress | [33,75,76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro-Lérida, I.; Aragay, A.M.; Asensio, A.; Ribas, C. Gq Signaling in Autophagy Control: Between Chemical and Mechanical Cues. Antioxidants 2022, 11, 1599. https://doi.org/10.3390/antiox11081599
Navarro-Lérida I, Aragay AM, Asensio A, Ribas C. Gq Signaling in Autophagy Control: Between Chemical and Mechanical Cues. Antioxidants. 2022; 11(8):1599. https://doi.org/10.3390/antiox11081599
Chicago/Turabian StyleNavarro-Lérida, Inmaculada, Anna M. Aragay, Alejandro Asensio, and Catalina Ribas. 2022. "Gq Signaling in Autophagy Control: Between Chemical and Mechanical Cues" Antioxidants 11, no. 8: 1599. https://doi.org/10.3390/antiox11081599
APA StyleNavarro-Lérida, I., Aragay, A. M., Asensio, A., & Ribas, C. (2022). Gq Signaling in Autophagy Control: Between Chemical and Mechanical Cues. Antioxidants, 11(8), 1599. https://doi.org/10.3390/antiox11081599