
Bioactive Components from Ampelopsis japonica with Antioxidant, Anti-α-Glucosidase, and Antiacetylcholinesterase Activities

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and General Procedures
2.2. Preparation of A. japonica Extracts
2.3. Preparation of Active Compounds
2.4. Reversed-Phase HPLC
2.5. Determination of Total Phenolic Content (TPC)
2.6. Determination of Total Flavonoid Content (TFC)
2.7. DPPH Radical Scavenging Activity
2.8. ABTS Radical Scavenging Assay
2.9. Superoxide Radical Scavenging Assay
2.10. Ferric Reducing Antioxidant Power (FRAP) Assay
2.11. α-Glucosidase Inhibitory Activity Assay
2.12. Acetylcholinesterase (AChE) Inhibitory Assay
2.13. Molecular Modeling Docking Study
2.14. Statistical Analysis
3. Results
3.1. Determination of TPC, TFC, and Yields in Each Solvent Extract
3.2. DPPH Radical Scavenging Activity
3.3. ABTS Free Radical Scavenging Effect
3.4. Superoxide Radical Scavenging Effect
3.5. Ferric Reducing Antioxidant Power (FRAP) Effect
3.6. Anti-α-Glucosidase Effect
3.7. AChE Inhibition Activity
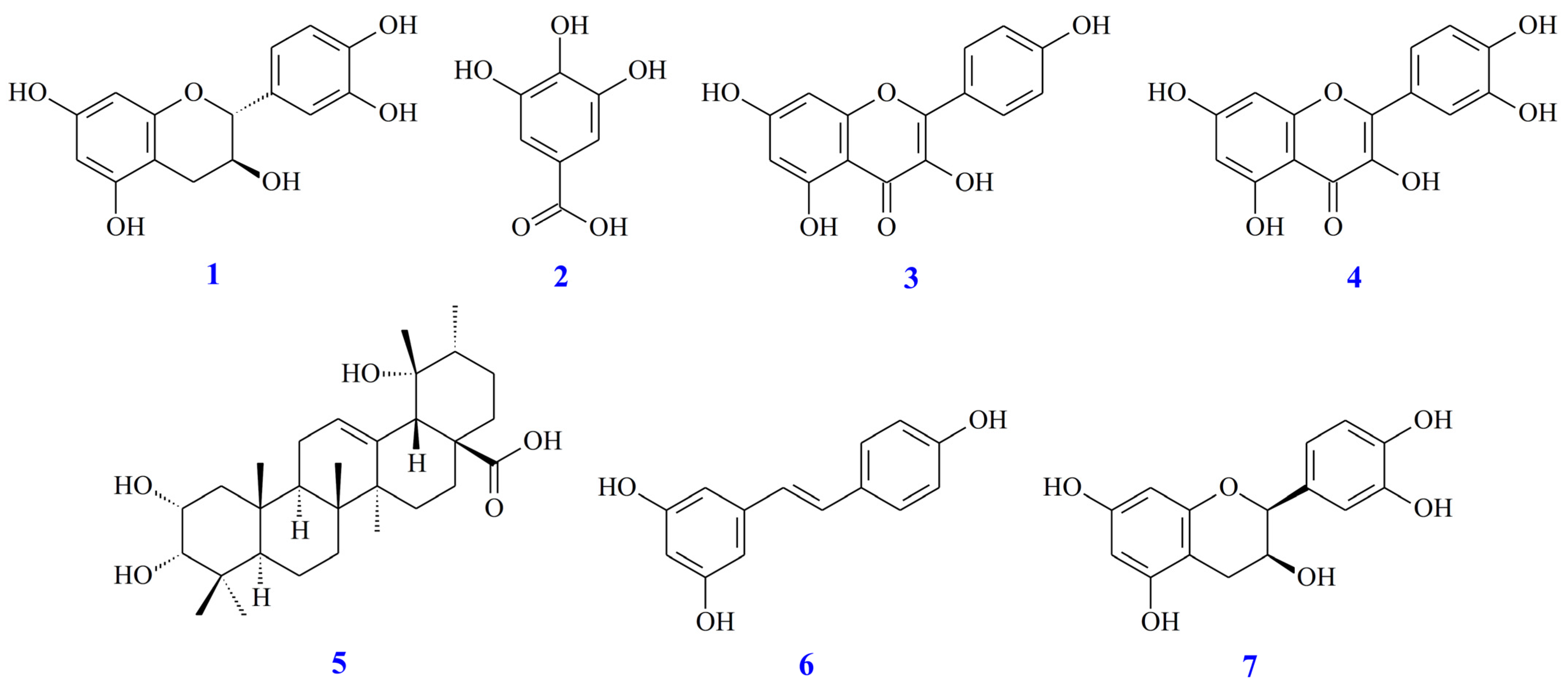
3.8. Quantitation of Active Components in Different Solvent Extracts
3.9. Antioxidant Activities of Isolated Components
3.10. Anti-α-Glucosidase Activities of Isolated Components
3.11. AChE Inhibition Assay of Isolated Components
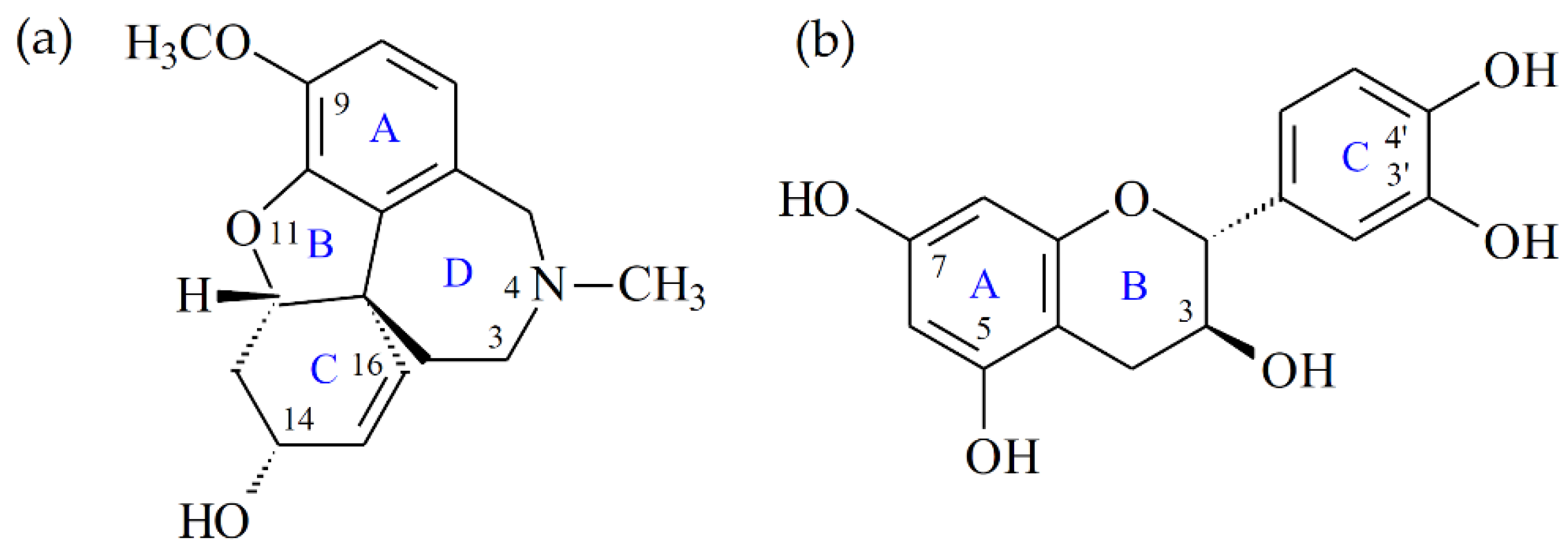
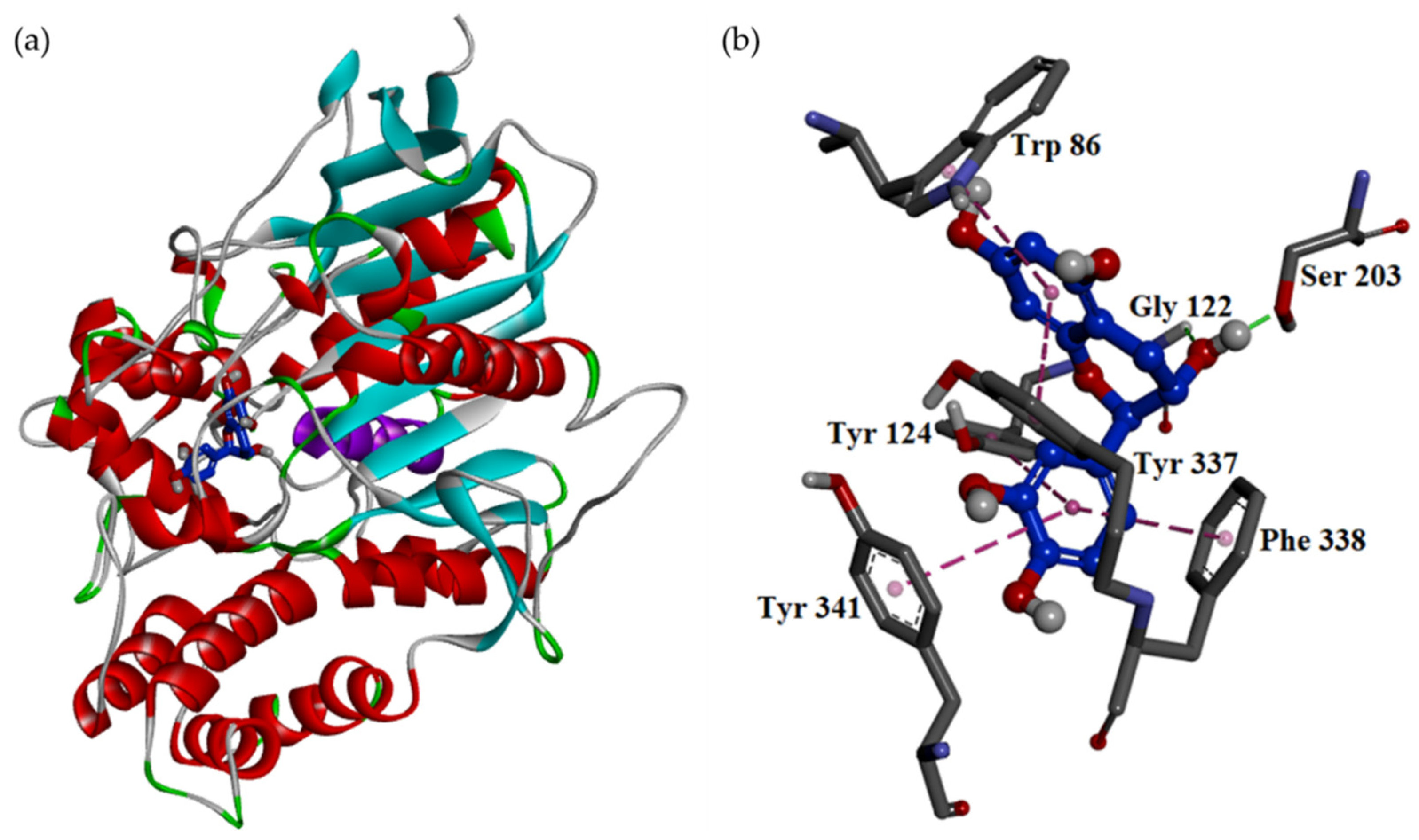
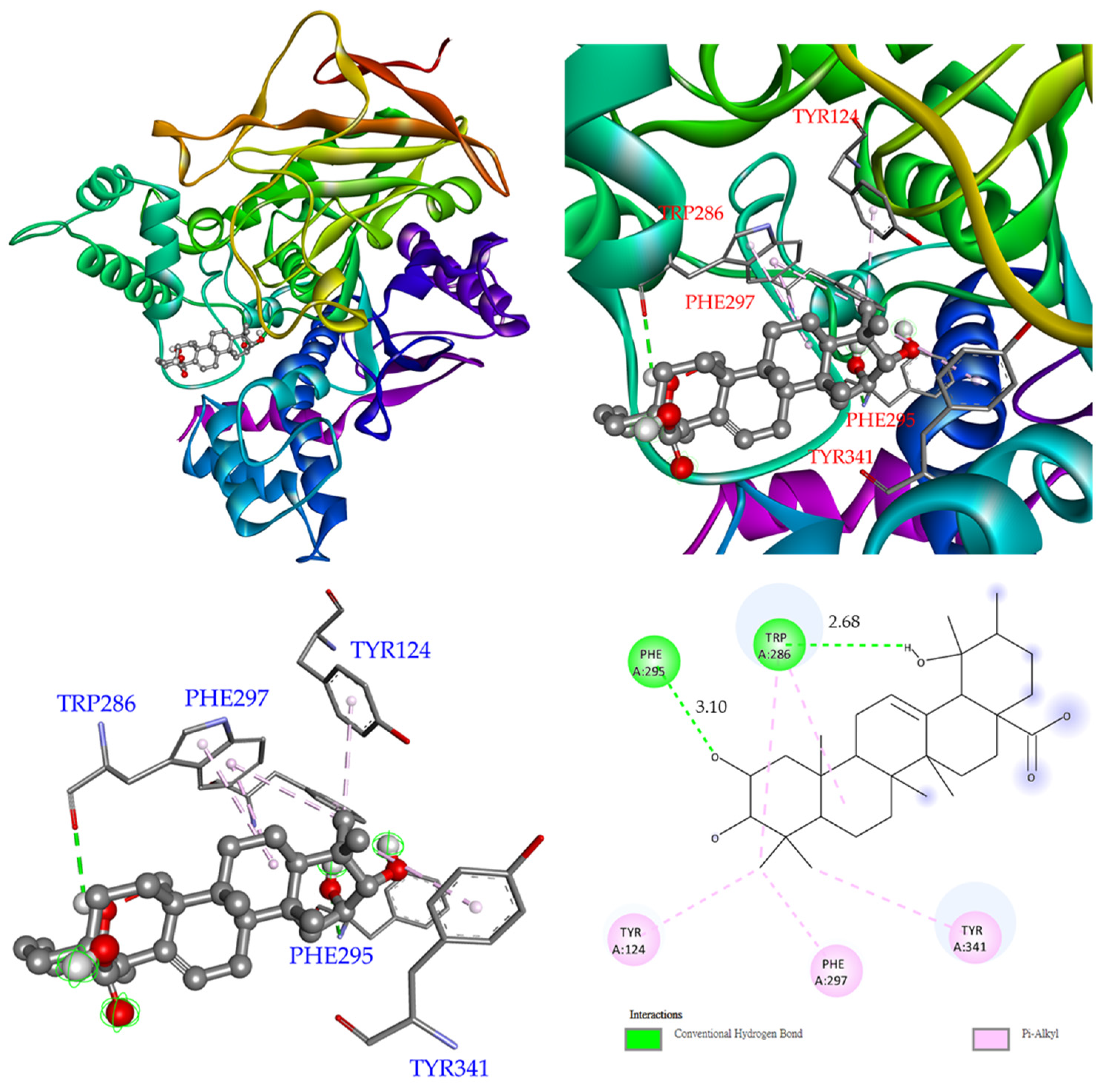
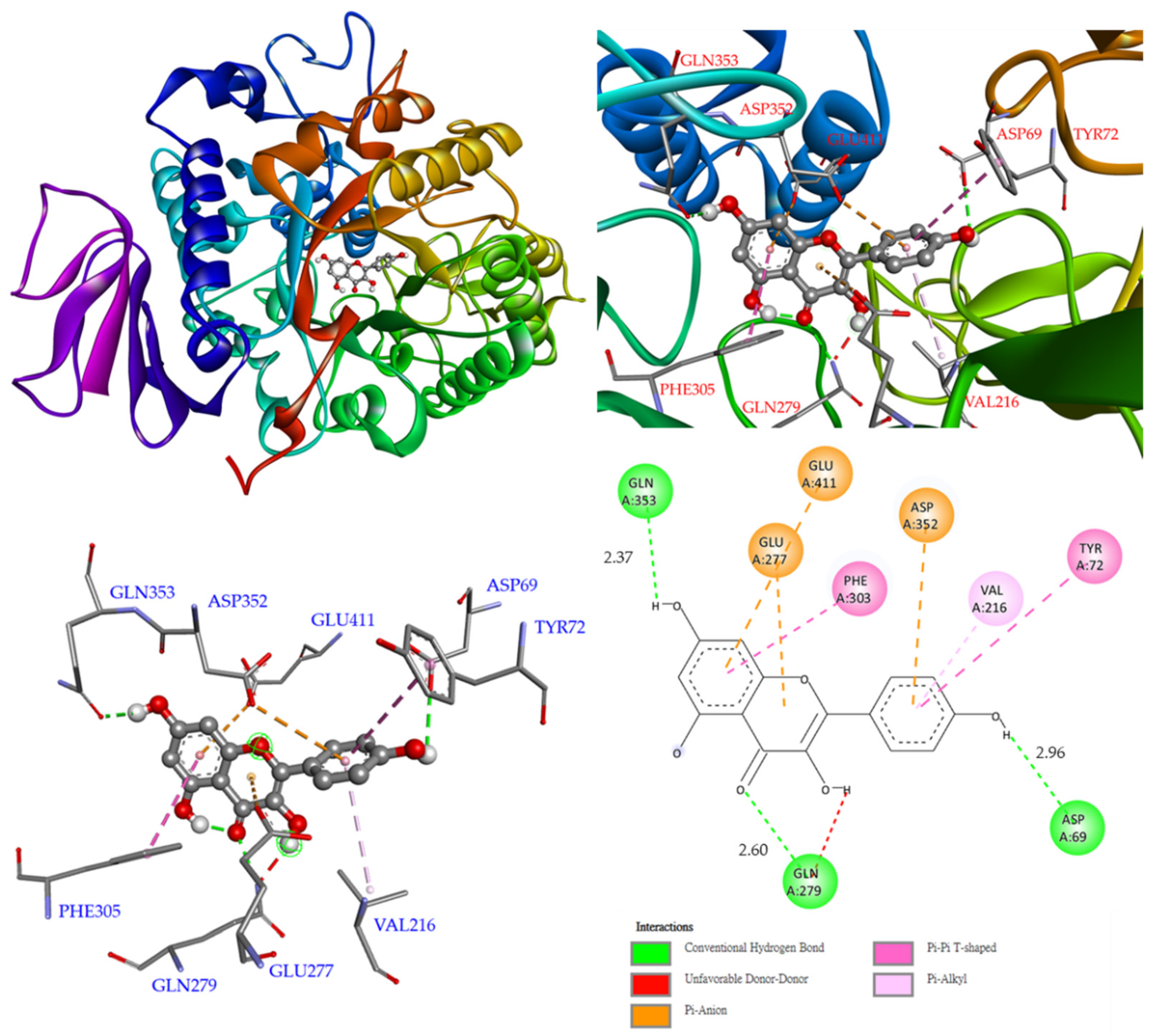
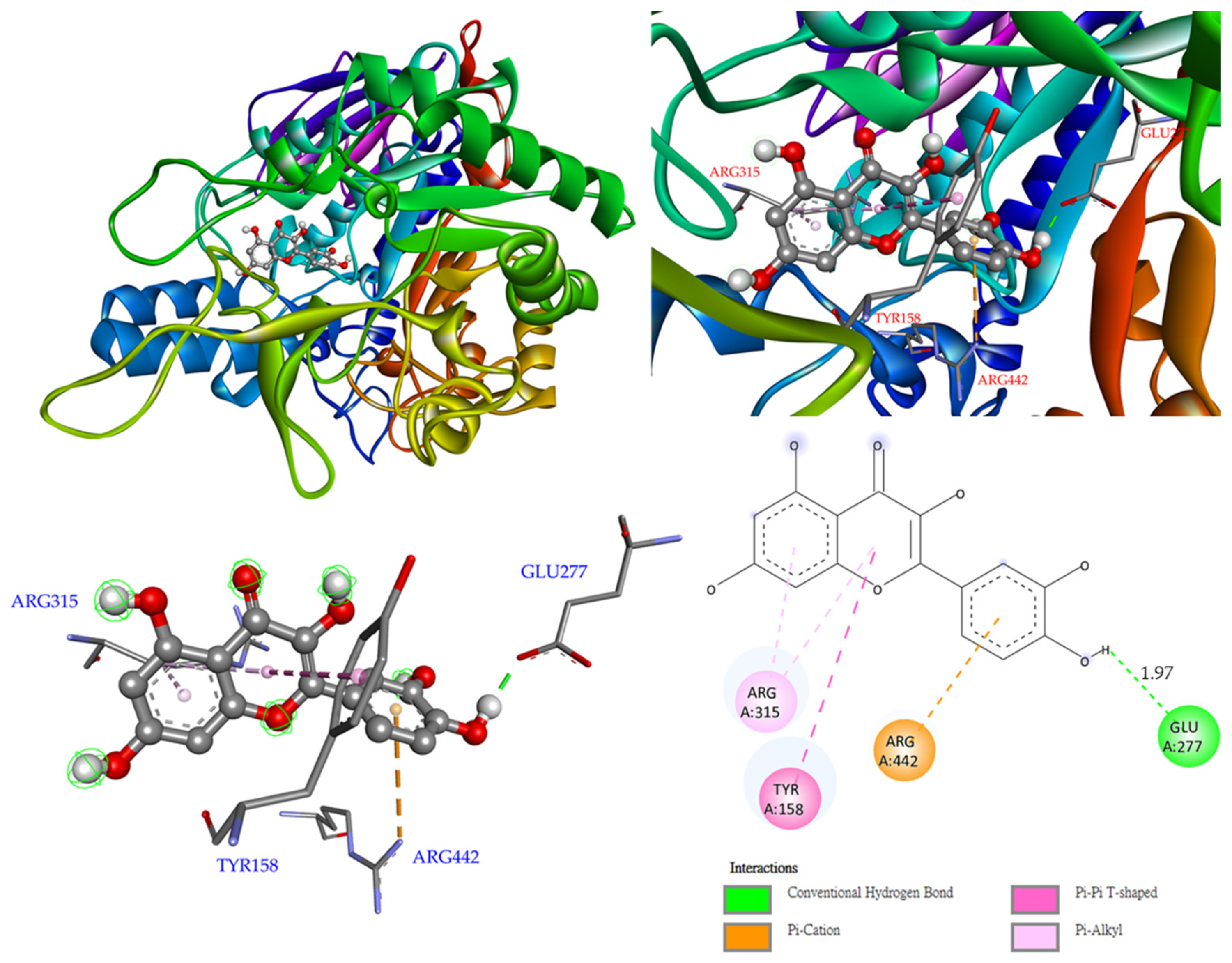
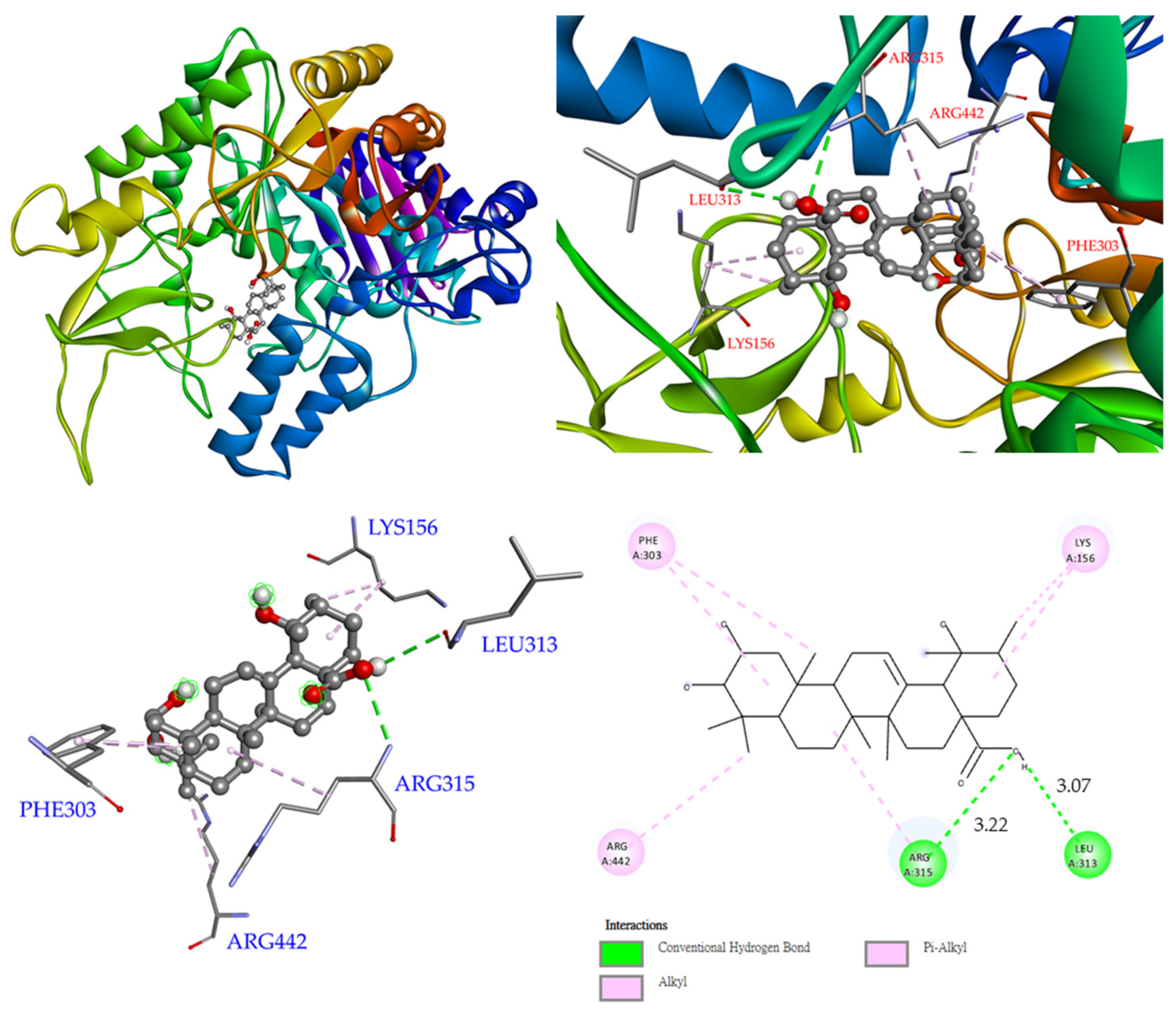
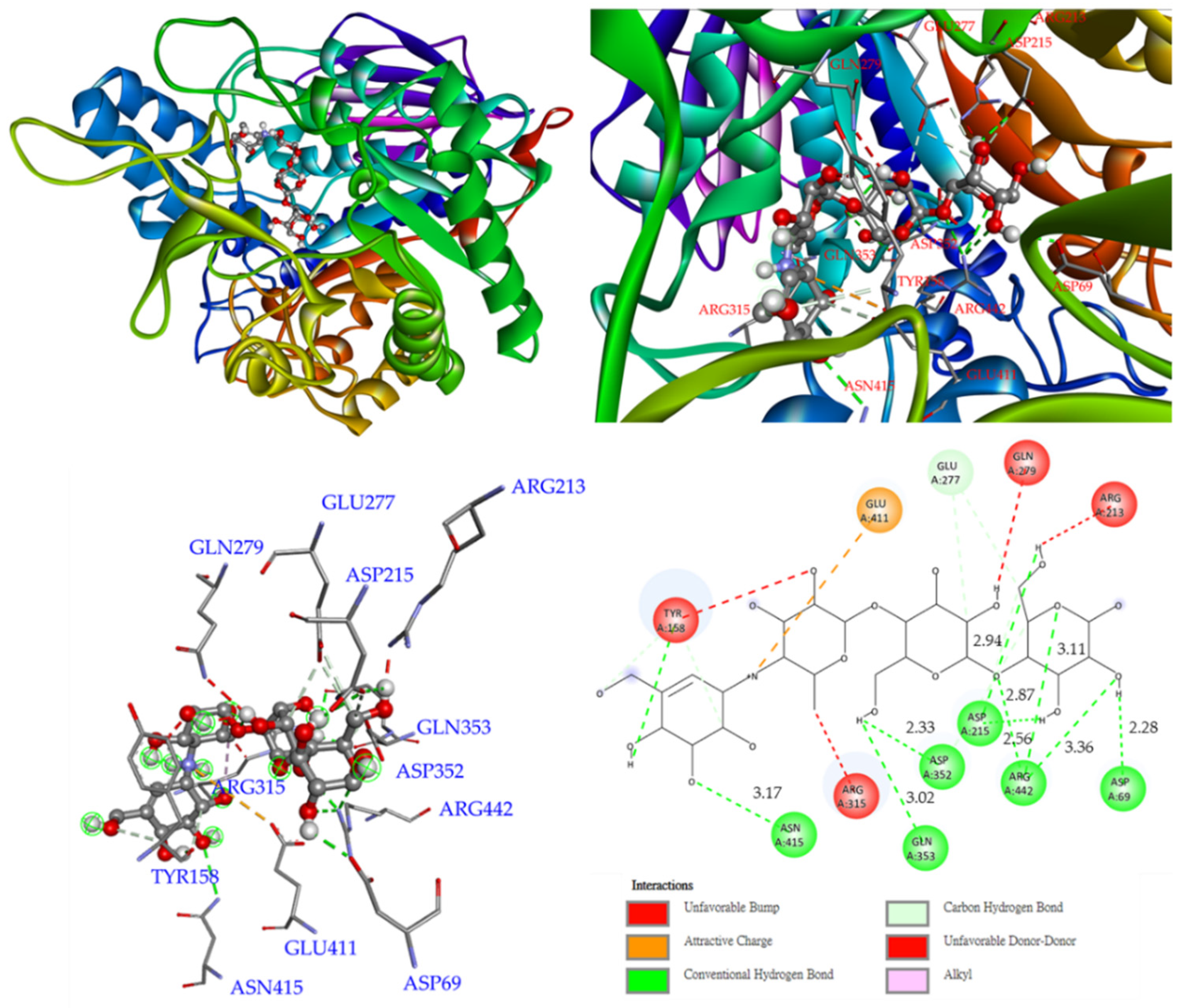
3.12. Molecular Modeling Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meena, D.K.; Sahoo, A.K.; Srivastava, P.P.; Sahu, N.P.; Jadhav, M.; Gandhi, M.; Swain, H.S.; Borah, S.; Das, B.K. On valorization of solvent extracts of Terminalia arjuna (arjuna) upon DNA scission and free radical scavenging improves coupling responses and cognitive functions under in vitro conditions. Sci. Rep. 2021, 11, 10656. [Google Scholar] [CrossRef] [PubMed]
- Yew, P.N.; Lee, L.W.L.; Lim, Y.Y. Antioxidant and intracellular reactive oxygen species/reactive nitrogen species scavenging activities of three porcupine bezoars from Hystrix brachyura. Phcog. Res. 2017, 9, 366–372. [Google Scholar] [PubMed]
- Ndhlala, A.R.; Moyo, M.; Van Staden, J. Natural antioxidants: Fascinating or mythical biomolecules? Molecules 2010, 15, 6905–6930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanvir, E.M.; Hossen, M.S.; Hossain, M.F.; Afroz, R.; Gan, S.H.; Khalil, M.I.; Karim, N. Antioxidant properties of popular turmeric (Curcuma longa) varieties from Bangladesh. J. Food Qual. 2017, 2017, 8471785. [Google Scholar] [CrossRef] [Green Version]
- Kaur, C.; Kapoor, H.C. Antioxidants in fruits and vegetables—The millennium’s health. Int. J. Food Sci. Technol. 2001, 36, 703–725. [Google Scholar] [CrossRef]
- Li, C.W.; Chu, Y.C.; Huang, C.Y.; Fu, S.L.; Chen, J.J. Evaluation of antioxidant and anti-α-glucosidase activities of various solvent extracts and major bioactive components from the seeds of Myristica fragrans. Molecules 2020, 25, 5198. [Google Scholar] [CrossRef]
- Guo, J.M.; Weng, X.C.; Wu, H.; Li, Q.H.; Bi, K.S. Antioxidants from a Chinese medicinal herb—Psoralea corylifolia L. Food Chem. 2005, 91, 287–292. [Google Scholar]
- Siebert, D.A.; Campos, J.S.; Alberton, M.D.; Vitali, L.; Micke, G.A. Dual electrophoretically-mediated microanalysis in multiple injection mode for the simultaneous determination of acetylcholinesterase and α-glucosidase activity applied to selected polyphenols. Talanta 2021, 224, 121773. [Google Scholar] [CrossRef]
- Zaidi, H.; Ouchemoukh, S.; Amessis-Ouchemoukh, N.; Debbache, N.; Pacheco, R.; Serralheiro, M.L.; Araujo, M.E. Biological properties of phenolic compound extracts in selected Algerian honeys—The inhibition of acetylcholinesterase and α-glucosidase activities. Eur. J. Integr. Med. 2019, 25, 77–84. [Google Scholar] [CrossRef]
- Etxeberria, U.; de la Garza, A.L.; Campión, J.; Martínez, J.A.; Milagro, F.I. Antidiabetic effects of natural plant extracts via inhibition of carbohydrate hydrolysis enzymes with emphasis on pancreatic alpha amylase. Expert Opin. Ther. Targets 2012, 16, 269–297. [Google Scholar] [CrossRef] [Green Version]
- Van De Laar, F.A. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manag. 2008, 4, 1189–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orhan, I.; Kartal, M.; Tosun, F.; Şener, B. Screening of various phenolic acids and flavonoid derivatives for their anticholinesterase potential. Z. Naturforsch. C 2014, 62, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, D.; Desseva, I.; Popova, A.; Dincheva, I.; Vrancheva, R.; Lante, A.; Krastanov, A. GC-MS metabolic profile and α-glucosidase-, α-amylase-, lipase-, and acetylcholinesterase-inhibitory activities of eight peach varieties. Molecules 2021, 26, 4183. [Google Scholar] [CrossRef] [PubMed]
- Abbas-Mohammadi, M.; Farimani, M.M.; Salehi, P.; Ebrahimi, S.N.; Sonboli, A.; Kelso, C.; Skropeta, D. Acetylcholinesterase-inhibitory activity of Iranian plants: Combined HPLC/bioassay-guided fractionation, molecular networking and docking strategies for the dereplication of active compounds. J. Pharm. Biomed. Anal. 2018, 158, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Park, C.; Rampogu, S.; Zeb, A.; Lee, K.W. Discovery of novel acetylcholinesterase inhibitors as potential candidates for the treatment of Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 1000. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.L.; Jhan, Y.L.; Chiang, H.M.; Chen, Y.H.; Chen, C.J.; Chang, Y.S. Characterization of chemical constituents with their antioxidant and anti-melanogenesis activities from the roots of Ampelopsis japonica. Nat. Prod. Res. 2021, 36, 1–5. [Google Scholar] [CrossRef]
- Chu, Y.C.; Yang, C.S.; Cheng, M.J.; Fu, S.L.; Chen, J.J. Comparison of various solvent extracts and major bioactive components from unsalt-fried and salt-fried rhizomes of Anemarrhena asphodeloides for antioxidant, anti-α-glucosidase, and anti-acetylcholinesterase activities. Antioxidants 2022, 11, 385. [Google Scholar] [CrossRef]
- Lin, Y.T.; Lin, H.R.; Yang, C.S.; Liaw, C.C.; Sung, P.J.; Kuo, Y.H.; Cheng, M.J.; Chen, J.J. Antioxidant and Anti-α-glucosidase activities of various solvent extracts and major bioactive components from the fruits of Crataegus pinnatifida. Antioxidants 2022, 11, 320. [Google Scholar] [CrossRef]
- Koivikko, R.; Loponen, J.; Honkanen, T.; Jormalainen, V. Contents of soluble, cell-wall-bound and exuded phlorotannins in the brown alga Fucus vesiculosus, with implications on their ecological functions. J. Chem. Ecol. 2005, 31, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Do, Q.D.; Angkawijaya, A.E.; Tran-Nguyen, P.L.; Huynh, L.H.; Soetaredjo, F.E.; Ismadji, S.; Ju, Y.H. Effect of extraction solvent on total phenol content, total flavonoid content, and antioxidant activity of Limnophila aromatica. J. Food Drug Anal. 2014, 22, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Takao, T.; Kitatani, F.; Watanabe, N.; Yagi, A.; Sakata, K. A simple screening method for antioxidants and isolation of several antioxidants produced by marine bacteria from fish and shellfish. Biosci. Biotechnol. Biochem. 1994, 58, 1780–1783. [Google Scholar] [CrossRef] [Green Version]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free. Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Zhang, Q.F.; Zhang, Z.R.; Cheung, H.Y. Antioxidant activity of Rhizoma Smilacis Glabrae extracts and its key constituent-astilbin. Food Chem. 2009, 115, 297–303. [Google Scholar] [CrossRef]
- Alvarez-Jubete, L.; Wijngaard, H.; Arendt, E.K.; Gallagher, E. Polyphenol composition and in vitro antioxidant activity of amaranth, quinoa buckwheat and wheat as affected by sprouting and baking. Food Chem. 2010, 119, 770–778. [Google Scholar] [CrossRef]
- Pujirahayu, N.; Bhattacharjya, D.K.; Suzuki, T.; Katayama, T. α-Glucosidase inhibitory activity of cycloartane-type triterpenes isolated from Indonesian stingless bee Propolis and their structure–activity relationship. Pharmaceuticals 2019, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Omena, C.M.B.; Valentim, I.B.; Guedes, G.D.; Rabelo, L.A.; Mano, C.M.; Bechara, E.J.H.; Sawaya, A.C.H.F.; Trevisan, M.T.S.; da Costa, J.G.; Ferreira, R.C.S.; et al. Antioxidant, anti-acetylcholinesterase and cytotoxic activities of ethanol extracts of peel, pulp and seeds of exotic Brazilian fruits. Food Res. Int. 2012, 49, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Raves, M.L.; Harel, M.; Pang, Y.P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (−)-Huperzine A. Nat. Struct. Biol. 1997, 4, 57–63. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Guillou, C.; Guénard, D.; Argaman, A.; Botti, S.; Badet, B.; Thal, C.; Silman, I.; Sussman, J.L. The complex of a bivalent derivative of galanthamine with Torpedo acetylcholinesterase displays drastic deformation of the active-site gorge: Implications for structure-based drug design. J. Am. Chem. Soc. 2004, 126, 15405–15411. [Google Scholar] [CrossRef]
- Bourne, Y.; Grassi, J.; Bougis, P.E.; Marchot, P. Conformational flexibility of the acetylcholinesterase tetramer suggested by X-ray crystallography. J. Biol. Chem. 1999, 274, 30370–30376. [Google Scholar] [CrossRef] [Green Version]
- Bartolucci, C.; Perola, E.; Pilger, C.; Fels, G.; Lamba, D. Three-dimensional structure of a complex of galanthamine (Nivalin®) with acetylcholinesterase from Torpedo californica: Implications for the design of new anti-Alzheimer drugs. Proteins 2001, 42, 182–191. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cheng, X.; Wang, L.; Wang, S.; Ren, G. A determination of potential α-glucosidase inhibitors from azuki beans (Vigna angularis). Int. J. Mol. Sci. 2011, 12, 6445–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, L.; Chen, W.H.; Park, H.; Ke, Z.; Wang, B. Binding mechanism and synergetic effects of xanthone derivatives as noncompetitive α-glucosidase inhibitors: A theoretical and experimental study. J. Phys. Chem. B 2013, 117, 13464–13471. [Google Scholar] [CrossRef]
- Gholamhoseinian, A.; Moradi, M.N.; Sharifi-Far, F. Screening the methanol extracts of some Iranian plants for acetylcholinesterase inhibitory activity. Res. Pharm. Sci. 2009, 4, 105–112. [Google Scholar]
- Suganthy, N.; Pandima Devi, K. In vitro antioxidant and anti-cholinesterase activities of Rhizophora mucronata. Pharm. Biol. 2016, 54, 118–129. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracting Solvents | Relative Polarity | TPC (mg/g) a (GAE) | TFC (mg/g) b (QE) | Yields (%) c |
|---|---|---|---|---|
| n-Hexane | 0.009 | 15.01 ± 0.44 *** | 56.36 ± 3.86 ** | 0.28 ± 0.08 |
| Chloroform | 0.259 | 20.92 ± 1.47 ** | 94.22 ± 1.34 *** | 0.50 ± 0.08 |
| Dichloromethane | 0.269 | 20.19 ± 1.99 ** | 62.60 ± 3.51 *** | 0.45 ± 0.04 |
| Ethyl acetate | 0.228 | 79.09 ± 8.45 ** | 80.05 ± 7.82 ** | 0.66 ± 0.10 |
| Acetone | 0.355 | 142.89 ± 4.07 *** | 71.72 ± 2.66 *** | 1.03 ± 0.27 |
| Methanol | 0.762 | 95.69 ± 7.80 ** | 5.72 ± 1.13 ** | 1.90 ± 0.50 |
| Ethanol | 0.654 | 95.98 ± 6.68 ** | 22.75 ± 1.24 *** | 4.87 ± 0.03 |
| Water | 1.000 | 47.38 ± 2.18 *** | 4.45 ± 1.21 * | 16.46 ± 0.31 |
| Extracting Solvents | SC50 (μg/mL) a | TE (mM/g) d | ||
|---|---|---|---|---|
| DPPH | ABTS | Superoxide | FRAP | |
| n-Hexane | >200 | >200 | >400 | 26.03 ± 1.90 ** |
| Chloroform | >200 | >200 | >400 | 94.07 ± 7.35 *** |
| Dichloromethane | >200 | >200 | >400 | 104.20 ± 9.18 *** |
| Ethyl acetate | 92.14 ± 8.12 * | 57.45 ± 4.74 * | >400 | 587.11 ± 20.61 *** |
| Acetone | 54.88 ± 4.40 * | 33.88 ± 2.31 ** | >400 | 1001.00 ± 46.17 *** |
| Methanol | 84.73 ± 7.82 * | 53.77 ± 4.65 ** | 290.83 ± 15.23 * | 712.56 ± 18.32 *** |
| Ethanol | 87.12 ± 6.45 * | 64.56 ± 4.80 ** | 307.20 ± 22.39 * | 736.95 ± 14.40 *** |
| Water | 98.54 ± 7.09 * | 99.30 ± 7.02 * | 313.84 ± 20.24 * | 413.34 ± 21.08 *** |
| BHT b | 33.04 ± 2.12 ** | 14.09 ± 0.24 ** | N.A. c | 4257.97 ± 145.90 *** |
| Extracting Solvents | IC50 (μg/mL) a | |
|---|---|---|
| α-Glucosidase | AChE | |
| n-Hexane | 28.43 ± 3.78 * | 83.97 ± 8.90 * |
| Chloroform | 34.16 ± 3.88 * | 91.64 ± 8.77 * |
| Dichloromethane | 28.00 ± 0.14 * | 91.47 ± 26.03 * |
| Ethyl acetate | 12.51 ± 2.42 * | 103.30 ± 2.15 * |
| Acetone | 8.30 ± 0.78 ** | 61.95 ± 5.54 ** |
| Methanol | 19.27 ± 1.12 * | 77.99 ± 5.08 * |
| Ethanol | 11.06 ± 2.07 * | 37.08 ± 7.67 * |
| Water | >400 | 85.82 ± 8.74 * |
| Acarbose b | 335.50 ± 2.14 * | — |
| Chlorogenic acid b | — | 66.69 ± 0.16 * |
| Extracting Solvents | mg/kg | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Total Amount | |
| Water | 6.27 ± 0.58 | 8.43 ± 0.73 | 4.64 ± 0.22 | 22.30 ± 1.62 | 0.68 ± 0.03 | N.D. | N.D. | 42.32 ± 3.18 |
| Methanol | 10.27 ± 1.08 | 12.73 ± 1.42 | 7.24 ± 0.64 | 9.30 ± 0.91 | 1.26 ± 0.08 | 3.01 ± 0.28 | 0.67 ± 0.03 | 44.48 ± 4.44 |
| Ethanol | 7.85 ± 0.52 | 16.43 ± 1.64 | 2.94 ± 0.03 | 3.71 ± 0.28 | 0.86 ± 0.07 | 11.22 ± 1.02 | 0.58 ± 0.04 | 43.59 ± 3.60 |
| Acetone | 8.43 ± 0.63 | 12.43 ± 1.72 | 3.21 ± 0.06 | 6.64 ± 0.74 | 1.21 ± 0.02 | 12.42 ± 1.44 | 3.41 ± 0.18 | 47.75 ± 4.79 |
| Ethyl acetate | 3.79 ± 0.16 | 8.86 ± 0.63 | 1.54 ± 0.08 | 3.62 ± 0.21 | 1.01 ± 0.08 | 2.60 ± 0.11 | 0.52 ± 0.09 | 21.94 ± 1.36 |
| Chloroform | 4.12 ± 0.34 | 2.63 ± 0.08 | 7.83 ± 0.55 | 4.02 ± 0.43 | 0.91 ± 0.06 | 0.70 ± 0.08 | 0.84 ± 0.07 | 21.05 ± 1.61 |
| Dichloromethane | 2.74 ± 0.16 | 1.63 ± 0.06 | 4.62 ± 0.43 | 2.66 ± 0.12 | 0.84 ± 0.08 | 1.83 ± 0.06 | 0.71 ± 0.07 | 15.03 ± 0.98 |
| n-Hexane | 1.43 ± 0.08 | 3.46 ± 0.26 | 1.12 ± 0.07 | 2.63 ± 0.22 | 0.69 ± 0.07 | 6.23 ± 0.52 | N.D. | 15.56 ± 1.22 |
| Compounds | SC50 (μg/mL) a | TE (mM/g) | ||
|---|---|---|---|---|
| DPPH | ABTS | Superoxide | FRAP | |
| 1 | 10.08 ± 3.09 ** | 2.23 ± 0.22 ** | 64.43 ± 7.73 * | 8729.33 ± 424.55 *** |
| 2 | 2.60 ± 0.67 * | 1.45 ± 0.14 ** | 47.40 ± 3.01 * | 28,512.82 ± 43.27 *** |
| 3 | 12.48 ± 3.01 ** | 5.24 ± 0.45 * | N.A. −b | 7912.47 ± 220.08 *** |
| 4 | 3.36 ± 0.58 ** | 3.15 ± 0.49 * | 31.89 ± 2.03 ** | 16,038.26 ± 86.89 *** |
| 5 | >400 | >400 | >400 | 8.67 ± 3.93 * |
| 6 | 13.19 ± 4.78 * | 2.81 ± 0.12 ** | 66.16 ± 5.23 * | 7453.94 ± 60.09 *** |
| 7 | 2.78 ± 0.25 * | 3.78 ± 0.03 ** | 41.76 ± 4.20 * | 13,122.77 ± 182.42 *** |
| BHT b | 36.99 ± 4.54 * | 17.36 ± 3.14 * | N.A. c | 3997.23 ± 144.35 *** |
| Compounds | IC50 (μg/mL) | |
|---|---|---|
| α-Glucosidase | AChE | |
| 1 | 81.78 ± 11.58 ** | 26.35 ± 9.55 ** |
| 2 | >400 | 41.59 ± 7.57 * |
| 3 | 5.81 ± 2.70 ** | 55.04 ± 8.57 ** |
| 4 | 14.39 ± 5.93 ** | 66.34 ± 5.09 ** |
| 5 | 20.38 ± 2.13 * | 11.64 ± 2.69 ** |
| 6 | 28.81 ± 5.65 * | 80.75 ± 9.21 ** |
| 7 | 88.73 ± 10.94 * | 53.38 ± 7.30 * |
| Acarbose a | 334.53 ± 2.22 * | — |
| Chlorogenic acid a | — | 64.42 ± 0.16 * |
| Galanthamine hydrobromide a | — | 0.57 ± 0.09 * |
| Compounds | Affinity (kcal/mol) |
|---|---|
| 1 | −8.5 |
| 2 | −8.3 |
| 3 | −8.1 |
| 4 | −7.8 |
| 5 | −8.7 |
| 6 | −7.6 |
| 7 | −8.1 |
| Chlorogenic acid a | −8.0 |
| Galanthamine a | −9.4 |
| Compounds | Affinity (kcal/mol) |
|---|---|
| 1 | −7.6 |
| 2 | −5.1 |
| 3 | −8.5 |
| 4 | −8.0 |
| 5 | −7.8 |
| 6 | −7.8 |
| 7 | −7.5 |
| Acarbose a | −5.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.-H.; Lin, H.-R.; Yang, C.-S.; Liaw, C.-C.; Wang, I.-C.; Chen, J.-J. Bioactive Components from Ampelopsis japonica with Antioxidant, Anti-α-Glucosidase, and Antiacetylcholinesterase Activities. Antioxidants 2022, 11, 1228. https://doi.org/10.3390/antiox11071228
Liang J-H, Lin H-R, Yang C-S, Liaw C-C, Wang I-C, Chen J-J. Bioactive Components from Ampelopsis japonica with Antioxidant, Anti-α-Glucosidase, and Antiacetylcholinesterase Activities. Antioxidants. 2022; 11(7):1228. https://doi.org/10.3390/antiox11071228
Chicago/Turabian StyleLiang, Jia-Hua, Hsiang-Ru Lin, Chang-Syun Yang, Chia-Ching Liaw, I-Chou Wang, and Jih-Jung Chen. 2022. "Bioactive Components from Ampelopsis japonica with Antioxidant, Anti-α-Glucosidase, and Antiacetylcholinesterase Activities" Antioxidants 11, no. 7: 1228. https://doi.org/10.3390/antiox11071228
APA StyleLiang, J.-H., Lin, H.-R., Yang, C.-S., Liaw, C.-C., Wang, I.-C., & Chen, J.-J. (2022). Bioactive Components from Ampelopsis japonica with Antioxidant, Anti-α-Glucosidase, and Antiacetylcholinesterase Activities. Antioxidants, 11(7), 1228. https://doi.org/10.3390/antiox11071228