
The Breast Cancer Protooncogenes HER2, BRCA1 and BRCA2 and Their Regulation by the iNOS/NOS2 Axis


 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Breast Cancer
2.1. Breast Cancer Protooncogenes
2.1.1. HER2
Transcriptional Regulation of HER2
Epigenetic Regulation of HER2
Post-Transcriptional Regulation of HER2
2.1.2. BRCA1
Wild-Type BRCA1
Mutated BRCA1
Transcriptional Regulation of Wild-Type BRCA1
Epigenetic Regulation of Wild-Type BRCA1
Post-Transcriptional Regulation of BRCA1
2.1.3. BRCA2
Transcriptional Regulation of BRCA2
Epigenetic Regulation of BRCA2
Post Transcriptional Regulation of BRCA2
3. iNOS/NO
3.1. General
3.1.1. Transcriptional Regulation of iNOS
3.1.2. Epigenetic Regulation
3.1.3. Post-Transcriptional Regulation
3.1.4. iNOS-Derived NO Levels in Cells
3.2. iNOS-Induced NO Effects
3.3. iNOS/NO Functions in Cancers
4. iNOS and Breast Cancer Implications
4.1. Triple-Negative Breast Cancer (TNBC)
4.2. ER Breast Cancer
5. iNOS and Breast Cancer Oncogenes HER2, BRCA1, and BRCA2
5.1. iNOS and HER2
5.2. iNOS and BRCA1
5.3. iNOS and BRCA2
6. Implication of iNOS, HER2, and BRCA1/2 in CSC Pathophysiology
7. Bioinformatic Analyses: Correlation between BRCA1/2 Mutations and Genes Involved in the NOS Pathway
8. Discussion and Perspectives
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-OH-dg | Deoxyguanosine |
| A549 | Adenocarcinomic human alveolar basal epithelial cells |
| ADR | Adriamycin |
| Akt | Automated turbidimetric-kinetic |
| AP-1 | Activating protein-1 |
| ATF 2 | Activating transcription factor 2 |
| ATM | Ataxia-telangiectasia mutated |
| ATR | ATM and Rad-3 related |
| β-hCG | Beta human chorionic gonadotropin |
| BACH1 | BTB domain and CNC homology 1 |
| BCL11A | B-cell leukemia/lymphoma |
| BCLC | Breast Cancer Linkage Consortium |
| BRCA1/2 | Breast cancer susceptibility gene |
| Bp | Base pair |
| BRCT | BRCA1 C terminus |
| Ca2+ | Calcium |
| CaM | Calmodomin |
| CAT | Chloramphenicol acetyltransferase |
| CBP | CREB binding protein |
| CCDC98 | Coiled-coil domain-containing protein |
| CD1 | Cluster of differentiation 1 |
| Cdk 2/4/6 | Cyclin-dependent kinase 2/4/6 |
| CEBPBs | CCAAT-enhancer-binding proteins |
| cGMP/PKG | Cyclic guanosine monophosphate/protein kinase G |
| ChIP | Chromatin immunoprecipitation |
| Chk1 | Checkpoint protein 1 |
| CK1 | Casein kinase 1 |
| C-myc | Cellular myc |
| cNOS | Constitutive nitric oxide synthase |
| COX-2 | Cyclooxygenase 2 |
| CREB | cAMP-response element binding protein |
| CREM | cAMP-responsive element modulator |
| CSC | Cancer stem cell |
| CtBP1 | Carboxyl-terminal binding protein 1 |
| CtIP | COUP-TF interacting protein |
| Cys | Cysteine |
| DNA | Deoxyribonucleic acid |
| E1A | Adenovirus early region 1A |
| E2/E3 | Estradiol 2/3 |
| E2F1/4 | E2 transcription factor 1/4 |
| EGCG | Epigallocatechin-3 |
| EGFR | Epidermal growth factor receptor |
| EMSA | Electrophoretic mobility shift assay |
| eNOS/NOS3 | Endothelial nitric oxide synthase |
| EOC | Epithelial ovarian carcinoma |
| ER | Estrogen receptor-negative breast cancer |
| ERα | Estrogen receptor |
| ER-β | Estrogen receptor-β |
| ErbB2 | Erythroblastic oncogene B2 |
| ERK | Extracellular-signal-regulated kinase |
| ERM | Ezrin, radixin, and moesin |
| EMT | Epithelial–mesenchymal transition |
| Ets-1 | Erythroblast transformation specific-1 |
| FISH | Fluorescence in situ hybridization |
| G2/M | Growth 2/mitosis |
| GABP-α/β | GA binding protein |
| GAPDH | Glyceraldehyde-3-phosphate-dehydrogenase |
| GATA-1 | GATA-binding factor-1/erythroid transcription factor |
| H3K9 | Histone H3 lysine 9 |
| HCC 38 | Hepatocellular carcinoma |
| HDAC | Histone deacetylases |
| Hdm2 | Human double minute 2 |
| HER2/neu | Human epidermal growth factor receptor 2 |
| HR | Homologous recombination |
| HT-29 | Human colon adenocarcinoma cell line |
| ID ¼ | Inhibitor of differentiation 1/4 |
| IFN-γ | Interferon gamma |
| IHC | Immunohistochemistry |
| IL-1/2/4/8 | Interleukin 1/2/4/8 |
| IL-1B | Interleukin 1 beta |
| iNOS | Inducible nitric oxide synthase |
| KRAS | Kristen rat sarcoma viral oncogene homolog |
| L. donovani | Leishmania donovani |
| lncRNA | Long non-coding RNA |
| L-NMMA | NG-monomethyl-L-arginine monoacetate |
| LOH | Loss of heterozygosity |
| MAPK | Mitogen-activated protein kinase |
| MCF-7 | Michigan Cancer Foundation-7 |
| MEK | Mitogen-activated extracellular signal-regulated kinase |
| miRNA | MicroRNA |
| MMC | Mitomycin C |
| MMP 0/2/9 | Matrix metalloproteinase 0/2/9 |
| mRNA | Messenger RNA |
| ncRNA | non-coding RNA |
| NF-kB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NFAT-5/C4 | Nuclear factor of activated T cells 5/C4 |
| NHEJ | Nonhomologous end-joining |
| NLS | Nuclear localization sequences |
| nNOS/NOS1 | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| NOSIP | Nitric oxide synthase interacting protein |
| NOS1AP | Nitric oxide synthase 1 adaptor protein |
| NOTCH-1 | NOTCH homolog 1 |
| Oct | Octamer factor |
| PANC-1 | Human pancreatic cancer cell line |
| p53 | Tumor suppressor p53 |
| PALB2 | Partner and localizer of BRCA2 |
| PARP | Poly(ADP-ribose) polymerase |
| PCAT-1 | Prostate cancer associated transcript 1 |
| PCR | Polymerase chain reaction |
| PDAC | Pancreatic adenocarcinoma |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphoinositide 3 kinase |
| qPCR | Quantitative polymerase chain reaction |
| Ras | Rat Sarcoma Virus |
| RB | Retinoblastoma protein |
| Rb-E2F | Retinoblastoma protein family-activator E2F |
| RBL2 | RB Transcriptional corepressor like 2 |
| RING | Really interesting new gene |
| RKO | Rectal carcinoma cell line |
| RNA | Ribonucleic acid |
| RNAseq | RNA sequence |
| RNS | Reactive nitrogen species |
| RR | Ribonucleotide reductase |
| RT-PCR | Reverse transcription PCR |
| RUNX1 | Runt-related transcription factor 1 |
| S phase | Synthesis phase |
| S100B | S100 calcium-binding protein B |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| Slug | Snail 1/2 |
| Sp-1 | Specificity protein 1 |
| SNAP | S-nitroso-N-acetylpenicillamine |
| STAT-1α | Signal transducer and activator of transcription 1 |
| SV40 | Simian vacuolating virus 40 |
| TCGA | The Cancer Genome Atlas |
| TGFBRII | Transforming growth factor beta receptor type II gene |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| TNF | Tumor necrosis factor |
| TNF-α | Tumor necrosis factor alpha |
| Trp53 | Transformation related protein 53 |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| USF | Upstream stimulatory factor |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
References
- Garcea, G.; Dennison, A.R.; Steward, W.P.; Berry, D.P. Role of Inflammation in Pancreatic Carcinogenesis and the Implications for Future Therapy. Pancreatology 2005, 5, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Singh, R.; Sen, S.; Chaudhuri, G. Dual Role of Nitric Oxide in Cancer Biology. In Nitric Oxide (NO) and Cancer: Prognosis, Prevention, and Therapy; Bonavida, B., Ed.; Cancer Drug Discovery and Development; Springer: New York, NY, USA, 2010; pp. 39–57. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The Dual Role of INOS in Cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kielbik, M.; Szulc-Kielbik, I.; Klink, M. The Potential Role of INOS in Ovarian Cancer Progression and Chemoresistance. Int. J. Mol. Sci. 2019, 20, 1751. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of INOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, S.; Falzone, L.; Basile, M.S.; Candido, S.; Libra, M. Nitric Oxide in Hematological Cancers: Partner or Rival? Antioxid. Redox Signal. 2021, 34, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, A.; Urbanski, S.J.; McCafferty, D.-M. Induction of Inducible Nitric Oxide Synthase: A Protective Mechanism in Colitis-Induced Adenocarcinoma. Carcinogenesis 2007, 28, 1122–1130. [Google Scholar] [CrossRef]
- Wink, D.A.; Ridnour, L.A.; Hussain, S.P.; Harris, C.C. The Reemergence of Nitric Oxide and Cancer. Nitric Oxide 2008, 19, 65–67. [Google Scholar] [CrossRef]
- Xie, K.; Huang, S.; Dong, Z.; Juang, S.H.; Gutman, M.; Xie, Q.W.; Nathan, C.; Fidler, I.J. Transfection with the Inducible Nitric Oxide Synthase Gene Suppresses Tumorigenicity and Abrogates Metastasis by K-1735 Murine Melanoma Cells. J. Exp. Med. 1995, 181, 1333–1343. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The Role of Nitric Oxide in Cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The Yin and Yang of Nitric Oxide in Cancer Progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef]
- Vane, J.R.; Mitchell, J.A.; Appleton, I.; Tomlinson, A.; Bishop-Bailey, D.; Croxtall, J.; Willoughby, D.A. Inducible Isoforms of Cyclooxygenase and Nitric-Oxide Synthase in Inflammation. Proc. Natl. Acad. Sci. USA 1994, 91, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, V.A. Nitric Oxide–Dependent Downregulation of BRCA1 Expression Promotes Genetic Instability. Cancer Res. 2013, 73, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of Inducible Nitric Oxide Synthase (INOS) Expression on Triple Negative Breast Cancer Outcome and Activation of EGFR and ERK Signaling Pathways. Oncotarget 2017, 8, 80568–80588. [Google Scholar] [CrossRef]
- Chen, D.-M.; Shi, Z.-Q.; Tan, L.-L.; Chen, Y.-P.; Li, C.-Q.; Wang, Q.; Li, H.; Zhang, M.-L.; Song, J.-P.; Xu, Q.; et al. Short-Hairpin RNA-Guided Single Gene Knockdown Reverses Triple-Negative Breast Cancer. bioRxiv 2018, 418764. [Google Scholar] [CrossRef]
- Mishra, D.; Patel, V.; Banerjee, D. Nitric Oxide and S-Nitrosylation in Cancers: Emphasis on Breast Cancer. Breast Cancer 2020, 14, 1178223419882688. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, L.L.; Miles, D.W.; Happerfield, L.; Bobrow, L.G.; Knowles, R.G.; Moncada, S. Nitric Oxide Synthase Activity in Human Breast Cancer. Br. J. Cancer 1995, 72, 41–44. [Google Scholar] [CrossRef]
- Thomsen, L.L.; Lawton, F.G.; Knowles, R.G.; Beesley, J.E.; Riveros-Moreno, V.; Moncada, S. Nitric Oxide Synthase Activity in Human Gynecological Cancer. Cancer Res. 1994, 54, 1352–1354. [Google Scholar]
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of Cancer Incidence, Mortality, and Prevalence across Five Continents: Defining Priorities to Reduce Cancer Disparities in Different Geographic Regions of the World. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef]
- Polyak, K. Breast Cancer: Origins and Evolution. J. Clin. Investig. 2007, 117, 3155–3163. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.; Wilson, P.; Tripathy, D. Oncogenes and Tumor Suppressor Genes in Breast Cancer: Potential Diagnostic and Therapeutic Applications. Oncologist 2004, 9, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Torry, D.S.; Cooper, G.M. Proto-Oncogenes in Development and Cancer. Am. J. Reprod. Immunol. 1991, 25, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayanajois, S.; Villalba, S.; Jianchao, L.; Lin, G.M. Design, Synthesis, and Docking Studies of Peptidomimetics Based on HER2–Herceptin Binding Site with Potential Antiproliferative Activity against Breast Cancer Cell Lines. Chem. Biol. Drug Des. 2009, 74, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-J.; Lee, K.-J.; Yu, H.W.; Chen, C.-Y.; Hsu, C.-H.; Chen, H.-Y.; Tsai, F.-J.; Chen, C.Y.-C. Structure-Based and Ligand-Based Drug Design for HER 2 Receptor. J. Biomol. Struct. Dyn. 2010, 28, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the HER-2/Neu Oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/Neu Proto-Oncogene in Human Breast and Ovarian Cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Tandon, A.K.; Clark, G.M.; Chamness, G.C.; Ullrich, A.; McGuire, W.L. HER-2/Neu Oncogene Protein and Prognosis in Breast Cancer. J. Clin. Oncol. 1989, 7, 1120–1128. [Google Scholar] [CrossRef]
- Seshadri, R.; Firgaira, F.A.; Horsfall, D.J.; McCaul, K.; Setlur, V.; Kitchen, P. Clinical Significance of HER-2/Neu Oncogene Amplification in Primary Breast Cancer. The South Australian Breast Cancer Study Group. J. Clin. Oncol. 1993, 11, 1936–1942. [Google Scholar] [CrossRef]
- Tsai, C.-M.; Chang, K.-T.; Perng, R.-P.; Mitsudomi, T.; Chen, M.-H.; Kadoyama, C.; Gazdar, A.F. Correlation of Intrinsic Chemoresistance of Non-Small-Cell Lung Cancer Cell Lines with HER-2/Neu Gene Expression but Not with Ras Gene Mutations. J. Natl. Cancer Inst. 1993, 85, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Huo, L.; Yang, J.-Y.; Xia, W.; Wei, Y.; Liao, Y.; Chang, C.-J.; Yang, Y.; Lai, C.-C.; Lee, D.-F.; et al. Down-Regulation of Myeloid Cell Leukemia-1 through Inhibiting Erk/Pin 1 Pathway by Sorafenib Facilitates Chemosensitization in Breast Cancer. Cancer Res. 2008, 68, 6109–6117. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.J.-C.; Hua, C.-H.; Wan, L.; Lin, Y.-J.; Lai, M.-T.; Tseng, H.-C.; Jinawath, N.; Tsai, M.-H.; Chang, N.-W.; Lin, C.-F.; et al. Functional Genomic Analysis Identified Epidermal Growth Factor Receptor Activation as the Most Common Genetic Event in Oral Squamous Cell Carcinoma. Cancer Res. 2009, 69, 2568–2576. [Google Scholar] [CrossRef]
- van de Vijver, M.J.; Peterse, J.L.; Mooi, W.J.; Wisman, P.; Lomans, J.; Dalesio, O.; Nusse, R. Neu-Protein Overexpression in Breast Cancer. N. Engl. J. Med. 1988, 319, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Hung, M. Regulation of her2/neu gene-expression (Review). Oncol. Rep. 1995, 2, 497–503. [Google Scholar] [CrossRef]
- Chen, H.; Hung, M.-C. Involvement of Co-Activator P300 in the Transcriptional Regulation of the HER-2/Neu Gene. J. Biol. Chem. 1997, 272, 6101–6104. [Google Scholar] [CrossRef]
- Dyson, N.; Harlow, E. Adenovirus E1A Targets Key Regulators of Cell Proliferation. Cancer Surv. 1992, 12, 161–195. [Google Scholar]
- Wang, H.G.; Rikitake, Y.; Carter, M.C.; Yaciuk, P.; Abraham, S.E.; Zerler, B.; Moran, E. Identification of Specific Adenovirus E1A N-Terminal Residues Critical to the Binding of Cellular Proteins and to the Control of Cell Growth. J. Virol. 1993, 67, 476–488. [Google Scholar] [CrossRef]
- Janknecht, R.; Hunter, T. Transcriptional Control: Versatile Molecular Glue. Curr. Biol. 1996, 6, 951–954. [Google Scholar] [CrossRef]
- Yu, D.; Suen, T.C.; Yan, D.H.; Chang, L.S.; Hung, M.C. Transcriptional Repression of the Neu Protooncogene by the Adenovirus 5 E1A Gene Products. Proc. Natl. Acad. Sci. USA 1990, 87, 4499–4503. [Google Scholar] [CrossRef]
- Matin, A.; Hung, M.C. Negative Regulation of the Neu Promoter by the SV40 Large T Antigen. Cell Growth Differ. 1993, 4, 1051–1056. [Google Scholar] [PubMed]
- Hung, M.-C.; Matin, A.; Zhang, Y.; Xing, X.; Sorgi, F.; Huang, L.; Yu, D. HER-2/Neu-Targeting Gene Therapy—A Review. Gene 1995, 159, 65–71. [Google Scholar] [CrossRef]
- Hudson, L.G.; Ertl, A.P.; Gill, G.N. Structure and Inducible Regulation of the Human C-Erb B2/Neu Promoter. J. Biol. Chem. 1990, 265, 4389–4393. [Google Scholar] [CrossRef]
- Suen, T.C.; Hung, M.C. Multiple Cis- and Trans-Acting Elements Involved in Regulation of the Neu Gene. Mol. Cell. Biol. 1990, 10, 6306–6315. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.H.; Chang, L.S.; Hung, M.C. Repressed Expression of the HER-2/c-ErbB-2 Proto-Oncogene by the Adenovirus E1a Gene Products. Oncogene 1991, 6, 343–345. [Google Scholar]
- White, M.R.; Hung, M.C. Cloning and Characterization of the Mouse Neu Promoter. Oncogene 1992, 7, 677–683. [Google Scholar]
- Suen, T.C.; Hung, M.C. C-Myc Reverses Neu-Induced Transformed Morphology by Transcriptional Repression. Mol. Cell. Biol. 1991, 11, 354–362. [Google Scholar] [CrossRef]
- Miller, S.; Hung, M. Her2 neu overexpression counteracts the growth effects of C-Myc in breast-cancer cells. Int. J. Oncol. 1994, 4, 965–969. [Google Scholar] [CrossRef]
- Pianetti, S.; Guo, S.; Kavanagh, K.T.; Sonenshein, G.E. Green Tea Polyphenol Epigallocatechin-3 Gallate Inhibits Her-2/Neu Signaling, Proliferation, and Transformed Phenotype of Breast Cancer Cells1. Cancer Res. 2002, 62, 652–655. [Google Scholar]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the Neu Protooncogene in the Mammary Epithelium of Transgenic Mice Induces Metastatic Disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef]
- Dreosti, I.E.; Wargovich, M.J.; Yang, C.S. Inhibition of Carcinogenesis by Tea: The Evidence from Experimental Studies. Crit. Rev. Food Sci. Nutr. 1997, 37, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Pianetti, S.; Arsura, M.; Romieu-Mourez, R.; Coffey, R.J.; Sonenshein, G.E. Her-2/Neu Overexpression Induces NF-ΚB via a PI3-Kinase/Akt Pathway Involving Calpain-Mediated Degradation of IκB-α That Can Be Inhibited by the Tumor Suppressor PTEN. Oncogene 2001, 20, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Berchuck, A.; Kamel, A.; Whitaker, R.; Kerns, B.; Olt, G.; Kinney, R.; Soper, J.T.; Dodge, R.; Clarke-Pearson, D.L.; Marks, P.; et al. Overexpression of HER-2/Neu Is Associated with Poor Survival in Advanced Epithelial Ovarian Cancer1. Cancer Res. 1990, 50, 4087–4091. [Google Scholar] [PubMed]
- Hattori, M.; Sakamoto, H.; Satoh, K.; Yamamoto, T. DNA Demethylase Is Expressed in Ovarian Cancers and the Expression Correlates with Demethylation of CpG Sites in the Promoter Region of C-ErbB-2 and Survivin Genes. Cancer Lett. 2001, 169, 155–164. [Google Scholar] [CrossRef]
- Ishii, S.; Imamoto, F.; Yamanashi, Y.; Toyoshima, K.; Yamamoto, T. Characterization of the Promoter Region of the Human C-ErbB-2 Protooncogene. Proc. Natl. Acad. Sci. USA 1987, 84, 4374–4378. [Google Scholar] [CrossRef]
- Nezu, M.; Sasaki, H.; Kuwahara, Y.; Ochiya, T.; Yamada, Y.; Sakamoto, H.; Tashiro, H.; Yamazaki, M.; Ikeuchi, T.; Saito, Y.; et al. Identification of a Novel Promoter and Exons of the C-ERBB-2Gene. Biochem. Biophys. Res. Commun. 1999, 258, 499–505. [Google Scholar] [CrossRef]
- Yang, G.; Cai, K.Q.; Thompson-Lanza, J.A.; Bast, R.C.; Liu, J. Inhibition of Breast and Ovarian Tumor Growth through Multiple Signaling Pathways by Using Retrovirus-Mediated Small Interfering RNA against Her-2/Neu Gene Expression. J. Biol. Chem. 2004, 279, 4339–4345. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Choudhury, A.; Charo, J.; Parapuram, S.K.; Hunt, R.C.; Hunt, D.M.; Seliger, B.; Kiessling, R. Small Interfering RNA (SiRNA) Inhibits the Expression of the Her2/Neu Gene, Upregulates HLA Class I and Induces Apoptosis of Her2/Neu Positive Tumor Cell Lines. Int. J. Cancer 2004, 108, 71–77. [Google Scholar] [CrossRef]
- Sengodan, S.K.; Sreelatha, K.H.; Nadhan, R.; Srinivas, P. Regulation of Epithelial to Mesenchymal Transition by BRCA1 in Breast Cancer. Crit. Rev. Oncol./Hematol. 2018, 123, 74–82. [Google Scholar] [CrossRef]
- Diaz-Cruz, E.S.; Cabrera, M.C.; Nakles, R.; Rutstein, B.H.; Furth, P.A. BRCA1 Deficient Mouse Models to Study Pathogenesis and Therapy of Triple Negative Breast Cancer. Breast Dis. 2011, 32, 85–97. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, T.C.; Veeck, J.; de Hoon, J.P.J.; van Engeland, M.; Tjan-Heijnen, V.C. Characteristics of Triple-Negative Breast Cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-X. BRCA1: Cell Cycle Checkpoint, Genetic Instability, DNA Damage Response and Cancer Evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Mullan, P.B.; Quinn, J.E.; Harkin, D.P. The Role of BRCA1 in Transcriptional Regulation and Cell Cycle Control. Oncogene 2006, 25, 5854–5863. [Google Scholar] [CrossRef]
- Di, L.-J.; Fernandez, A.G.; De Siervi, A.; Longo, D.L.; Gardner, K. Transcriptional Regulation of BRCA1 Expression by a Metabolic Switch. Nat. Struct. Mol. Biol. 2010, 17, 1406–1413. [Google Scholar] [CrossRef]
- Paull, T.T.; Cortez, D.; Bowers, B.; Elledge, S.J.; Gellert, M. Direct DNA Binding by Brca1. Proc. Natl. Acad. Sci. USA 2001, 98, 6086–6091. [Google Scholar] [CrossRef]
- Mueller, C.R.; Roskelley, C.D. Regulation of BRCA1 Expression and Its Relationship to Sporadic Breast Cancer. Breast Cancer Res. 2002, 5, 45. [Google Scholar] [CrossRef]
- Yarden, R.I.; Pardo-Reoyo, S.; Sgagias, M.; Cowan, K.H.; Brody, L.C. BRCA1 Regulates the G2/M Checkpoint by Activating Chk1 Kinase upon DNA Damage. Nat. Genet. 2002, 30, 285–289. [Google Scholar] [CrossRef]
- Ye, Q.; Hu, Y.-F.; Zhong, H.; Nye, A.C.; Belmont, A.S.; Li, R. BRCA1-Induced Large-Scale Chromatin Unfolding and Allele-Specific Effects of Cancer-Predisposing Mutations. J. Cell Biol. 2001, 155, 911–922. [Google Scholar] [CrossRef]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular Basis for Estrogen Receptor α Deficiency in BRCA1-Linked Breast Cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef]
- Clucas, J.; Valderrama, F. ERM Proteins in Cancer Progression. J. Cell Sci. 2015, 128, 1253. [Google Scholar] [CrossRef] [PubMed]
- Coene, E.D.; Gadelha, C.; White, N.; Malhas, A.; Thomas, B.; Shaw, M.; Vaux, D.J. A Novel Role for BRCA1 in Regulating Breast Cancer Cell Spreading and Motility. J. Cell Biol. 2011, 192, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, A.; Liu, W.; Dekhil, H.; Kassab, A.; Aloyz, R.; Foulkes, W.D.; Al Moustafa, A.-E. BRCA1 Mutations Contribute to Cell Motility and Invasion by Affecting Its Main Regulators. Cell Cycle 2008, 7, 3781–3783. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lindeman, G.J.; Visvader, J.E. Cell Fate Takes a Slug in BRCA1-Associated Breast Cancer. Breast Cancer Res. 2011, 13, 306. [Google Scholar] [CrossRef]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic Predisposition Directs Breast Cancer Phenotype by Dictating Progenitor Cell Fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef]
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.-W.; Harris, C.C.; Ried, T.; Deng, C.-X. Centrosome Amplification and a Defective G2–M Cell Cycle Checkpoint Induce Genetic Instability in BRCA1 Exon 11 Isoform–Deficient Cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Irwin, J.J. Community Benchmarks for Virtual Screening. J. Comput. Aided Mol. Des. 2008, 22, 193–199. [Google Scholar] [CrossRef]
- Clark, S.L.; Rodriguez, A.M.; Snyder, R.R.; Hankins, G.D.V.; Boehning, D. Structure-function of the tumor suppressor BRCA1. Comput. Struct. Biotechnol. J. 2012, 1, e201204005. [Google Scholar] [CrossRef]
- Greenberg, R.A. BRCA1, Everything but the RING? Science 2011, 334, 459–460. [Google Scholar] [CrossRef]
- Shakya, R.; Reid, L.J.; Reczek, C.R.; Cole, F.; Egli, D.; Lin, C.-S.; deRooij, D.G.; Hirsch, S.; Ravi, K.; Hicks, J.B.; et al. BRCA1 Tumor Suppression Depends on BRCT Phosphoprotein Binding, But Not Its E3 Ligase Activity. Science 2011, 334, 525–528. [Google Scholar] [CrossRef]
- Deng, C.-X.; Brodie, S.G. Roles of BRCA1 and Its Interacting Proteins. BioEssays 2000, 22, 728–737. [Google Scholar] [CrossRef]
- Mohammad, D.H.; Yaffe, M.B. 14-3-3 Proteins, FHA Domains and BRCT Domains in the DNA Damage Response. DNA Repair 2009, 8, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires CtIP, a Phosphorylation-Dependent Binding Partner of BRCA1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Huang, J.; Chen, J. CCDC98 Is a BRCA1-BRCT Domain–Binding Protein Involved in the DNA Damage Response. Nat. Struct. Mol. Biol. 2007, 14, 710–715. [Google Scholar] [CrossRef]
- Lane, T.F.; Deng, C.; Elson, A.; Lyu, M.S.; Kozak, C.A.; Leder, P. Expression of Brca1 Is Associated with Terminal Differentiation of Ectodermally and Mesodermally Derived Tissues in Mice. Genes Dev. 1995, 9, 2712–2722. [Google Scholar] [CrossRef]
- Marquis, S.T.; Rajan, J.V.; Wynshaw-Boris, A.; Xu, J.; Yin, G.-Y.; Abel, K.J.; Weber, B.L.; Chodosh, L.A. The Developmental Pattern of Brca1 Expression Implies a Role in Differentiation of the Breast and Other Tissues. Nat. Genet. 1995, 11, 17–26. [Google Scholar] [CrossRef]
- Gorski, J.J.; Kennedy, R.D.; Hosey, A.M.; Harkin, D.P. The Complex Relationship between BRCA1 and ERα in Hereditary Breast Cancer. Clin. Cancer Res. 2009, 15, 1514–1518. [Google Scholar] [CrossRef]
- Jeffy, B.D.; Hockings, J.K.; Kemp, M.Q.; Morgan, S.S.; Hager, J.A.; Beliakoff, J.; Whitesell, L.J.; Bowden, G.T.; Romagnolo, D.F. An Estrogen Receptor-α/P300 Complex Activates the BRCA-1 Promoter at an AP-1 Site That Binds Jun/Fos Transcription Factors: Repressive Effects of P53 on BRCA-1 Transcription. Neoplasia 2005, 7, 873–882. [Google Scholar] [CrossRef]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.-Q.; Meng, Q.; Wang, J.-A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. P300 Modulates the BRCA1 Inhibition of Estrogen Receptor Activity. Cancer Res. 2002, 62, 141–151. [Google Scholar]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.-C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic Heterogeneity and Penetrance Analysis of the BRCA1 and BRCA2 Genes in Breast Cancer Families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Bishop, D.T.; Ford, D.; Crockford, G.P. Genetic Linkage Analysis in Familial Breast and Ovarian Cancer: Results from 214 Families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1993, 52, 678–701. [Google Scholar] [PubMed]
- Narod, S.A.; Ford, D.; Devilee, P.; Barkardottir, R.B.; Lynch, H.T.; Smith, S.A.; Ponder, B.A.J.; Weber, B.L.; Garber, J.E.; Birch, J.M.; et al. An Evaluation of Genetic Heterogeneity in 145 Breast-Ovarian Cancer Families. Am. J. Hum. Genet. 1995, 56, 254–264. [Google Scholar]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Gatekeepers and Caretakers. Nature 1997, 386, 761–763. [Google Scholar] [CrossRef]
- Crook, T.; Crossland, S.; Crompton, M.R.; Osin, P.; Gusterson, B.A. P53 Mutations in BRCA1-Associated Familial Breast Cancer. Lancet 1997, 350, 638–639. [Google Scholar] [CrossRef]
- Crook, T.; Brooks, L.A.; Crossland, S.; Osin, P.; Barker, K.T.; Waller, J.; Philp, E.; Smith, P.D.; Yulug, I.; Peto, J.; et al. P53 Mutation with Frequent Novel Codons but Not a Mutator Phenotype in BRCA1- and BRCA2-Associated Breast Tumours. Oncogene 1998, 17, 1681–1689. [Google Scholar] [CrossRef]
- Palacios, J.; Honrado, E.; Osorio, A.; Cazorla, A.; Sarrió, D.; Barroso, A.; Rodríguez, S.; Cigudosa, J.C.; Diez, O.; Alonso, C.; et al. Phenotypic Characterization of BRCA1 and BRCA2 Tumors Based in a Tissue Microarray Study with 37 Immunohistochemical Markers. Breast Cancer Res. Treat. 2005, 90, 5–14. [Google Scholar] [CrossRef]
- Aaltonen, K.; Blomqvist, C.; Amini, R.-M.; Eerola, H.; Aittomäki, K.; Heikkilä, P.; Nevanlinna, H. Familial Breast Cancers without Mutations in BRCA1 or BRCA2 Have Low Cyclin E and High Cyclin D1 in Contrast to Cancers in BRCA Mutation Carriers. Clin. Cancer Res. 2008, 14, 1976–1983. [Google Scholar] [CrossRef]
- Sengodan, S.K.; Nadhan, R.; Nair, R.S.; Hemalatha, S.K.; Somasundaram, V.; Sushama, R.R.; Rajan, A.; Latha, N.R.; Varghese, G.R.; Thankappan, R.K.; et al. BRCA1 Regulation on β-HCG: A Mechanism for Tumorigenicity in BRCA1 Defective Breast Cancer. Oncogenesis 2017, 6, e376. [Google Scholar] [CrossRef] [PubMed]
- Stenman, U.-H.; Alfthan, H.; Hotakainen, K. Human Chorionic Gonadotropin in Cancer. Clin. Biochem. 2004, 37, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, P.; Grankvist, K.; Wulff, M.; Chen, T.; Johansson, R.; Schock, H.; Lenner, P.; Hallmans, G.; Lehtinen, M.; Kaaks, R.; et al. Human Chorionic Gonadotropin in Pregnancy and Maternal Risk of Breast Cancer. Cancer Res. 2010, 70, 6779–6786. [Google Scholar] [CrossRef]
- Gillott, D.J.; Iles, R.K.; Chard, T. The Effects of Beta-Human Chorionic Gonadotrophin on the in Vitro Growth of Bladder Cancer Cell Lines. Br. J. Cancer 1996, 73, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.; Gunderson, S.I.; Andrusiewicz, M.; Burczynska, B.; Szczerba, A.; Jarmolowski, A.; Nowak-Markwitz, E.; Warchol, J.B. Reduction of Human Chorionic Gonadotropin Beta Subunit Expression by Modified U1 SnRNA Caused Apoptosis in Cervical Cancer Cells. Mol. Cancer 2008, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, G.; Schauer, I.G.; Yang, G.; Mercado-Uribe, I.; Yang, F.; Zhang, S.; He, Y.; Liu, J. Overexpression of the β Subunit of Human Chorionic Gonadotropin Promotes the Transformation of Human Ovarian Epithelial Cells and Ovarian Tumorigenesis. Am. J. Pathol. 2011, 179, 1385–1393. [Google Scholar] [CrossRef]
- Wan, H.; Versnel, M.A.; Leijten, L.M.E.; van Helden-Meeuwsen, C.G.; Fekkes, D.; Leenen, P.J.M.; Khan, N.A.; Benner, R.; Kiekens, R.C.M. Chorionic Gonadotropin Induces Dendritic Cells to Express a Tolerogenic Phenotype. J. Leukoc. Biol. 2008, 83, 894–901. [Google Scholar] [CrossRef]
- Tanizaki, J.; Ercan, D.; Capelletti, M.; Dodge, M.; Xu, C.; Bahcall, M.; Tricker, E.M.; Butaney, M.; Calles, A.; Sholl, L.M.; et al. Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res. 2015, 75, 3139–3146. [Google Scholar] [CrossRef]
- Deng, Y.; Deng, H.; Liu, J.; Han, G.; Malkoski, S.; Liu, B.; Zhao, R.; Wang, X.-J.; Zhang, Q. Transcriptional Down-Regulation of Brca1 and E-Cadherin by CtBP1 in Breast Cancer. Mol. Carcinog. 2012, 51, 500–507. [Google Scholar] [CrossRef]
- Wang, A.; Schneider-Broussard, R.; Kumar, A.P.; MacLeod, M.C.; Johnson, D.G. Regulation of BRCA1 Expression by the Rb-E2F Pathway. J. Biol. Chem. 2000, 275, 4532–4536. [Google Scholar] [CrossRef]
- Tao, Y.; Kassatly, R.F.; Cress, W.D.; Horowitz, J.M. Subunit Composition Determines E2F DNA-Binding Site Specificity. Mol. Cell. Biol. 1997, 17, 6994–7007. [Google Scholar] [CrossRef] [PubMed]
- Almasan, A.; Yin, Y.; Kelly, R.E.; Lee, E.Y.; Bradley, A.; Li, W.; Bertino, J.R.; Wahl, G.M. Deficiency of Retinoblastoma Protein Leads to Inappropriate S-Phase Entry, Activation of E2F-Responsive Genes, and Apoptosis. Proc. Natl. Acad. Sci. USA 1995, 92, 5436–5440. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.E.; Sah, V.P.; Williams, B.O.; Mäkelä, T.P.; Weinberg, R.A.; Jacks, T. Altered Cell Cycle Kinetics, Gene Expression, and G1 Restriction Point Regulation in Rb-Deficient Fibroblasts. Mol. Cell. Biol. 1996, 16, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Hurford, R.K.; Cobrinik, D.; Lee, M.H.; Dyson, N. PRB and P107/P130 Are Required for the Regulated Expression of Different Sets of E2F Responsive Genes. Genes Dev. 1997, 11, 1447–1463. [Google Scholar] [CrossRef]
- Rice, J.C.; Massey-Brown, K.S.; Futscher, B.W. Aberrant Methylation of the BRCA1 CpG Island Promoter Is Associated with Decreased BRCA1 MRNA in Sporadic Breast Cancer Cells. Oncogene 1998, 17, 1807–1812. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A Gene Hypermethylation Profile of Human Cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar]
- Jones, P.A.; Laird, P.W. Cancer-Epigenetics Comes of Age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA Hypermethylation in Tumorigenesis: Epigenetics Joins Genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Lu, Y.; Chu, A.; Turker, M.S.; Glazer, P.M. Hypoxia-Induced Epigenetic Regulation and Silencing of the BRCA1 Promoter. Mol. Cell. Biol. 2011, 31, 3339–3350. [Google Scholar] [CrossRef]
- Bosviel, R.; Durif, J.; Déchelotte, P.; Bignon, Y.-J.; Bernard-Gallon, D. Epigenetic Modulation of BRCA1 and BRCA2 Gene Expression by Equol in Breast Cancer Cell Lines. Br. J. Nutr. 2012, 108, 1187–1193. [Google Scholar] [CrossRef]
- Garcia, A.I.; Buisson, M.; Bertrand, P.; Rimokh, R.; Rouleau, E.; Lopez, B.S.; Lidereau, R.; Mikaélian, I.; Mazoyer, S. Down-regulation of BRCA1 Expression by MiR-146a and MiR-146b-5p in Triple Negative Sporadic Breast Cancers. EMBO Mol. Med. 2011, 3, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, J.R.; Wang, W.-X.; Hébert, S.S.; Wilfred, B.R.; Mao, G.; Nelson, P.T. The MiR-15/107 Group of MicroRNA Genes: Evolutionary Biology, Cellular Functions, and Roles in Human Diseases. J. Mol. Biol. 2010, 402, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Quann, K.; Jing, Y.; Rigoutsos, I. Post-Transcriptional Regulation of BRCA1 through Its Coding Sequence by the MiR-15/107 Group of MiRNAs. Front. Genet. 2015, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian MicroRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a Big Role in Gene Regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of Post-Transcriptional Regulation by MicroRNAs: Are the Answers in Sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Simard, J.; Rommens, J.; Couch, F.; Shattuck-Eidens, D.; Neuhausen, S.; Merajver, S.; Thorlacius, S.; Offit, K.; Stoppa-Lyonnet, D.; et al. The Complete BRCA2 Gene and Mutations in Chromosome 13q-Linked Kindreds. Nat. Genet. 1996, 12, 333–337. [Google Scholar] [CrossRef]
- Bertwistle, D.; Swift, S.; Marston, N.J.; Jackson, L.E.; Crossland, S.; Crompton, M.R.; Marshall, C.J.; Ashworth, A. Nuclear Location and Cell Cycle Regulation of the BRCA2 Protein1. Cancer Res. 1997, 57, 5485–5488. [Google Scholar]
- Milner, J.; Ponder, B.; Hughes-Davies, L.; Seltmann, M.; Kouzarides, T. Transcriptional Activation Functions in BRCA2. Nature 1997, 386, 772–773. [Google Scholar] [CrossRef]
- Chapman, M.S.; Verma, I.M. Transcriptional Activation by BRCA1. Nature 1996, 382, 678–679. [Google Scholar] [CrossRef]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic Lethality and Radiation Hypersensitivity Mediated by Rad51 in Mice Lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, P.-L.; Riley, D.J.; Lee, W.-H.; Allred, D.C.; Osborne, C.K. Response: Location of BRCA1 in Human Breast and Ovarian Cancer Cells. Science 1996, 272, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Bièche, I.; Noguès, C.; Lidereau, R. Overexpression of BRCA2 Gene in Sporadic Breast Tumours. Oncogene 1999, 18, 5232–5238. [Google Scholar] [CrossRef] [PubMed]
- Callens, N.; Dumont, M.; Begue, A.; Lint, C.; Baert, J.-L.; Simard, J.; Launoit, Y. Genomic Organization and Expression of the Mouse Brca2 Gene. Mamm. Genome 2002, 13, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Callens, N.; Baert, J.-L.; Monté, D.; Sunesen, M.; Van Lint, C.; de Launoit, Y. Transcriptional Regulation of the Murine Brca2 Gene by CREB/ATF Transcription Factors. Biochem. Biophys. Res. Commun. 2003, 312, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Jiang, S.-W.; Couch, F.J. P53 Mediates Repression of the BRCA2 Promoter and Down-Regulation of BRCA2 MRNA and Protein Levels in Response to DNA Damage. J. Biol. Chem. 2003, 278, 15652–15660. [Google Scholar] [CrossRef]
- Lobanova, O.E.; Rossokha, Z.I.; Medvedieva, N.L.; Cheshuk, V.E.; Vereshchako, R.I.; Vershyhora, V.O.; Fishchuk, L.Y.; Zakhartseva, L.M.; Gorovenko, N.G. Prevalence of BRCA1 and BRCA2 Genes Promoter Hypermethylation in Breast Cancer Tissue. Exp. Oncol. 2021, 43, 56–60. [Google Scholar] [CrossRef]
- Dworkin, A.M.; Spearman, A.D.; Tseng, S.Y.; Sweet, K.; Toland, A.E. Methylation Not a Frequent “Second Hit” in Tumors with Germline BRCA Mutations. Fam. Cancer 2009, 8, 339–346. [Google Scholar] [CrossRef]
- Mogilyansky, E.; Clark, P.; Quann, K.; Zhou, H.; Londin, E.; Jing, Y.; Rigoutsos, I. Post-Transcriptional Regulation of BRCA2 through Interactions with MiR-19a and MiR-19b. Front. Genet. 2016, 7, 143. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chen, W.; Iyer, M.K.; Cao, Q.; Ma, T.; Han, S.; Sahu, A.; Malik, R.; Wilder-Romans, K.; Navone, N.; et al. PCAT-1, a Long Noncoding RNA, Regulates BRCA2 and Controls Homologous Recombination in Cancer. Cancer Res. 2014, 74, 1651–1660. [Google Scholar] [CrossRef]
- Wan, G.; Hu, X.; Liu, Y.; Han, C.; Sood, A.K.; Calin, G.A.; Zhang, X.; Lu, X. A Novel Noncoding RNA LncRNA-JADE Connects DNA Damage Signaling to Histone H4 Acetylation. EMBO J. 2013, 32, 2833–2847. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Mathur, R.; Hu, X.; Liu, Y.; Zhang, X.; Peng, G.; Lu, X. Long Non-Coding RNA ANRIL (CDKN2B-AS) Is Induced by the ATM-E2F1 Signaling Pathway. Cell. Signal. 2013, 25, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. The 1991 Ulf von Euler Lecture:The l-Arginine: Nitric Oxide Pathway. Acta Physiol. Scand. 1992, 145, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P. Inducible Nitric Oxide Synthase: A Little Bit of Good in All of Us. Gut 2000, 47, 6–9. [Google Scholar] [CrossRef]
- Marletta, M.A. Nitric Oxide Synthase: Aspects Concerning Structure and Catalysis. Cell 1994, 78, 927–930. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible Nitric Oxide Synthase: Regulation, Structure, and Inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Schmidlin, A.; Wiesinger, H. Transport of L-Arginine in Cultured Glial Cells. Glia 1994, 11, 262–268. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric Oxide and the Immune Response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Aktan, F. INOS-Mediated Nitric Oxide Production and Its Regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef]
- Mayer, B.; Hemmens, B. Biosynthesis and Action of Nitric Oxide in Mammalian Cells. Trends Biochem. Sci. 1997, 22, 477–481. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem. J. 2001, 37, 593–615. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Yoho, L.L.; Stuehr, D.J. Calmodulin Controls Neuronal Nitric-Oxide Synthase by a Dual Mechanism. Activation of Intra- and Interdomain Electron Transfer. J. Biol. Chem. 1994, 269, 32047–32050. [Google Scholar] [CrossRef]
- Kone, B.C.; Kuncewicz, T.; Zhang, W.; Yu, Z.-Y. Protein Interactions with Nitric Oxide Synthases: Controlling the Right Time, the Right Place, and the Right Amount of Nitric Oxide. Am. J. Physiol.-Ren. Physiol. 2003, 285, F178–F190. [Google Scholar] [CrossRef] [PubMed]
- Venema, R.C.; Sayegh, H.S.; Kent, J.D.; Harrison, D.G. Identification, Characterization, and Comparison of the Calmodulin-Binding Domains of the Endothelial and Inducible Nitric Oxide Synthases. J. Biol. Chem. 1996, 271, 6435–6440. [Google Scholar] [CrossRef]
- Kwon, N.S.; Stuehr, D.J.; Nathan, C.F. Inhibition of Tumor Cell Ribonucleotide Reductase by Macrophage-Derived Nitric Oxide. J. Exp. Med. 1991, 174, 761–767. [Google Scholar] [CrossRef]
- Mĕlková, Z.; Esteban, M. Inhibition of Vaccinia Virus DNA Replication by Inducible Expression of Nitric Oxide Synthase. J. Immunol. 1995, 155, 5711–5718. [Google Scholar]
- Roy, B.; Lepoivre, M.; Henry, Y.; Fontecave, M. Inhibition of Ribonucleotide Reductase by Nitric Oxide Derived from Thionitrites: Reversible Modifications of Both Subunits. Biochemistry 1995, 34, 5411–5418. [Google Scholar] [CrossRef]
- Karupiah, G.; Harris, N. Inhibition of Viral Replication by Nitric Oxide and Its Reversal by Ferrous Sulfate and Tricarboxylic Acid Cycle Metabolites. J. Exp. Med. 1995, 181, 2171–2179. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.; Nathan, C. Nitric Oxide and Macrophage Function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Buttery, L.D.; Evans, T.J.; Springall, D.R.; Carpenter, A.; Cohen, J.; Polak, J.M. Immunochemical Localization of Inducible Nitric Oxide Synthase in Endotoxin-Treated Rats. Lab. Investig. 1994, 71, 755–764. [Google Scholar]
- Luoma, J.S.; Strålin, P.; Marklund, S.L.; Hiltunen, T.P.; Särkioja, T.; Ylä-Herttuala, S. Expression of Extracellular SOD and INOS in Macrophages and Smooth Muscle Cells in Human and Rabbit Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, A.K.; Giardina, C. Regulation of ICAM-1 Expression in Mouse Macrophages. Inflammation 2000, 24, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Nitric Oxide as a Secretory Product of Mammalian Cells. FASEB J. 1992, 6, 3051–3064. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Rosenfeld, M.E.; Ylä-Herttuala, S.; Gurtner, G.C.; Socher, S.S.; Butler, S.W.; Parthasarathy, S.; Carew, T.E.; Steinberg, D.; Witztum, J.L. Low Density Lipoprotein Undergoes Oxidative Modification in Vivo. Proc. Natl. Acad. Sci. USA 1989, 86, 1372–1376. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E.; Lundberg, J.M.; Alving, K. Intragastric Nitric Oxide Production in Humans: Measurements in Expelled Air. Gut 1994, 35, 1543–1546. [Google Scholar] [CrossRef]
- Pereira, C.; Ferreira, N.R.; Rocha, B.S.; Barbosa, R.M.; Laranjinha, J. The Redox Interplay between Nitrite and Nitric Oxide: From the Gut to the Brain. Redox Biol. 2013, 1, 276–284. [Google Scholar] [CrossRef]
- Hu, Y.; Xiang, J.; Su, L.; Tang, X. The Regulation of Nitric Oxide in Tumor Progression and Therapy. J. Int. Med. Res. 2020, 48, 0300060520905985. [Google Scholar] [CrossRef]
- Kleinert, H.; Pautz, A.; Linker, K.; Schwarz, P.M. Regulation of the Expression of Inducible Nitric Oxide Synthase. Eur. J. Pharmacol. 2004, 500, 255–266. [Google Scholar] [CrossRef]
- Taylor-Robinson, A.W.; Liew, F.Y.; Severn, A.; Xu, D.; McSorley, S.J.; Garside, P.; Padron, J.; Phillips, R.S. Regulation of the Immune Response by Nitric Oxide Differentially Produced by T Helper Type 1 and T Helper Type 2 Cells. Eur. J. Immunol. 1994, 24, 980–984. [Google Scholar] [CrossRef]
- Kröncke, K.-D.; Fehsel, K.; Kolb-Bachofen, V. Inducible Nitric Oxide Synthase in Human Diseases. Clin. Exp. Immunol. 1998, 113, 147–156. [Google Scholar] [CrossRef]
- Chartrain, N.A.; Geller, D.A.; Koty, P.P.; Sitrin, N.F.; Nussler, A.K.; Hoffman, E.P.; Billiar, T.R.; Hutchinson, N.I.; Mudgett, J.S. Molecular Cloning, Structure, and Chromosomal Localization of the Human Inducible Nitric Oxide Synthase Gene. J. Biol. Chem. 1994, 269, 6765–6772. [Google Scholar] [CrossRef]
- Morris, S.M.; Billiar, T.R. New Insights into the Regulation of Inducible Nitric Oxide Synthesis. Am. J. Physiol.-Endocrinol. Metab. 1994, 266, E829–E839. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.A.; Lowenstein, C.J.; Shapiro, R.A.; Nussler, A.K.; Di Silvio, M.; Wang, S.C.; Nakayama, D.K.; Simmons, R.L.; Snyder, S.H.; Billiar, T.R. Molecular Cloning and Expression of Inducible Nitric Oxide Synthase from Human Hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, H.; Schwarz, P.M.; Förstermann, U. Regulation of the Expression of Inducible Nitric Oxide Synthase. Biol. Chem. 2003, 384, 1343–1364. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-ΚB and rel proteins: Evolutionarily Conserved Mediators of Immune Responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Tedeschi, E.; Menegazzi, M.; Margotto, D.; Suzuki, H.; Förstermann, U.; Kleinert, H. Anti-Inflammatory Actions of St. John’s Wort: Inhibition of Human Inducible Nitric-Oxide Synthase Expression by Down-Regulating Signal Transducer and Activator of Transcription-1α (STAT-1α) Activation. J. Pharm. Exp. 2003, 307, 254–261. [Google Scholar] [CrossRef]
- Tedeschi, E.; Menegazzi, M.; Yao, Y.; Suzuki, H.; Förstermann, U.; Kleinert, H. Green Tea Inhibits Human Inducible Nitric-Oxide Synthase Expression by Down-Regulating Signal Transducer and Activator of Transcription-1α Activation. Mol. Pharm. 2004, 65, 111–120. [Google Scholar] [CrossRef]
- Yao, Y.; Hausding, M.; Erkel, G.; Anke, T.; Förstermann, U.; Kleinert, H. Sporogen, S14-95, and S-Curvularin, Three Inhibitors of Human Inducible Nitric-Oxide Synthase Expression Isolated from Fungi. Mol. Pharm. 2003, 63, 383–391. [Google Scholar] [CrossRef]
- Eberhardt, W.; Plüss, C.; Hummel, R.; Pfeilschifter, J. Molecular Mechanisms of Inducible Nitric Oxide Synthase Gene Expression by IL-1β and CAMP in Rat Mesangial Cells. J. Immunol. 1998, 160, 4961–4969. [Google Scholar]
- Goldring, C.E.P.; Reveneau, S.; Algarté, M.; Jeannin, J.-F. In Vivo Footprinting of the Mouse Inducible Nitric Oxide Synthase Gene: Inducible Protein Occupation of Numerous Sites Including Oct and NF-IL6. Nucleic Acids Res. 1996, 24, 1682–1687. [Google Scholar] [CrossRef][Green Version]
- Xie, Q. A Novel Lipopolysaccharide-Response Element Contributes to Induction of Nitric Oxide Synthase. J. Biol. Chem. 1997, 272, 14867–14872. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Ko, C.B.; Park, Y.P.; Kim, Y.J.; Paik, S.G. Octamer Motif Is Required for the NF-KappaB-Mediated Induction of the Inducible Nitric Oxide Synthase Gene Expression in RAW 264.7 Macrophages. Mol Cells 1999, 9, 99–109. [Google Scholar] [PubMed]
- Boyault, S.; Simonin, M.-A.; Bianchi, A.; Compe, E.; Liagre, B.; Mainard, D.; Bécuwe, P.; Dauça, M.; Netter, P.; Terlain, B.; et al. 15-Deoxy-Δ12,14-PGJ2, but Not Troglitazone, Modulates IL-1β Effects in Human Chondrocytes by Inhibiting NF-ΚB and AP-1 Activation Pathways. FEBS Lett. 2001, 501, 24–30. [Google Scholar] [CrossRef]
- Fahmi, H.; Di Battista, J.A.; Pelletier, J.-P.; Mineau, F.; Ranger, P.; Martel-Pelletier, J. Peroxisome Proliferator–Activated Receptor γ Activators Inhibit Interleukin-1β–Induced Nitric Oxide and Matrix Metalloproteinase 13 Production in Human Chondrocytes. Arthritis Rheum. 2001, 44, 595–607. [Google Scholar] [CrossRef]
- Li, M.; Pascual, G.; Glass, C.K. Peroxisome Proliferator-Activated Receptor γ-Dependent Repression of the Inducible Nitric Oxide Synthase Gene. Mol. Cell. Biol. 2000, 20, 4699–4707. [Google Scholar] [CrossRef]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric Oxide-Induced P53 Accumulation and Regulation of Inducible Nitric Oxide Synthase Expression by Wild-Type P53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef]
- Ambs, S.; Ogunfusika, M.O.; Merriam, W.G.; Bennett, W.P.; Billiar, T.R.; Harris, C.C. Up-Regulation of Inducible Nitric Oxide Synthase Expression in Cancer-Prone P53 Knockout Mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8823–8828. [Google Scholar] [CrossRef]
- Melillo, G.; Musso, T.; Sica, A.; Taylor, L.S.; Cox, G.W.; Varesio, L. A Hypoxia-Responsive Element Mediates a Novel Pathway of Activation of the Inducible Nitric Oxide Synthase Promoter. J. Exp. Med. 1995, 182, 1683–1693. [Google Scholar] [CrossRef]
- Jung, F.; Palmer, L.A.; Zhou, N.; Johns, R.A. Hypoxic Regulation of Inducible Nitric Oxide Synthase via Hypoxia Inducible Factor-1 in Cardiac Myocytes. Circ. Res. 2000, 86, 319–325. [Google Scholar] [CrossRef]
- Sirsjö, A.; Gidlöf, A.C.; Olsson, A.; Törmä, H.; Ares, M.; Kleinert, H.; Förstermann, U.; Hansson, G.K. Retinoic Acid Inhibits Nitric Oxide Synthase-2 Expression through the Retinoic Acid Receptor-α. Biochem. Biophys. Res. Commun. 2000, 270, 846–851. [Google Scholar] [CrossRef]
- Uchimura, K.; Nakamuta, M.; Enjoji, M.; Irie, T.; Sugimoto, R.; Muta, T.; Iwamoto, H.; Nawata, H. Activation of Retinoic X Receptor and Peroxisome Proliferator–Activated Receptor-γ Inhibits Nitric Oxide and Tumor Necrosis Factor-α Production in Rat Kupffer Cells. Hepatology 2001, 33, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nuedling, S.; Karas, R.H.; Mendelsohn, M.E.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; Meyer, R.; Vetter, H.; Grohé, C. Activation of Estrogen Receptor β Is a Prerequisite for Estrogen-Dependent Upregulation of Nitric Oxide Synthases in Neonatal Rat Cardiac Myocytes. FEBS Lett. 2001, 502, 103–108. [Google Scholar] [CrossRef]
- You, H.J.; Kim, J.Y.; Jeong, H.G. 17β-Estradiol Increases Inducible Nitric Oxide Synthase Expression in Macrophages. Biochem. Biophys. Res. Commun. 2003, 303, 1129–1134. [Google Scholar] [CrossRef]
- Mathy, N.W.; Burleigh, O.; Kochvar, A.; Whiteford, E.R.; Behrens, M.; Marta, P.; Tian, C.; Gong, A.-Y.; Drescher, K.M.; Steyger, P.S.; et al. A Novel Long Intergenic Non-Coding RNA, Nostrill, Regulates INOS Gene Transcription and Neurotoxicity in Microglia. J. Neuroinflammation 2021, 18, 16. [Google Scholar] [CrossRef]
- Yamamoto, M.; Okamoto, T.; Takeda, K.; Sato, S.; Sanjo, H.; Uematsu, S.; Saitoh, T.; Yamamoto, N.; Sakurai, H.; Ishii, K.J.; et al. Key Function for the Ubc13 E2 Ubiquitin-Conjugating Enzyme in Immune Receptor Signaling. Nat. Immunol. 2006, 7, 962–970. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-ΚB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Hu, G.; Gong, A.-Y.; Wang, Y.; Ma, S.; Chen, X.; Chen, J.; Su, C.-J.; Shibata, A.; Strauss-Soukup, J.K.; Drescher, K.M.; et al. LincRNA-Cox2 Promotes Late Inflammatory Gene Transcription in Macrophages through Modulating SWI/SNF-Mediated Chromatin Remodeling. J. Immunol. 2016, 196, 2799–2808. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Bogdan, C.; Paik, J.; Xie, Q.W.; Nathan, C. Mechanisms of Suppression of Macrophage Nitric Oxide Release by Transforming Growth Factor Beta. J. Exp. Med. 1993, 178, 605–613. [Google Scholar] [CrossRef]
- Pautz, A.; Art, J.; Hahn, S.; Nowag, S.; Voss, C.; Kleinert, H. Regulation of the Expression of Inducible Nitric Oxide Synthase. Nitric Oxide 2010, 23, 75–93. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.-J.; Lee, J.T. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological Organization of Multichromosomal Regions by the Long Intergenic Noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Cai, M.; Liu, M.; Su, G.; An, D.; Moon, B.; Lyu, G.; Si, Y.; Chen, L.; Lu, W. LncRNA 5430416N02Rik Promotes the Proliferation of Mouse Embryonic Stem Cells by Activating Mid1 Expression through 3D Chromatin Architecture. Stem Cell Rep. 2020, 14, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.C.; Fish, J.E.; Mawji, I.A.; Leung, D.D.; Rachlis, A.C.; Marsden, P.A. Epigenetic Basis for the Transcriptional Hyporesponsiveness of the Human Inducible Nitric Oxide Synthase Gene in Vascular Endothelial Cells. J. Immunol. 2005, 175, 3846–3861. [Google Scholar] [CrossRef] [PubMed]
- Matouk, C.C.; Marsden, P.A. Epigenetic Regulation of Vascular Endothelial Gene Expression. Circ. Res. 2008, 102, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-G.; Wu, K.K. Regulation of Inducible Nitric Oxide Synthase Expression by P300 and P50 Acetylation. J. Immunol. 2003, 171, 6581–6588. [Google Scholar] [CrossRef]
- Yu, Z.; Kone, B.C. Hypermethylation of the Inducible Nitric-Oxide Synthase Gene Promoter Inhibits Its Transcription. J. Biol. Chem. 2004, 279, 46954–46961. [Google Scholar] [CrossRef]
- de Andres, M.C.; Takahashi, A.; Oreffo, R.O. Demethylation of an Nuclear Factor-Kb (NF-Kb) Enhancer Element Orchestrates INOS Induction in Osteoarthritis via Cell Cycle Regulation. Osteoarthr. Cartil. 2015, 23, A191. [Google Scholar] [CrossRef][Green Version]
- Linker, K.; Pautz, A.; Fechir, M.; Hubrich, T.; Greeve, J.; Kleinert, H. Involvement of KSRP in the Post-Transcriptional Regulation of Human INOS Expression–Complex Interplay of KSRP with TTP and HuR. Nucleic Acids Res. 2005, 33, 4813–4827. [Google Scholar] [CrossRef]
- Stefano, G.B.; Salzet, M.; Magazine, H.I.; Bilfinger, T.V. Antagonism of LPS and IFN-γ Induction of INOS in Human Saphenous Vein Endothelium by Morphine and Anandamide by Nitric Oxide Inhibition of Adenylate Cyclase. J. Cardiovasc. Pharmacol. 1998, 31, 813–820. [Google Scholar] [CrossRef]
- Stefano, G.B.; Goumon, Y.; Bilfinger, T.V.; Welters, I.D.; Cadet, P. Basal Nitric Oxide Limits Immune, Nervous and Cardiovascular Excitation: Human Endothelia Express a Mu Opiate Receptor. Prog. Neurobiol. 2000, 60, 513–530. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 1990, 87, 10043–10047. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and Ugly. Am. J. Physiol.-Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Kelm, M. Nitric Oxide Metabolism and Breakdown. Biochim. Biophys. Acta Bioenerg. 1999, 1411, 273–289. [Google Scholar] [CrossRef]
- Farrell, A.J.; Blake, D.R. Nitric Oxide. Ann. Rheum. Dis. 1996, 55, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Lamas, S.; Fang, F.C. Nitrosylation: The Prototypic Redox-Based Signaling Mechanism. Cell 2001, 106, 675–683. [Google Scholar] [CrossRef]
- Marshall, H.E.; Hess, D.T.; Stamler, J.S. S-Nitrosylation: Physiological Regulation of NF-ΚB. Proc. Natl. Acad. Sci. USA 2004, 101, 8841–8842. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.M.; van der Vliet, A.; Janssen-Heininger, Y.M.W. Nitric Oxide Represses Inhibitory ΚB Kinase through S-Nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950. [Google Scholar] [CrossRef]
- Matthews, J.R.; Botting, C.H.; Panico, M.; Morris, H.R.; Hay, R.T. Inhibition of NF-ΚB DNA Binding by Nitric Oxide. Nucleic Acids Res. 1996, 24, 2236–2242. [Google Scholar] [CrossRef]
- Marshall, H.E.; Stamler, J.S. Inhibition of NF-ΚB by S-Nitrosylation. Biochemistry 2001, 40, 1688–1693. [Google Scholar] [CrossRef]
- King, M.-C.; Marks, J.H.; Mandell, J.B. Breast and Ovarian Cancer Risks Due to Inherited Mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Saad, A.F.; Hu, W.; Sood, A.K. Microenvironment and Pathogenesis of Epithelial Ovarian Cancer. Horm. Cancer 2010, 1, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.M.; Saed, G.M.; Diamond, M.P.; Sokol, R.J.; Munkarah, A.R. The Effects of the Inhibition of Inducible Nitric Oxide Synthase on Angiogenesis of Epithelial Ovarian Cancer. Am. J. Obstet. Gynecol. 2006, 194, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Saed, G.M.; Ali-Fehmi, R.; Jiang, Z.L.; Fletcher, N.M.; Diamond, M.P.; Abu-Soud, H.M.; Munkarah, A.R. Myeloperoxidase Serves as a Redox Switch That Regulates Apoptosis in Epithelial Ovarian Cancer. Gynecol. Oncol. 2010, 116, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Fletcher, N.M.; Ali-Fehmi, R.; Diamond, M.P.; Abu-Soud, H.M.; Munkarah, A.R.; Saed, G.M. Modulation of Redox Signaling Promotes Apoptosis in Epithelial Ovarian Cancer Cells. Gynecol. Oncol. 2011, 122, 418–423. [Google Scholar] [CrossRef]
- Saed, G.M.; Diamond, M.P.; Fletcher, N.M. Updates of the Role of Oxidative Stress in the Pathogenesis of Ovarian Cancer. Gynecol. Oncol. 2017, 145, 595–602. [Google Scholar] [CrossRef]
- Sha, Y.; Marshall, H.E. S-Nitrosylation in the Regulation of Gene Transcription. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 701–711. [Google Scholar] [CrossRef]
- van Dieck, J.; Teufel, D.P.; Jaulent, A.M.; Fernandez-Fernandez, M.R.; Rutherford, T.J.; Wyslouch-Cieszynska, A.; Fersht, A.R. Posttranslational Modifications Affect the Interaction of S100 Proteins with Tumor Suppressor P53. J. Mol. Biol. 2009, 394, 922–930. [Google Scholar] [CrossRef]
- Schonhoff, C.M.; Daou, M.-C.; Jones, S.N.; Schiffer, C.A.; Ross, A.H. Nitric Oxide-Mediated Inhibition of Hdm2−p53 Binding. Biochemistry 2002, 41, 13570–13574. [Google Scholar] [CrossRef]
- Padgett, C.M.; Whorton, A.R. S-Nitrosoglutathione Reversibly Inhibits GAPDH by S-Nitrosylation. Am. J. Physiol.-Cell Physiol. 1995, 269, C739–C749. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-Selective Protein S-Nitrosylation by Sequence Motif Recognition. Cell 2014, 159, 623–634. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Willard, B.; Smith, J.D.; Stuehr, D.J.; Hazen, S.L.; Fox, P.L. Protection of Extraribosomal RPL13a by GAPDH and Dysregulation by S-Nitrosylation. Mol. Cell 2012, 47, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The Chemical Biology of Nitric Oxide: Implications in Cellular Signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric Oxide and Redox Mechanisms in the Immune Response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A.; Martin, D.N.; Switzer, C.H.; Hudson, R.S.; Wink, D.A.; et al. Increased NOS2 Predicts Poor Survival in Estrogen Receptor–Negative Breast Cancer Patients. J. Clin. Investig. 2010, 120, 3843–3854. [Google Scholar] [CrossRef]
- Switzer, C.H.; Cheng, R.Y.-S.; Ridnour, L.A.; Glynn, S.A.; Ambs, S.; Wink, D.A. Ets-1 Is a Transcriptional Mediator of Oncogenic Nitric Oxide Signaling in Estrogen Receptor-Negative Breast Cancer. Breast Cancer Res. 2012, 14, R125. [Google Scholar] [CrossRef]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase Regulates Angiogenesis Induced by Colon Cancer Cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef]
- Brown, J.R.; DuBois, R.N. COX-2: A Molecular Target for Colorectal Cancer Prevention. J. Clin. Oncol. 2005, 23, 2840–2855. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Inflammation: A Driving Force Speeds Cancer Metastasis. Cell Cycle 2009, 8, 3267–3273. [Google Scholar] [CrossRef]
- Grellner, W.; Georg, T.; Wilske, J. Quantitative Analysis of Proinflammatory Cytokines (IL-1β, IL-6, TNF-α) in Human Skin Wounds. Forensic Sci. Int. 2000, 113, 251–264. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Zidi, I.; Mestiri, S.; Bartegi, A.; Amor, N.B. TNF-α and Its Inhibitors in Cancer. Med. Oncol. 2010, 27, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 Signaling Plays a Critical Role in the Epithelial–Mesenchymal Transition of Human Carcinoma Cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning Inflammation and Immunity by Chemokine Sequestration: Decoys and More. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour Necrosis Factor and Cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-Derived IL-1β Stimulates Wnt Signaling and Growth of Colon Cancer Cells: A Crosstalk Interrupted by Vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef]
- Ben-Baruch, A. The Tumor-Promoting Flow of Cells Into, Within and Out of the Tumor Site: Regulation by the Inflammatory Axis of TNFα and Chemokines. Cancer Microenviron. 2012, 5, 151–164. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of INOS as a Novel Effective Targeted Therapy against Triple-Negative Breast Cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma Stem Cell Proliferation and Tumor Growth Are Promoted by Nitric Oxide Synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef]
- Okayama, H.; Saito, M.; Oue, N.; Weiss, J.M.; Stauffer, J.; Takenoshita, S.; Wiltrout, R.H.; Hussain, S.P.; Harris, C.C. NOS2 Enhances KRAS-Induced Lung Carcinogenesis, Inflammation and MicroRNA-21 Expression. Int. J. Cancer 2013, 132, 9–18. [Google Scholar] [CrossRef]
- Dittmer, J. The Biology of the Ets1 Proto-Oncogene. Mol. Cancer 2003, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, D.W., 2nd; Bove, K. The Transcription Factor Ets-1 in Breast Cancer. Front. Biosci.-Landmark 2005, 10, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.P.; Findlay, V.J.; Moussa, O.; Watson, D.K. Defining ETS Transcription Regulatory Networks and Their Contribution to Breast Cancer Progression. J. Cell. Biochem. 2007, 102, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.U.; Kumar, R.; Singh, A.; Khan, A.; Tanwar, P.; Tripathi, R.; Mehrotra, R.; Hussain, S. Breast Cancer Invasion and Progression by MMP-9 through Ets-1 Transcription Factor. Gene 2019, 711, 143952. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Jung, H.H.; Ahn, J.S.; Im, Y.-H. Ets-1 Upregulates HER2-Induced MMP-1 Expression in Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2008, 377, 389–394. [Google Scholar] [CrossRef]
- Kim, S.; Han, J.; Shin, I.; Kil, W.H.; Lee, J.E.; Nam, S.J. A Functional Comparison between the HER2high/HER3 and the HER2low/HER3 Dimers on Heregulin-Β1-Induced MMP-1 and MMP-9 Expression in Breast Cancer Cells. Exp. Mol. Med. 2012, 44, 473–482. [Google Scholar] [CrossRef]
- Haynes, M.P.; Li, L.; Sinha, D.; Russell, K.S.; Hisamoto, K.; Baron, R.; Collinge, M.; Sessa, W.C.; Bender, J.R. Src Kinase Mediates Phosphatidylinositol 3-Kinase/Akt-Dependent Rapid Endothelial Nitric-Oxide Synthase Activation by Estrogen. J. Biol. Chem. 2003, 278, 2118–2123. [Google Scholar] [CrossRef]
- Switzer, C.H.; Glynn, S.A.; Ridnour, L.A.; Cheng, R.Y.-S.; Vitek, M.P.; Ambs, S.; Wink, D.A. Nitric Oxide and Protein Phosphatase 2A Provide Novel Therapeutic Opportunities in ER-Negative Breast Cancer. Trends Pharmacol. Sci. 2011, 32, 644–651. [Google Scholar] [CrossRef]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA Deaminating Ability and Genotoxicity of Nitric Oxide and Its Progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef]
- Nguyen, T.; Brunson, D.; Crespi, C.L.; Penman, B.W.; Wishnok, J.S.; Tannenbaum, S.R. DNA Damage and Mutation in Human Cells Exposed to Nitric Oxide in Vitro. Proc. Natl. Acad. Sci. USA 1992, 89, 3030–3034. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory Cytokines Induce DNA Damage and Inhibit DNA Repair in Cholangiocarcinoma Cells by a Nitric Oxide-Dependent Mechanism1. Cancer Res. 2000, 60, 184–190. [Google Scholar] [PubMed]
- Hussain, S.P.; He, P.; Subleski, J.; Hofseth, L.J.; Trivers, G.E.; Mechanic, L.; Hofseth, A.B.; Bernard, M.; Schwank, J.; Nguyen, G.; et al. Nitric Oxide Is a Key Component in Inflammation-Accelerated Tumorigenesis. Cancer Res. 2008, 68, 7130–7136. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Katafuchi, A.; Shimizu, R.; Terato, H.; Suzuki, T.; Tauchi, H.; Makino, K.; Skorvaga, M.; Van Houten, B.; Ide, H. Repair Activity of Base and Nucleotide Excision Repair Enzymes for Guanine Lesions Induced by Nitrosative Stress. Nucleic Acids Res. 2005, 33, 2181–2191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marth, C.; Fiegl, H.; Zeimet, A.G.; Müller-Holzner, E.; Deibl, M.; Doppler, W.; Daxenbichler, G. Interferon-γ Expression Is an Independent Prognostic Factor in Ovarian Cancer. Am. J. Obstet. Gynecol. 2004, 191, 1598–1605. [Google Scholar] [CrossRef]
- Marth, C.; Müller-Holzner, E.; Greiter, E.; Cronauer, M.V.; Zeimet, A.G.; Doppler, W.; Eibl, B.; Hynes, N.E.; Daxenbichler, G. γ-Interferon Reduces Expression of the Protooncogene c-ErbB-2 in Human Ovarian Carcinoma Cells. Cancer Res. 1990, 50, 7037–7041. [Google Scholar]
- Ganster, R.W.; Taylor, B.S.; Shao, L.; Geller, D.A. Complex Regulation of Human Inducible Nitric Oxide Synthase Gene Transcription by Stat 1 and NF-ΚB. Proc. Natl. Acad. Sci. USA 2001, 98, 8638–8643. [Google Scholar] [CrossRef]
- Rieder, J.; Jahnke, R.; Schloesser, M.; Seibel, M.; Czechowski, M.; Marth, C.; Hoffmann, G. Nitric Oxide-Dependent Apoptosis in Ovarian Carcinoma Cell Lines. Gynecol. Oncol. 2001, 82, 172–176. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-Induced down-Regulation of BRCA1 Expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef]
- Bindra, R.S.; Glazer, P.M. Repression of RAD51 Gene Expression by E2F4/P130 Complexes in Hypoxia. Oncogene 2007, 26, 2048–2057. [Google Scholar] [CrossRef]
- Plenchette, S.; Paul, C.; Bettaieb, A. Chapter 6—Nitric Oxide and Platinum-Derivative-Based Regimens for Cancer Treatment: From Preclinical Studies to Clinical Trials. In Nitric Oxide (Donor/Induced) in Chemosensitizing; Bonavida, B., Ed.; Cancer Sensitizing Agents for Chemotherapy; Academic Press: Cambridge, MA, USA, 2017; Volume 1, pp. 91–103. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Shapiro, R.A.; Billiar, T.R.; Gores, G.J. Nitric Oxide–Mediated Inhibition of DNA Repair Potentiates Oxidative DNA Damage in Cholangiocytes. Gastroenterology 2001, 120, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Le Page, F.; Randrianarison, V.; Marot, D.; Cabannes, J.; Perricaudet, M.; Feunteun, J.; Sarasin, A. BRCA1 and BRCA2 Are Necessary for the Transcription-Coupled Repair of the Oxidative 8-Oxoguanine Lesion in Human Cells1. Cancer Res. 2000, 60, 5548–5552. [Google Scholar] [PubMed]
- Correia, C.; Weiskittel, T.M.; Ung, C.Y.; Villasboas Bisneto, J.C.; Billadeau, D.D.; Kaufmann, S.H.; Li, H. Uncovering Pharmacological Opportunities for Cancer Stem Cells—A Systems Biology View. Front. Cell Dev. Biol. 2022, 10, 752326. [Google Scholar] [CrossRef] [PubMed]
- Yongsanguanchai, N.; Pongrakhananon, V.; Mutirangura, A.; Rojanasakul, Y.; Chanvorachote, P. Nitric Oxide Induces Cancer Stem Cell-like Phenotypes in Human Lung Cancer Cells. Am. J. Physiol.-Cell Physiol. 2015, 308, C89–C100. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. INOS Promotes CD24+CD133+ Liver Cancer Stem Cell Phenotype through a TACE/ADAM17-Dependent Notch Signaling Pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef]
- Maiuthed, A.; Chantarawong, W.; Chanvorachote, P. Lung Cancer Stem Cells and Cancer Stem Cell-Targeting Natural Compounds. Anticancer Res. 2018, 38, 3797–3809. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Grimm, E.A.; Roszik, J. Targeting INOS to Increase Efficacy of Immunotherapies. Hum. Vaccines Immunother. 2017, 13, 1105–1108. [Google Scholar] [CrossRef]
- López-Sánchez, L.M.; Mena, R.; Guil-Luna, S.; Mantrana, A.; Peñarando, J.; Toledano-Fonseca, M.; Conde, F.; De la Haba-Rodríguez, J.R.; Aranda, E.; Rodríguez-Ariza, A. Nitric Oxide-Targeted Therapy Inhibits Stemness and Increases the Efficacy of Tamoxifen in Estrogen Receptor-Positive Breast Cancer Cells. Lab. Investig. 2021, 101, 292–303. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Wang, W.-H.; Wu, W.-Y.; Hsu, C.-C.; Wei, L.-R.; Wang, S.-F.; Hsu, Y.-W.; Liaw, C.-C.; Tsai, W.-C. Novel Histone Deacetylase Inhibitor AR-42 Exhibits Antitumor Activity in Pancreatic Cancer Cells by Affecting Multiple Biochemical Pathways. PLoS ONE 2017, 12, e0183368. [Google Scholar] [CrossRef]
- Peñarando, J.; López-Sánchez, L.M.; Mena, R.; Guil-Luna, S.; Conde, F.; Hernández, V.; Toledano, M.; Gudiño, V.; Raponi, M.; Billard, C.; et al. A Role for Endothelial Nitric Oxide Synthase in Intestinal Stem Cell Proliferation and Mesenchymal Colorectal Cancer. BMC Biol. 2018, 16, 3. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Yun, Z. BRCA1 Regulates the Cancer Stem Cell Fate of Breast Cancer Cells in the Context of Hypoxia and Histone Deacetylase Inhibitors. Sci. Rep. 2019, 9, 9702. [Google Scholar] [CrossRef] [PubMed]
- Pupa, S.M.; Ligorio, F.; Cancila, V.; Franceschini, A.; Tripodo, C.; Vernieri, C.; Castagnoli, L. HER2 Signaling and Breast Cancer Stem Cells: The Bridge behind HER2-Positive Breast Cancer Aggressiveness and Therapy Refractoriness. Cancers 2021, 13, 4778. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; D’Elia, G.; Caliendo, G.; Albanese, L.; Signoriello, G.; Napoli, C.; Molinari, A.M. Pancreatic Cancer with Mutation in BRCA1/2, MLH1, and APC Genes: Phenotype Correlation and Detection of a Novel Germline BRCA2 Mutation. Genes 2022, 13, 321. [Google Scholar] [CrossRef]
- Han, S.-H.; Ryu, K.H.; Kwon, A.-Y. The Prognostic Impact of HER2 Genetic and Protein Expression in Pancreatic Carcinoma-HER2 Protein and Gene in Pancreatic Cancer. Diagnostics 2021, 11, 653. [Google Scholar] [CrossRef]
- Porcelli, L.; Quatrale, A.E.; Mantuano, P.; Leo, M.G.; Silvestris, N.; Rolland, J.F.; Carioggia, E.; Lioce, M.; Paradiso, A.; Azzariti, A. Optimize Radiochemotherapy in Pancreatic Cancer: PARP Inhibitors a New Therapeutic Opportunity. Mol. Oncol. 2013, 7, 308–322. [Google Scholar] [CrossRef]
- Komoto, M.; Nakata, B.; Nishii, T.; Kawajiri, H.; Shinto, O.; Amano, R.; Yamada, N.; Yashiro, M.; Hirakawa, K. In Vitro and in Vivo Evidence That a Combination of Lapatinib plus S-1 Is a Promising Treatment for Pancreatic Cancer. Cancer Sci. 2010, 101, 468–473. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V. The High-Throughput Analyses Era: Are We Ready for the Data Struggle? High-Throughput 2018, 7, 8. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Zhao, N.; Guo, M.; Wang, K.; Zhang, C.; Liu, X. Identification of Pan-Cancer Prognostic Biomarkers through Integration of Multi-Omics Data. Front. Bioeng. Biotechnol. 2020, 8, 268. [Google Scholar] [CrossRef]
- Candido, S.; Tomasello, B.M.R.; Lavoro, A.; Falzone, L.; Gattuso, G.; Libra, M. Novel Insights into Epigenetic Regulation of Il6 Pathway: In Silico Perspective on Inflammation and Cancer Relationship. Int. J. Mol. Sci. 2021, 22, 10172. [Google Scholar] [CrossRef]
- Zheng, Q.; Min, S.; Zhou, Q. Identification of Potential Diagnostic and Prognostic Biomarkers for LUAD Based on TCGA and GEO Databases. Biosci. Rep. 2021, 41, BSR20204370. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The UCSC Xena Platform for Public and Private Cancer Genomics Data Visualization and Interpretation. bioRxiv 2019, 326470. [Google Scholar] [CrossRef]
- Schleicher, M.; Brundin, F.; Gross, S.; Müller-Esterl, W.; Oess, S. Cell Cycle-Regulated Inactivation of Endothelial NO Synthase through NOSIP-Dependent Targeting to the Cytoskeleton. Mol. Cell. Biol. 2005, 25, 8251–8258. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, F.; Alttoa, A.; Reif, A. Neuronal Nitric Oxide Synthase (NOS1) and Its Adaptor, NOS1AP, as a Genetic Risk Factors for Psychiatric Disorders. Genes Brain Behav. 2015, 14, 46–63. [Google Scholar] [CrossRef]
- Ambs, S.; Glynn, S.A. Candidate Pathways Linking Inducible Nitric Oxide Synthase to a Basal-like Transcription Pattern and Tumor Progression in Human Breast Cancer. Cell Cycle 2011, 10, 619–624. [Google Scholar] [CrossRef]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxid Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible Nitric Oxide Synthase (INOS) in Tumor Biology: The Two Sides of the Same Coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. CRO 2016, 21, 333–351. [Google Scholar] [CrossRef]
- Foulkes, W.D. BRCA1 Functions as a Breast Stem Cell Regulator. J. Med. Genet. 2004, 41, 1–5. [Google Scholar] [CrossRef][Green Version]
- Belgorosky, D.; Girouard, J.; Langle, Y.; Hamelin Morrissette, J.; Marino, L.; Agüero, E.; Malagrino, H.; Reyes-Moreno, C.; Eiján, A.M. Relevance of INOS Expression in Tumor Growth and Maintenance of Cancer Stem Cells in a Bladder Cancer Model. J. Mol. Med. 2020, 98, 1615–1627. [Google Scholar] [CrossRef]
- Li, X.; Zou, Z.; Tang, J.; Zheng, Y.; Liu, Y.; Luo, Y.; Liu, Q.; Wang, Y. NOS1 Upregulates ABCG2 Expression Contributing to DDP Chemoresistance in Ovarian Cancer Cells. Oncol. Lett. 2019, 17, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Li, X.; Sun, Y.; Li, L.; Zhang, Q.; Zhu, L.; Zhong, Z.; Wang, M.; Wang, Q.; Liu, Z.; et al. NOS1 Expression Promotes Proliferation and Invasion and Enhances Chemoresistance in Ovarian Cancer. Oncol. Lett. 2020, 19, 2989–2995. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ye, S.; Li, K.; Huang, M.; Wang, Q.; Zeng, S.; Chen, X.; Gao, W.; Chen, J.; Zhang, Q.; et al. NOS1 Inhibits the Interferon Response of Cancer Cells by S-Nitrosylation of HDAC2. J. Exp. Clin. Cancer Res. 2019, 38, 483. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zou, Z.; Wang, Q.; Gao, W.; Zeng, S.; Ye, S.; Xu, P.; Huang, M.; Li, K.; Chen, J.; et al. Inhibition of NOS1 Promotes the Interferon Response of Melanoma Cells. J. Transl. Med. 2022, 20, 205. [Google Scholar] [CrossRef]
- Anastas, J.N.; Biechele, T.L.; Robitaille, M.; Muster, J.; Allison, K.H.; Angers, S.; Moon, R.T. A Protein Complex of SCRIB, NOS1AP and VANGL1 Regulates Cell Polarity and Migration, and Is Associated with Breast Cancer Progression. Oncogene 2012, 31, 3696–3708. [Google Scholar] [CrossRef]
- Li, L.-L.; de Mera, R.M.M.-F.; Chen, J.; Ba, W.; Kasri, N.N.; Zhang, M.; Courtney, M.J. Unexpected Heterodivalent Recruitment of NOS1AP to NNOS Reveals Multiple Sites for Pharmacological Intervention in Neuronal Disease Models. J. Neurosci. 2015, 35, 7349–7364. [Google Scholar] [CrossRef]
- Roselló-Lletí, E.; Tarazón, E.; Ortega, A.; Gil-Cayuela, C.; Carnicer, R.; Lago, F.; González-Juanatey, J.R.; Portolés, M.; Rivera, M. Protein Inhibitor of NOS1 Plays a Central Role in the Regulation of NOS1 Activity in Human Dilated Hearts. Sci. Rep. 2016, 6, 30902. [Google Scholar] [CrossRef]
- Sadaf, S.; Nagarkoti, S.; Awasthi, D.; Singh, A.K.; Srivastava, R.N.; Kumar, S.; Barthwal, M.K.; Dikshit, M. NNOS Induction and NOSIP Interaction Impact Granulopoiesis and Neutrophil Differentiation by Modulating Nitric Oxide Generation. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119018. [Google Scholar] [CrossRef]








{kind=link}
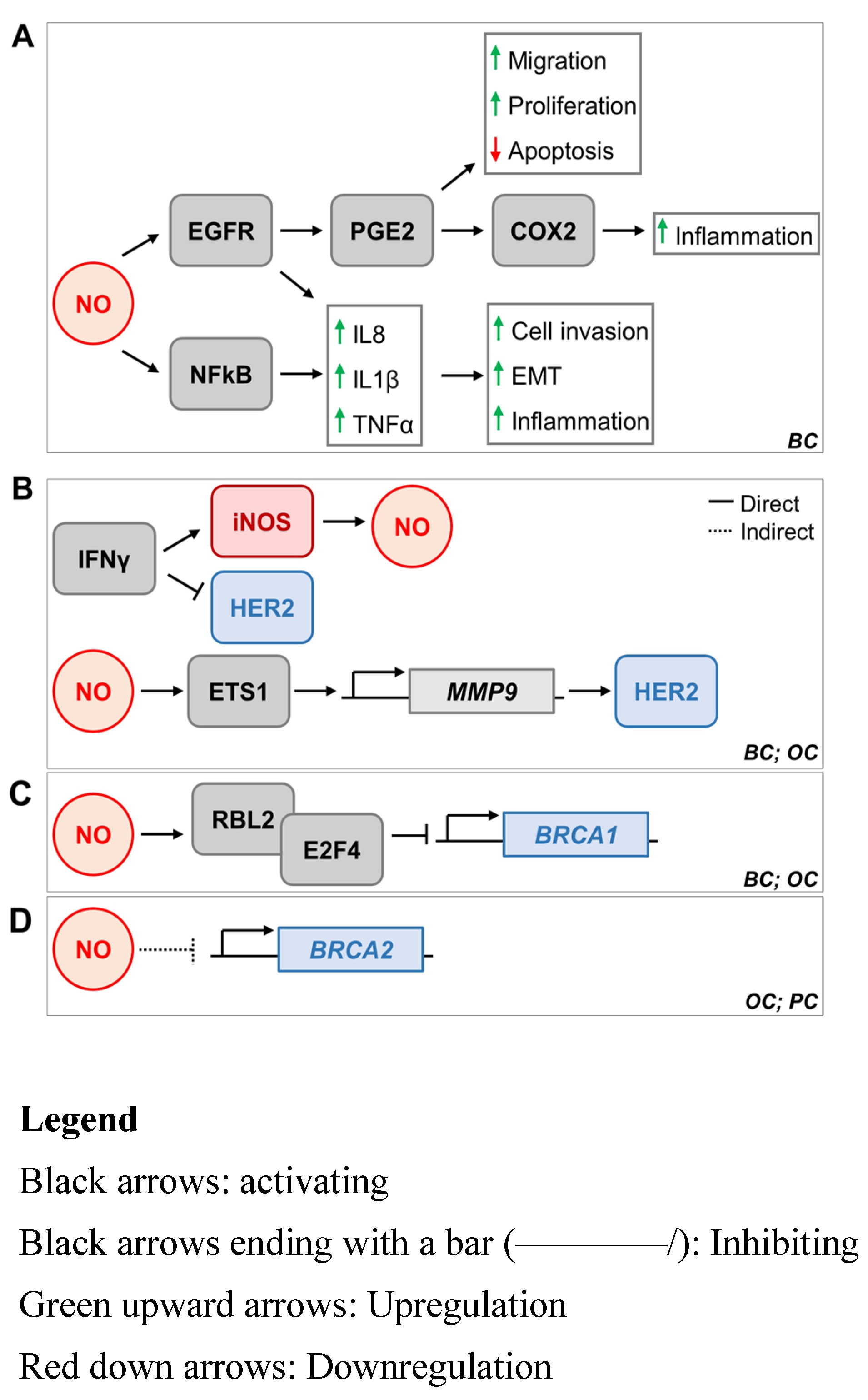{kind=link}
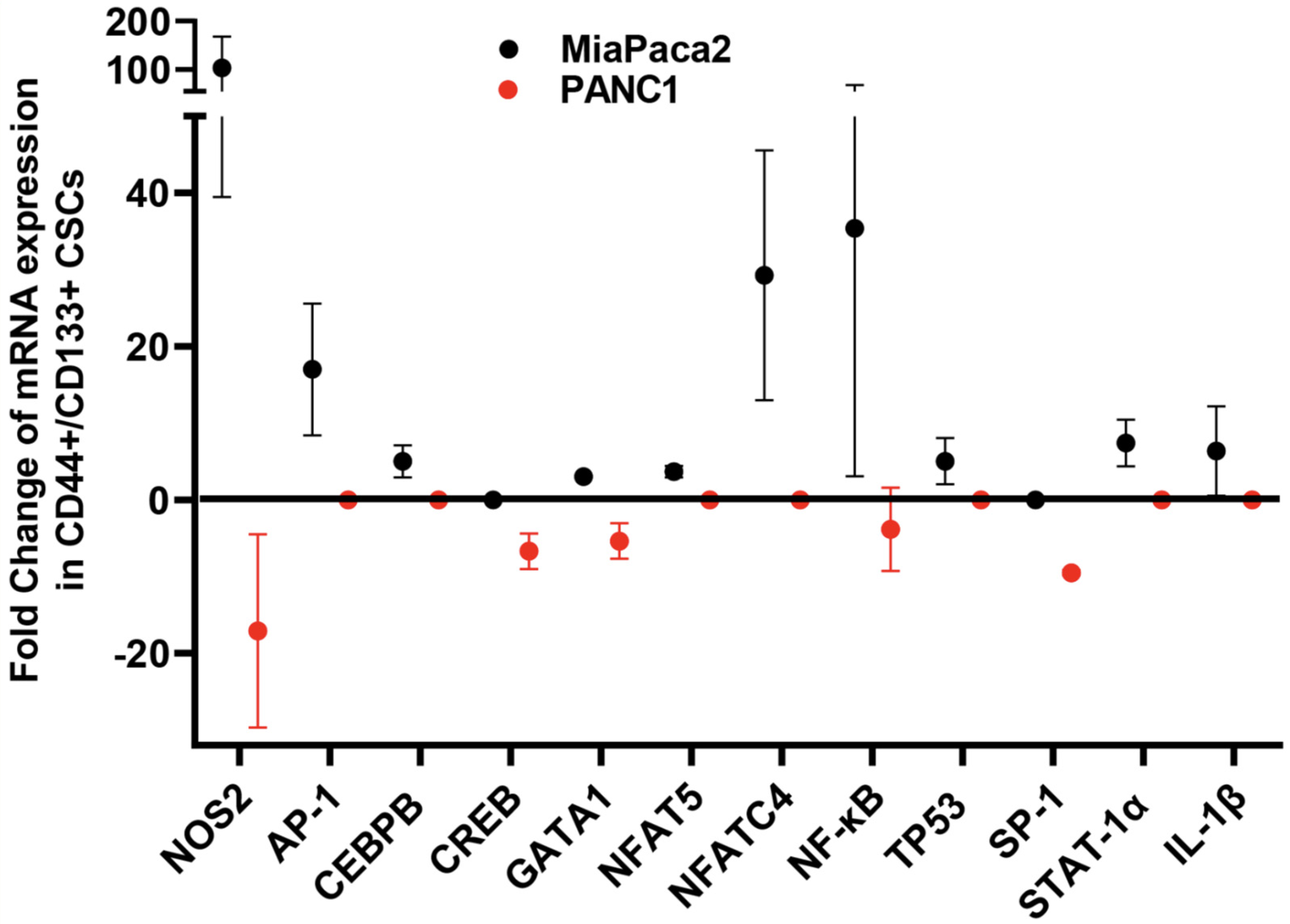{kind=link}
{kind=link}
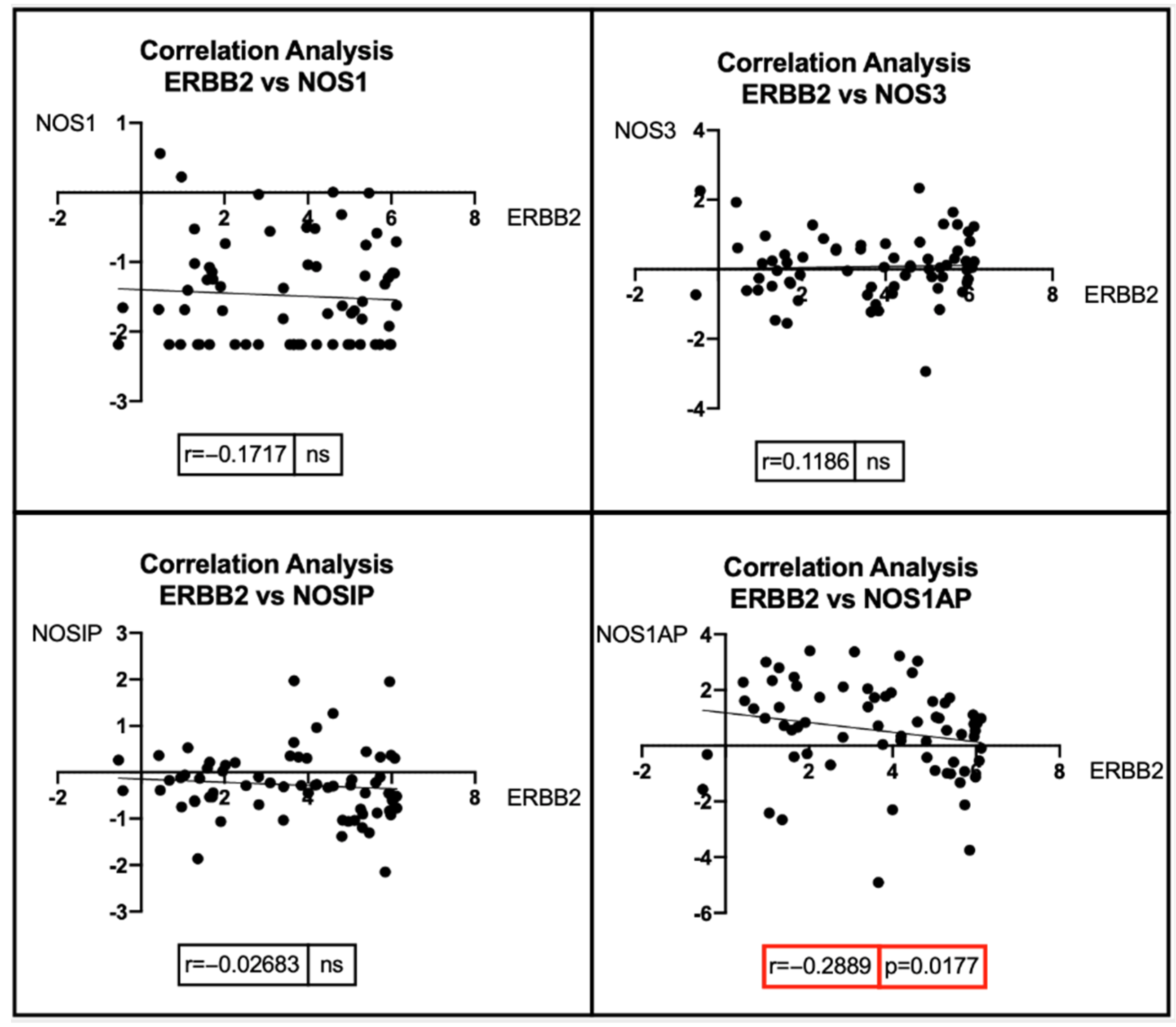{kind=link}
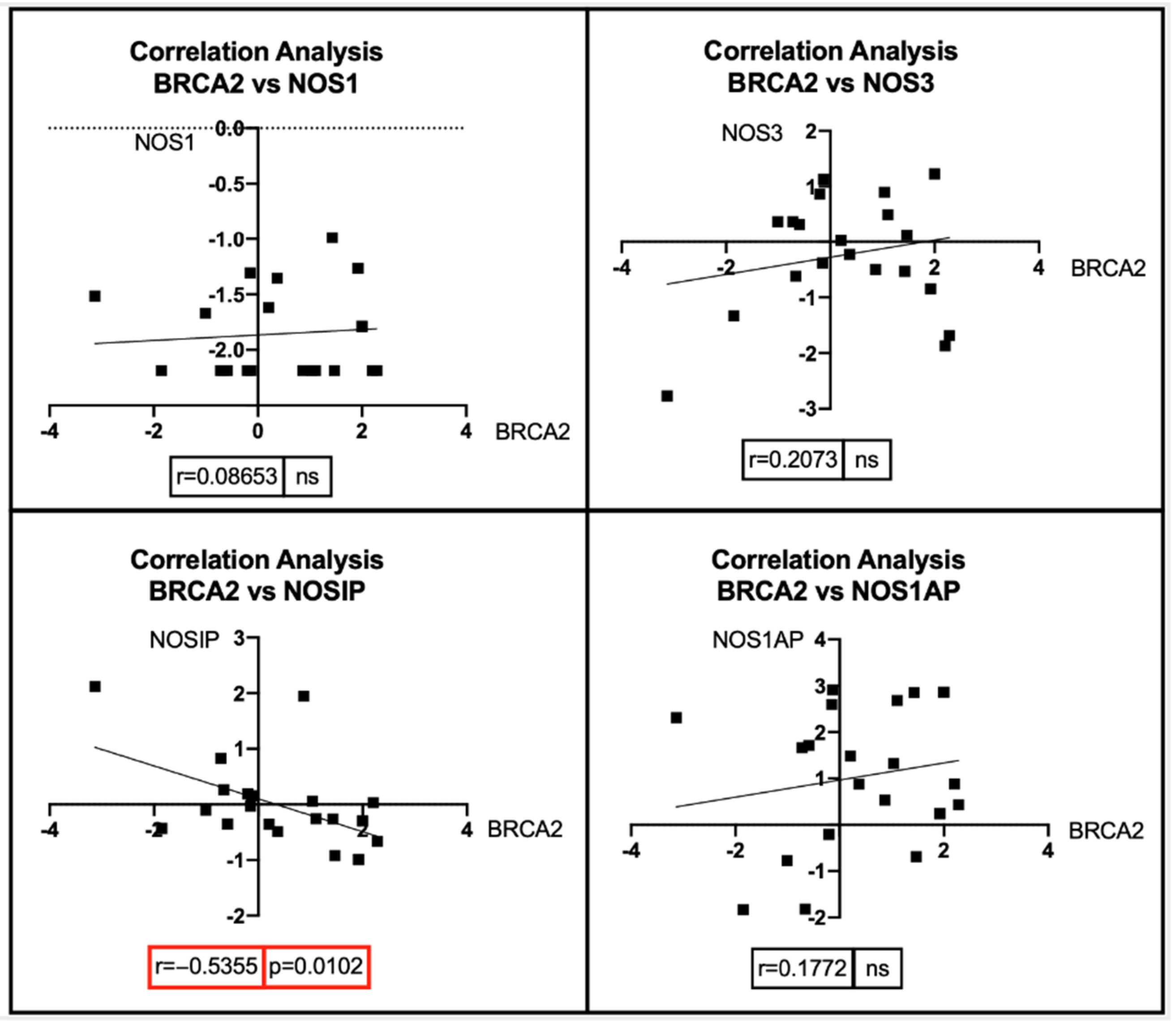{kind=link}
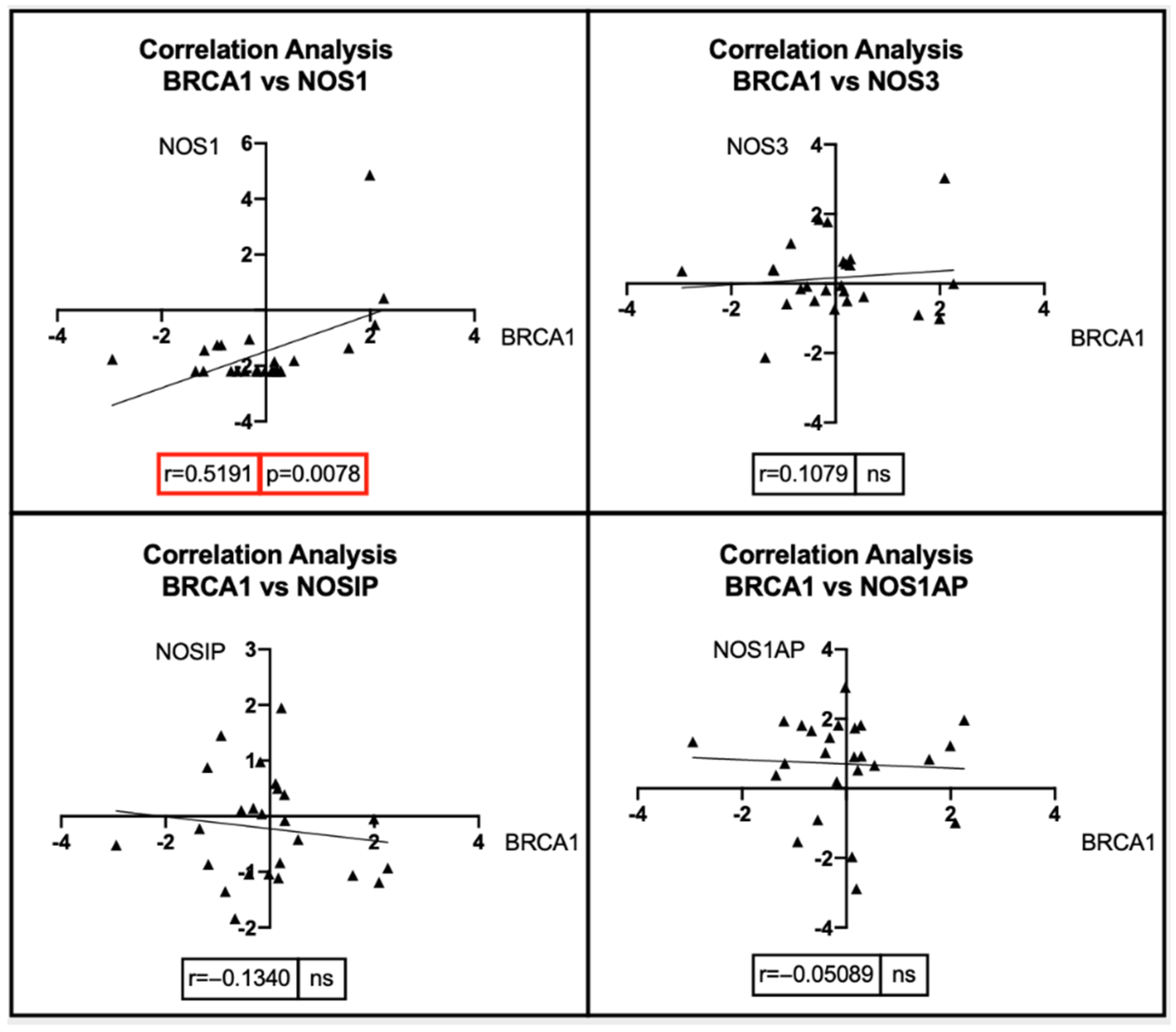{kind=link}
| Transcriptional Factors: Name | Type of Factor | Expression |
|---|---|---|
| NF-kB | Pre-transcriptional factor | Upregulation |
| STAT-1α | Pre-transcriptional factor | Upregulation |
| cAMP-induced transcription factors | Pre-transcriptional factors | Upregulation |
| AP-1 | Pre-transcriptional factors | Downregulation in human iNOSUpregulation in murine iNOS |
| Octamer factor | Pre-transcriptional factors | Upregulation |
| PPAR | Pre-transcriptional factors | Downregulation |
| p53 | Pre-transcriptional factors | Downregulation |
| HIF-1 | Pre-transcriptional factors | Upregulation |
| RAR-α | Pre-transcriptional factors | Downregulation |
| ER-β | Pre-transcriptional factors | Upregulation |
| Nostrill (lncRNA) | Transcription factor | Upregulation |
| DNA methylation | Epigenetic post-transcriptional regulation | Downregulation |
| Histone H3K9 methylation | Epigenetic post-transcriptional regulation | Downregulation |
| Hypermethylation | Epigenetic post-transcriptional regulation | Downregulation |
| Demethylation | Epigenetic post-transcriptional regulation | Upregulation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, K.; Baritaki, S.; Vivarelli, S.; Falzone, L.; Scalisi, A.; Libra, M.; Bonavida, B. The Breast Cancer Protooncogenes HER2, BRCA1 and BRCA2 and Their Regulation by the iNOS/NOS2 Axis. Antioxidants 2022, 11, 1195. https://doi.org/10.3390/antiox11061195
Lin K, Baritaki S, Vivarelli S, Falzone L, Scalisi A, Libra M, Bonavida B. The Breast Cancer Protooncogenes HER2, BRCA1 and BRCA2 and Their Regulation by the iNOS/NOS2 Axis. Antioxidants. 2022; 11(6):1195. https://doi.org/10.3390/antiox11061195
Chicago/Turabian StyleLin, Katie, Stavroula Baritaki, Silvia Vivarelli, Luca Falzone, Aurora Scalisi, Massimo Libra, and Benjamin Bonavida. 2022. "The Breast Cancer Protooncogenes HER2, BRCA1 and BRCA2 and Their Regulation by the iNOS/NOS2 Axis" Antioxidants 11, no. 6: 1195. https://doi.org/10.3390/antiox11061195
APA StyleLin, K., Baritaki, S., Vivarelli, S., Falzone, L., Scalisi, A., Libra, M., & Bonavida, B. (2022). The Breast Cancer Protooncogenes HER2, BRCA1 and BRCA2 and Their Regulation by the iNOS/NOS2 Axis. Antioxidants, 11(6), 1195. https://doi.org/10.3390/antiox11061195