
Strategies of Pathogens to Escape from NO-Based Host Defense




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
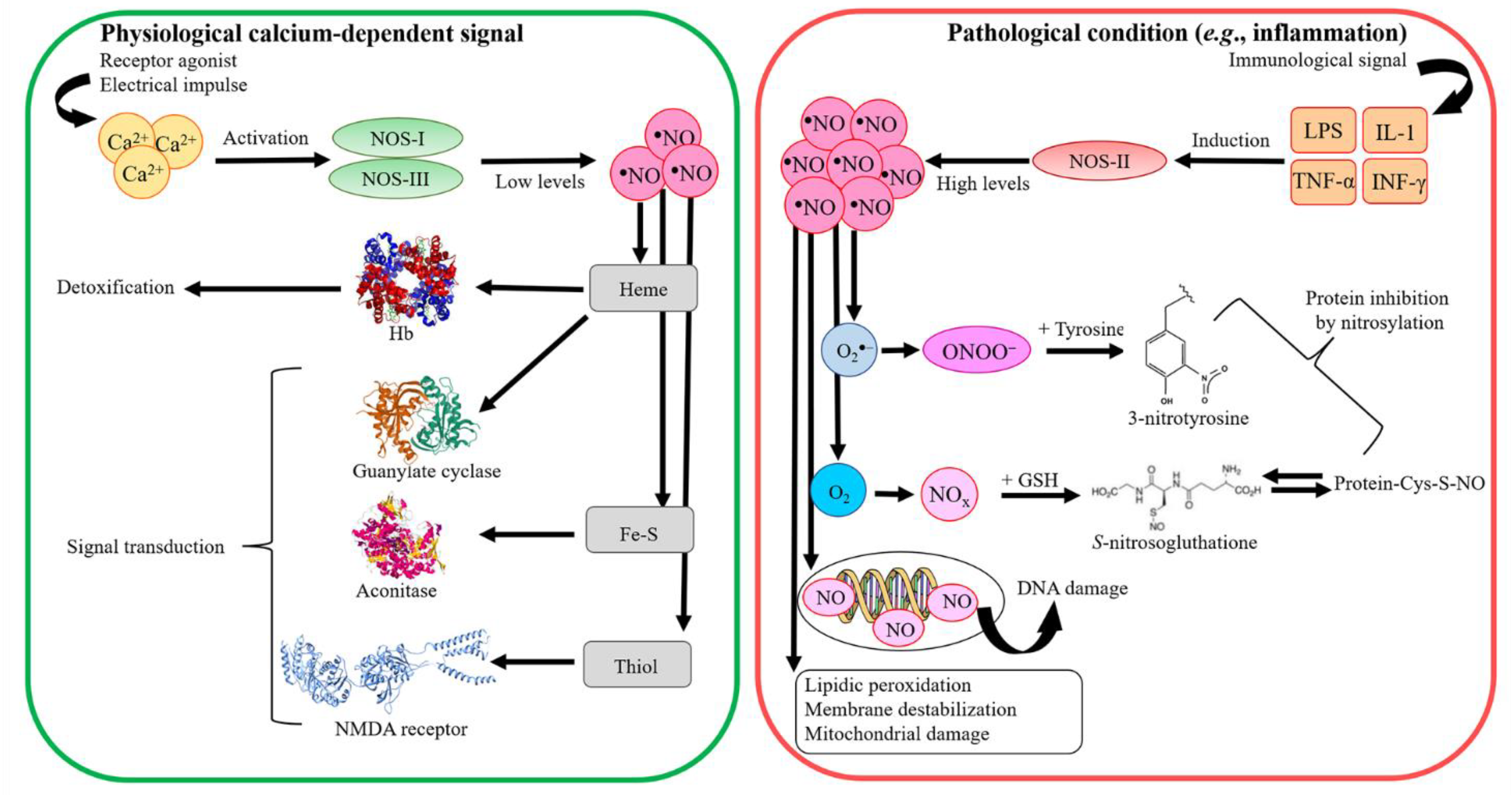
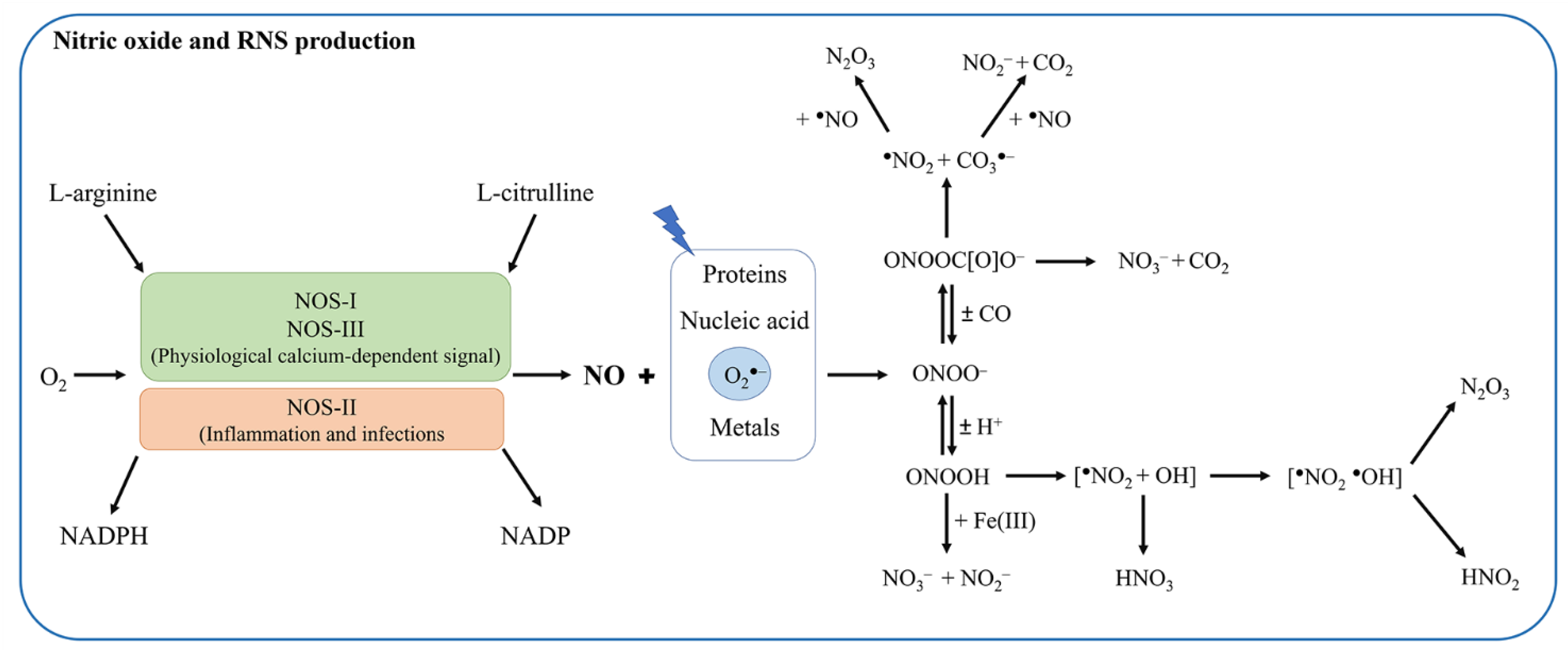
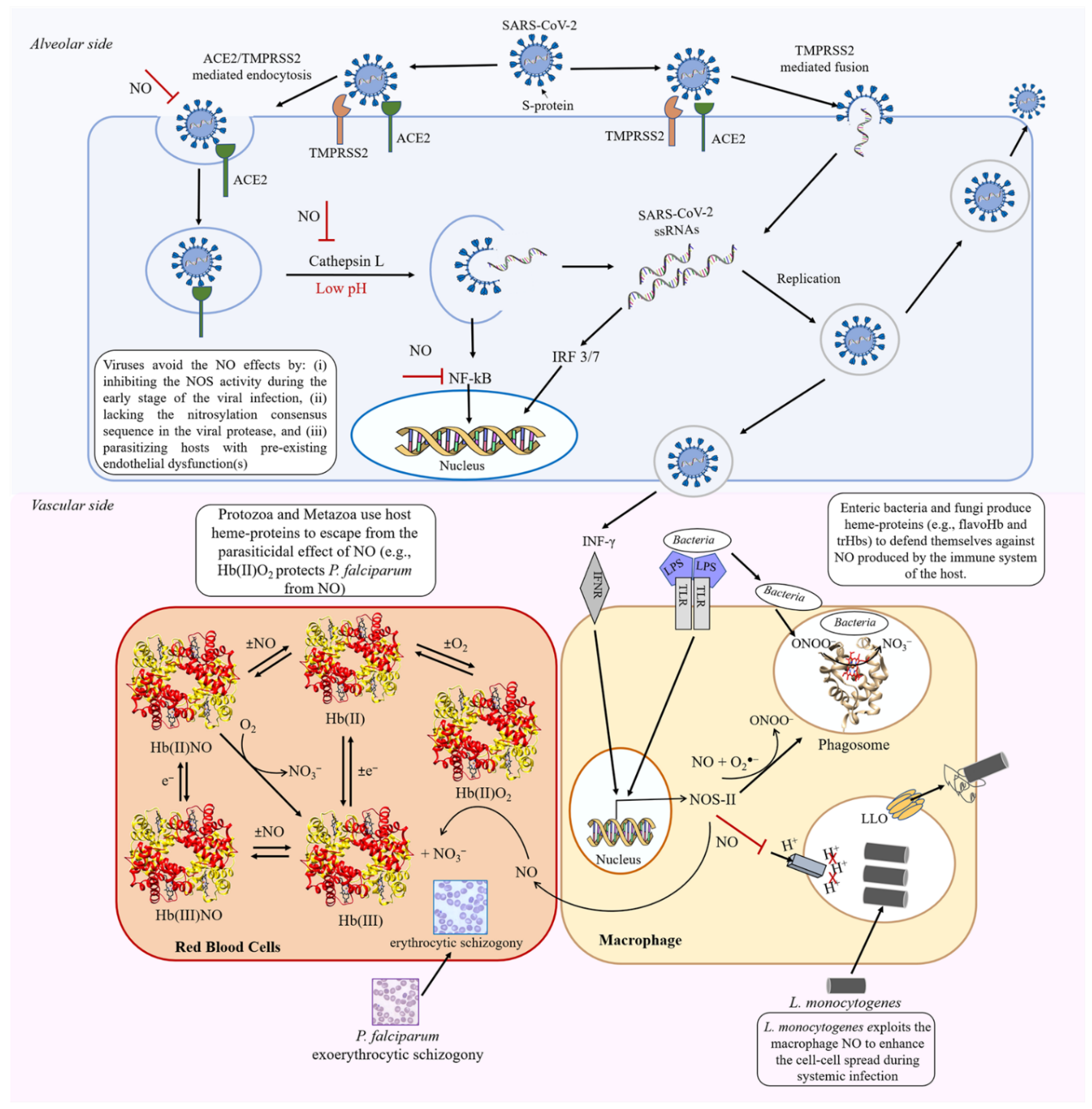
2. Differences in Macrophage NO Production and Regulation in Mice and Humans
3. Antiviral Action of NO
4. Effects of NO on Pathogenic Bacteria
4.1. NO Detoxification in Enteric Bacteria
4.2. NO Detoxification in M. leprae and M. tuberculosis
4.3. Lysteria Monocytogenes Escapes from NO
5. Antiparasitic Effects of NO on Protozoa and Metazoa
5.1. Trypanosoma cruzi
5.2. Toxoplasma gondii
5.3. Plasmodium falciparum
5.4. Schistosoma
5.5. Ascaris lumbricoides and Ascaris suum
6. Effects of NO on Fungal Infection
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 1983, 306, 174–176. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, T.J.; Hawkins, R.D.; Kandel, E.R.; Arancio, O. Tests of the roles of two diffusible substances in long-term potentiation: Evidence for nitric oxide as a possible early retrograde messenger. Proc. Natl. Acad. Sci. USA 1991, 88, 11285–11289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuman, E.M.; Madison, D.V. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 1991, 254, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, C.; Kilcoyne, C.M.; Cannon, R.O., 3rd; Panza, J.A. Interactions between nitric oxide and endothelin in the regulation of vascular tone of human resistance vessels in vivo. Hypertension 2000, 35, 1237–1241. [Google Scholar] [CrossRef] [Green Version]
- Stuart-Smith, K. Demystified. Nitric oxide. Mol. Pathol. 2002, 55, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Korbut, R.; Adamek-Guzik, T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 2003, 54, 469–487. [Google Scholar]
- Neuman, R.B.; Hayek, S.S.; Poole, J.C.; Rahman, A.; Menon, V.; Kavtaradze, N.; Polhemus, D.; Veledar, E.; Lefer, D.J.; Quyyumi, A.A. Nitric Oxide Contributes to Vasomotor Tone in Hypertensive African Americans Treated with Nebivolol and Metoprolol. J. Clin. Hypertens. 2016, 18, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Ghimire, K.; Altmann, H.M.; Straub, A.C.; Isenberg, J.S. Nitric oxide: What’s new to NO? Am. J. Physiol. Cell Physiol. 2017, 312, C254–C262. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M. Nitric oxide: Antidepressant mechanisms and inflammation. Adv. Pharmacol. 2019, 86, 121–152. [Google Scholar]
- Tewari, D.; Sah, A.N.; Bawari, S.; Nabavi, S.F.; Dehpour, A.R.; Shirooie, S.; Braidy, N.; Fiebich, B.L.; Vacca, R.A.; Nabavi, S.M. Role of Nitric Oxide in Neurodegeneration: Function, Regulation, and Inhibition. Curr. Neuropharmacol. 2021, 19, 114–126. [Google Scholar] [CrossRef]
- Kumar, R.; Coggan, A.R.; Ferreira, L.F. Nitric oxide and skeletal muscle contractile function. Nitric Oxide Biol. Chem. 2022, 122–123, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Dimmeler, S.; Molina y Vedia, L.; Lapetina, E.G. Nitric oxide: A signal for ADP-ribosylation of proteins. Life Sci. 1994, 54, 61–70. [Google Scholar] [CrossRef]
- Brune, B.; Lapetina, E.G. Protein thiol modification of glyceraldehyde-3-phosphate dehydrogenase as a target for nitric oxide signaling. Genet. Eng. 1995, 17, 149–164. [Google Scholar]
- Fillebeen, C.; Pantopoulos, K. Redox control of iron regulatory proteins. Redox Rep. 2002, 7, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.I.; Medvedeva, M.V.; Sevostyanova, I.A.; Schmalhausen, E.V. Modification of Glyceraldehyde-3-Phosphate Dehydrogenase with Nitric Oxide: Role in Signal Transduction and Development of Apoptosis. Biomolecules 2021, 11, 1656. [Google Scholar] [CrossRef]
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992, 6, 3051–3064. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Stuehr, D.J. Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 339–359. [Google Scholar] [CrossRef] [Green Version]
- Geller, D.A.; Billiar, T.R. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998, 17, 7–23. [Google Scholar] [CrossRef]
- Marletta, M.A.; Hurshman, A.R.; Rusche, K.M. Catalysis by nitric oxide synthase. Curr. Opin. Chem. Biol. 1998, 2, 656–663. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anavi, S.; Tirosh, O. iNOS as a metabolic enzyme under stress conditions. Free Radic. Biol. Med. 2020, 146, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Mader, M.M.; Boger, R.; Appel, D.; Schwedhelm, E.; Haddad, M.; Mohme, M.; Lamszus, K.; Westphal, M.; Czorlich, P.; Hannemann, J. Intrathecal and systemic alterations of L-arginine metabolism in patients after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2021, 41, 1964–1977. [Google Scholar] [CrossRef]
- Moncada, S. Nitric oxide in the vasculature: Physiology and pathophysiology. Ann. N. Y. Acad. Sci. 1997, 811, 60–67. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Gradoni, L. The anti-parasitic effects of nitric oxide. IUBMB Life 2003, 55, 573–578. [Google Scholar] [CrossRef]
- De Groote, M.A.; Fang, F.C. NO inhibitions: Antimicrobial properties of nitric oxide. Clin. Infect. Dis. 1995, 21 (Suppl. 2), S162–S165. [Google Scholar] [CrossRef]
- Clark, I.A.; Rockett, K.A. Nitric oxide and parasitic disease. Adv. Parasitol. 1996, 37, 1–56. [Google Scholar]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Garren, M.R.; Ashcraft, M.; Qian, Y.; Douglass, M.; Brisbois, E.J.; Handa, H. Nitric oxide and viral infection: Recent developments in antiviral therapies and platforms. Appl. Mater. Today 2021, 22, 100887. [Google Scholar] [CrossRef]
- Ignarro, L.J. Inhaled NO and COVID-19. Br. J. Pharmacol. 2020, 177, 3848–3849. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J. Lifestyle-mediated nitric oxide boost to prevent SARS-CoV-2 infection: A perspective. Nitric Oxide Biol. Chem. 2021, 115, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, S.; Lind, J.; Merenyi, G. Chemistry of peroxynitrites as compared to peroxynitrates. Chem. Rev. 2005, 105, 2457–2470. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Merenyi, G. The chemistry of peroxynitrite: Implications for biological activity. Methods Enzymol. 2008, 436, 49–61. [Google Scholar]
- Ahmad, R.; Hussain, A.; Ahsan, H. Peroxynitrite: Cellular pathology and implications in autoimmunity. J. Immunoass. Immunochem. 2019, 40, 123–138. [Google Scholar] [CrossRef]
- Augusto, O.; Goldstein, S.; Hurst, J.K.; Lind, J.; Lymar, S.V.; Merenyi, G.; Radi, R. Carbon dioxide-catalyzed peroxynitrite reactivity—The resilience of the radical mechanism after two decades of research. Free Radic. Biol. Med. 2019, 135, 210–215. [Google Scholar] [CrossRef]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef]
- Stamler, J.S. S-nitrosothiols and the bioregulatory actions of nitrogen oxides through reactions with thiol groups. Curr. Top. Microbiol. Immunol. 1995, 196, 19–36. [Google Scholar]
- Stamler, J.S.; Hausladen, A. Oxidative modifications in nitrosative stress. Nat. Struct. Biol. 1998, 5, 247–249. [Google Scholar] [CrossRef]
- Smith, B.C.; Marletta, M.A. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr. Opin. Chem. Biol. 2012, 16, 498–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, T.A.; da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; Wink, D.A.; Fukuto, J.M. Biological nitric oxide signalling: Chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Singel, D.J.; Stamler, J.S. The enzymatic function of the honorary enzyme: S-nitrosylation of hemoglobin in physiology and medicine. Mol. Asp. Med. 2022, 84, 101056. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Gradoni, L. Nitric oxide limits parasite development in vectors and in invertebrate intermediate hosts. IUBMB Life 2002, 53, 121–123. [Google Scholar]
- Ascenzi, P.; Bocedi, A.; Bolognesi, M.; Fabozzi, G.; Milani, M.; Visca, P. Nitric oxide scavenging by Mycobacterium leprae GlbO involves the formation of the ferric heme-bound peroxynitrite intermediate. Biochem. Biophys. Res. Commun. 2006, 339, 450–456. [Google Scholar] [CrossRef]
- Ascenzi, P.; Visca, P. Scavenging of reactive nitrogen species by mycobacterial truncated hemoglobins. Methods Enzymol. 2008, 436, 317–337. [Google Scholar]
- Arkenberg, A.; Runkel, S.; Richardson, D.J.; Rowley, G. The production and detoxification of a potent cytotoxin, nitric oxide, by pathogenic enteric bacteria. Biochem. Soc. Trans. 2011, 39, 1876–1879. [Google Scholar] [CrossRef]
- Bogdan, C. Listeria monocytogenes: No spreading without NO. Immunity 2012, 36, 697–699. [Google Scholar] [CrossRef] [Green Version]
- Cole, C.; Thomas, S.; Filak, H.; Henson, P.M.; Lenz, L.L. Nitric oxide increases susceptibility of Toll-like receptor-activated macrophages to spreading Listeria monocytogenes. Immunity 2012, 36, 807–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, J.; Sen, U.; Choudhury, D.; Datta, P.; Chakrabarti, A.; Chakrabarty, S.B.; Chakrabarty, A.; Dattagupta, J.K. Crystallization and preliminary X-ray structural studies of hemoglobin A2 and hemoglobin E, isolated from the blood samples of beta-thalassemic patients. Biochem. Biophys. Res. Commun. 2003, 303, 619–623. [Google Scholar] [CrossRef]
- Seeger, F.; Quintyn, R.; Tanimoto, A.; Williams, G.J.; Tainer, J.A.; Wysocki, V.H.; Garcin, E.D. Interfacial residues promote an optimal alignment of the catalytic center in human soluble guanylate cyclase: Heterodimerization is required but not sufficient for activity. Biochemistry 2014, 53, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, J.; Volbeda, A.; Carpentier, P.; Darnault, C.; Moulis, J.M.; Fontecilla-Camps, J.C. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure 2006, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.B.; Chang, S.; Xu, P.; Miao, M.; Wu, H.; Zhang, Y.; Zhang, T.; Wang, H.; Zhang, J.; Xie, C.; et al. Structural Basis of the Proton Sensitivity of Human GluN1-GluN2A NMDA Receptors. Cell Rep. 2018, 25, 3582–3590.e4. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Reade, M.C.; Young, J.D. Of mice and men (and rats): Implications of species and stimulus differences for the interpretation of studies of nitric oxide in sepsis. Br. J. Anaesth. 2003, 90, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Schneemann, M.; Schoedon, G.; Hofer, S.; Blau, N.; Guerrero, L.; Schaffner, A. Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J. Infect. Dis. 1993, 167, 1358–11363. [Google Scholar] [CrossRef]
- Schoedon, G.; Schneemann, M.; Hofer, S.; Guerrero, L.; Blau, N.; Schaffner, A. Regulation of the L-arginine-dependent and tetrahydrobiopterin-dependent biosynthesis of nitric oxide in murine macrophages. Eur. J. Biochem. 1993, 213, 833–839. [Google Scholar] [CrossRef]
- Albina, J.E.; Reichner, J.S. Oxygen and the regulation of gene expression in wounds. Wound Repair Regen. 2003, 11, 445–451. [Google Scholar] [CrossRef]
- Schneemann, M.; Schoeden, G. Macrophage biology and immunology: Man is not a mouse. J. Leukoc. Biol. 2007, 81, 579; discussion 580. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Mattila, J.T. “Of mice and men”: Arginine metabolism in macrophages. Front. Immunol. 2014, 5, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayeh, M.A.; Marletta, M.A. Macrophage oxidation of L-arginine to nitric oxide, nitrite, and nitrate. Tetrahydrobiopterin is required as a cofactor. J. Biol. Chem. 1989, 264, 19654–19658. [Google Scholar] [CrossRef]
- Pfister, H.; Remer, K.A.; Brcic, M.; Fatzer, R.; Christen, S.; Leib, S.; Jungi, T.W. Inducible nitric oxide synthase and nitrotyrosine in listeric encephalitis: A cross-species study in ruminants. Vet. Pathol. 2002, 39, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, L.E.; Chandrasekar, B.; Saldarriaga, O.A.; Zhao, W.; Arteaga, L.T.; Travi, B.L.; Melby, P.C. Reduced nitric oxide synthase 2 (NOS2) promoter activity in the Syrian hamster renders the animal functionally deficient in NOS2 activity and unable to control an intracellular pathogen. J. Immunol. 2006, 176, 5519–5528. [Google Scholar] [CrossRef] [Green Version]
- Kleinert, H.; Pautz, A.; Linker, K.; Schwarz, P.M. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol. 2004, 500, 255–266. [Google Scholar] [CrossRef]
- Lima-Junior, D.S.; Costa, D.L.; Carregaro, V.; Cunha, L.D.; Silva, A.L.; Mineo, T.W.; Gutierrez, F.R.; Bellio, M.; Bortoluci, K.R.; Flavell, R.A.; et al. Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nat. Med. 2013, 19, 909–915. [Google Scholar] [CrossRef]
- Pautz, A.; Art, J.; Hahn, S.; Nowag, S.; Voss, C.; Kleinert, H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide Biol. Chem. 2010, 23, 75–93. [Google Scholar] [CrossRef]
- Gross, T.J.; Kremens, K.; Powers, L.S.; Brink, B.; Knutson, T.; Domann, F.E.; Philibert, R.A.; Milhem, M.M.; Monick, M.M. Epigenetic silencing of the human NOS2 gene: Rethinking the role of nitric oxide in human macrophage inflammatory responses. J. Immunol. 2014, 192, 2326–2338. [Google Scholar] [CrossRef]
- Chesrown, S.E.; Monnier, J.; Visner, G.; Nick, H.S. Regulation of inducible nitric oxide synthase mRNA levels by LPS, INF-gamma, TGF-beta, and IL-10 in murine macrophage cell lines and rat peritoneal macrophages. Biochem. Biophys. Res. Commun. 1994, 200, 126–134. [Google Scholar] [CrossRef]
- Weinberg, J.B.; Misukonis, M.A.; Shami, P.J.; Mason, S.N.; Sauls, D.L.; Dittman, W.A.; Wood, E.R.; Smith, G.K.; McDonald, B.; Bachus, K.E.; et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 1995, 86, 1184–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colasanti, M.; Persichini, T.; Venturini, G.; Ascenzi, P. S-nitrosylation of viral proteins: Molecular bases for antiviral effect of nitric oxide. IUBMB Life 1999, 48, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Colasanti, M.; Gradoni, L.; Mattu, M.; Persichini, T.; Salvati, L.; Venturini, G.; Ascenzi, P. Molecular bases for the anti-parasitic effect of NO (Review). Int. J. Mol. Med. 2002, 9, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.R.; Ashkar, A.A.; Mossman, K.L. The nitric oxide pathway provides innate antiviral protection in conjunction with the type I interferon pathway in fibroblasts. PLoS ONE 2012, 7, e31688. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Cader, M.S.; Amarasinghe, A.; Abdul-Careem, M.F. Activation of toll-like receptor signaling pathways leading to nitric oxide-mediated antiviral responses. Arch. Virol. 2016, 161, 2075–2086. [Google Scholar] [CrossRef]
- Lisi, F.; Zelikin, A.N.; Chandrawati, R. Nitric Oxide to Fight Viral Infections. Adv. Sci. 2021, 8, 2003895. [Google Scholar] [CrossRef]
- Powell, K.L.; Baylis, S.A. The antiviral effects of nitric oxide. Trends Microbiol. 1995, 3, 81–82. [Google Scholar] [CrossRef]
- Peterhans, E. Reactive oxygen species and nitric oxide in viral diseases. Biol. Trace Elem. Res. 1997, 56, 107–116. [Google Scholar] [CrossRef]
- Maeda, H.; Akaike, T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry 1998, 63, 854–865. [Google Scholar]
- Olcott, M.C.; Andersson, J.; Sjoberg, B.M. Localization and characterization of two nucleotide-binding sites on the anaerobic ribonucleotide reductase from bacteriophage T4. J. Biol. Chem. 1998, 273, 24853–24860. [Google Scholar] [CrossRef]
- Reiss, C.S.; Komatsu, T. Does nitric oxide play a critical role in viral infections? J. Virol. 1998, 72, 4547–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Gravitt, P.E.; Song, H.; Maldonado, A.M.; Ozbun, M.A. Nitric oxide induces early viral transcription coincident with increased DNA damage and mutation rates in human papillomavirus-infected cells. Cancer Res. 2009, 69, 4878–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Zheng, S.; Dweik, R.A.; Erzurum, S.C. Role of epithelial nitric oxide in airway viral infection. Free Radic. Biol. Med. 2006, 41, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Adler, H.; Beland, J.L.; Del-Pan, N.C.; Kobzik, L.; Brewer, J.P.; Martin, T.R.; Rimm, I.J. Suppression of herpes simplex virus type 1 (HSV-1)-induced pneumonia in mice by inhibition of inducible nitric oxide synthase (iNOS, NOS2). J. Exp. Med. 1997, 185, 1533–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihm, S.; Fayyazi, A.; Ramadori, G. Hepatic expression of inducible nitric oxide synthase transcripts in chronic hepatitis C virus infection: Relation to hepatic viral load and liver injury. Hepatology 1997, 26, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Kandemir, O.; Polat, A.; Kaya, A. Inducible nitric oxide synthase expression in chronic viral hepatitis and its relation with histological severity of disease. J. Viral Hepat. 2002, 9, 419–423. [Google Scholar] [CrossRef]
- Valero, N.; Espina, L.M.; Anez, G.; Torres, E.; Mosquera, J.A. Short report: Increased level of serum nitric oxide in patients with dengue. Am. J. Trop. Med. Hyg. 2002, 66, 762–764. [Google Scholar] [CrossRef] [Green Version]
- Gamba, G.; Cavalieri, H.; Courreges, M.C.; Massouh, E.J.; Benencia, F. Early inhibition of nitric oxide production increases HSV-1 intranasal infection. J. Med. Virol. 2004, 73, 313–322. [Google Scholar] [CrossRef]
- Sanders, S.P.; Siekierski, E.S.; Richards, S.M.; Porter, J.D.; Imani, F.; Proud, D. Rhinovirus infection induces expression of type 2 nitric oxide synthase in human respiratory epithelial cells in vitro and in vivo. J. Allergy Clin. Immunol. 2001, 107, 235–243. [Google Scholar] [CrossRef]
- Sanders, S.P.; Proud, D.; Permutt, S.; Siekierski, E.S.; Yachechko, R.; Liu, M.C. Role of nasal nitric oxide in the resolution of experimental rhinovirus infection. J. Allergy Clin. Immunol. 2004, 113, 697–702. [Google Scholar] [CrossRef]
- McGill, J.; Heusel, J.W.; Legge, K.L. Innate immune control and regulation of influenza virus infections. J. Leukoc. Biol. 2009, 86, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasson, J.; Weidenfeld, J.; Mizrachi-Kol, R.; Ben-Hur, T.; Ovadia, H. The effect of herpes simplex virus-1 on nitric oxide synthase in the rat brain: The role of glucocorticoids. Neuroimmunomodulation 2011, 18, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Toone, E.J.; Lipton, S.A.; Sucher, N.J. (S)NO signals: Translocation, regulation, and a consensus motif. Neuron 1997, 18, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Saura, M.; Zaragoza, C.; McMillan, A.; Quick, R.A.; Hohenadl, C.; Lowenstein, J.M.; Lowenstein, C.J. An antiviral mechanism of nitric oxide: Inhibition of a viral protease. Immunity 1999, 10, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, N.S.; Moore, D.W.; Wang, H.K.; Broker, T.R.; Chow, L.T. NVN1000, a novel nitric oxide-releasing compound, inhibits HPV-18 virus production by interfering with E6 and E7 oncoprotein functions. Antivir. Res. 2019, 170, 104559. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Lynch, J.B.; Del Rio, C. Mild or Moderate COVID-19. N. Engl. J. Med. 2020, 383, 1757–1766. [Google Scholar] [CrossRef]
- Li, H.; Tian, S.; Chen, T.; Cui, Z.; Shi, N.; Zhong, X.; Qiu, K.; Zhang, J.; Zeng, T.; Chen, L.; et al. Newly diagnosed diabetes is associated with a higher risk of mortality than known diabetes in hospitalized patients with COVID-19. Diabetes Obes. Metab. 2020, 22, 1897–1906. [Google Scholar] [CrossRef]
- Cao, W.; Bao, C.; Lowenstein, C.J. Inducible nitric oxide synthase expression inhibition by adenovirus E1A. Proc. Natl. Acad. Sci. USA 2003, 100, 7773–7778. [Google Scholar] [CrossRef] [Green Version]
- Mannick, J.B. The antiviral role of nitric oxide. Res. Immunol. 1995, 146, 693–697. [Google Scholar] [CrossRef]
- Akerstrom, S.; Gunalan, V.; Keng, C.T.; Tan, Y.J.; Mirazimi, A. Dual effect of nitric oxide on SARS-CoV replication: Viral RNA production and palmitoylation of the S protein are affected. Virology 2009, 395, 1–9. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaberi, D.; Krambrich, J.; Ling, J.; Luni, C.; Hedenstierna, G.; Jarhult, J.D.; Lennerstrand, J.; Lundkvist, A. Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol. 2020, 37, 101734. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Gioia, M.; Ciaccio, C.; Calligari, P.; De Simone, G.; Sbardella, D.; Tundo, G.; Fasciglione, G.F.; Di Masi, A.; Di Pierro, D.; Bocedi, A.; et al. Role of proteolytic enzymes in the COVID-19 infection and promising therapeutic approaches. Biochem. Pharmacol. 2020, 182, 114225. [Google Scholar] [CrossRef] [PubMed]
- Kidde, J.; Sahebkar, A. From Foe to Friend in COVID-19: Renin-Angiotensin System Inhibitors. J. Infect. Dis. 2021, 223, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, P.C.; Lanctot, P.M.; Auger-Messier, M.; Escher, E.; Leduc, R.; Guillemette, G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br. J. Pharmacol. 2006, 148, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Pinheiro, L.C.; Oliveira-Paula, G.H.; Ferreira, G.C.; Dal-Cin de Paula, T.; Duarte, D.A.; Costa-Neto, C.M.; Tanus-Santos, J.E. Oral nitrite treatment increases S-nitrosylation of vascular protein kinase C and attenuates the responses to angiotensin II. Redox Biol. 2021, 38, 101769. [Google Scholar] [CrossRef]
- Forstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010, 459, 923–939. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A.; et al. Effect of SARS-CoV-2 proteins on vascular permeability. Elife 2021, 10, e69314. [Google Scholar] [CrossRef] [PubMed]
- Egashira, K.; Inou, T.; Hirooka, Y.; Kai, H.; Sugimachi, M.; Suzuki, S.; Kuga, T.; Urabe, Y.; Takeshita, A. Effects of age on endothelium-dependent vasodilation of resistance coronary artery by acetylcholine in humans. Circulation 1993, 88, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, G.; Bernini, G.; Magagna, A.; Salvetti, A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001, 38, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Torregrossa, A.C.; Aranke, M.; Bryan, N.S. Nitric oxide and geriatrics: Implications in diagnostics and treatment of the elderly. J. Geriatr. Cardiol. 2011, 8, 230–242. [Google Scholar] [PubMed] [Green Version]
- Nevzati, E.; Shafighi, M.; Bakhtian, K.D.; Treiber, H.; Fandino, J.; Fathi, A.R. Estrogen induces nitric oxide production via nitric oxide synthase activation in endothelial cells. Acta Neurochir. Suppl. 2015, 120, 141–145. [Google Scholar] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilchrist, M.; Winyard, P.G.; Benjamin, N. Dietary nitrate-good or bad? Nitric Oxide Biol. Chem. 2010, 22, 104–109. [Google Scholar] [CrossRef]
- Fabozzi, G.; Ascenzi, P.; Renzi, S.D.; Visca, P. Truncated hemoglobin GlbO from Mycobacterium leprae alleviates nitric oxide toxicity. Microb. Pathog. 2006, 40, 211–220. [Google Scholar] [CrossRef]
- Nardini, M.; Pesce, A.; Bolognesi, M. Truncated (2/2) hemoglobin: Unconventional structures and functional roles in vivo and in human pathogenesis. Mol. Asp. Med. 2022, 84, 101049. [Google Scholar] [CrossRef]
- Crawford, M.J.; Goldberg, D.E. Role for the Salmonella flavohemoglobin in protection from nitric oxide. J. Biol. Chem. 1998, 273, 12543–12547. [Google Scholar] [CrossRef]
- Mills, P.C.; Rowley, G.; Spiro, S.; Hinton, J.C.D.; Richardson, D.J. A combination of cytochrome c nitrite reductase (NrfA) and flavorubredoxin (NorV) protects Salmonella enterica serovar Typhimurium against killing by NO in anoxic environments. Microbiology 2008, 154, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, M.; Ghosh, T.; Tucker, N.; Zhang, X.; Dixon, R. Transcriptional regulation by the dedicated nitric oxide sensor, NorR: A route towards NO detoxification. Biochem. Soc. Trans. 2011, 39, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, R.K. Flavohaemoglobin: The pre-eminent nitric oxide-detoxifying machine of microorganisms. F1000Research 2020, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Membrillo-Hernandez, J.; Ioannidis, N.; Poole, R.K. The flavohaemoglobin (HMP) of Escherichia coli generates superoxide in vitro and causes oxidative stress in vivo. FEBS Lett. 1996, 382, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Membrillo-Hernandez, J.; Coopamah, M.D.; Anjum, M.F.; Stevanin, T.M.; Kelly, A.; Hughes, M.N.; Poole, R.K. The flavohemoglobin of Escherichia coli confers resistance to a nitrosating agent, a “Nitric oxide Releaser,” and paraquat and is essential for transcriptional responses to oxidative stress. J. Biol. Chem. 1999, 274, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausladen, A.; Gow, A.J.; Stamler, J.S. Nitrosative stress: Metabolic pathway involving the flavohemoglobin. Proc. Natl. Acad. Sci. USA 1998, 95, 14100–14105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonamore, A.; Boffi, A. Flavohemoglobin: Structure and reactivity. IUBMB Life 2008, 60, 19–28. [Google Scholar] [CrossRef]
- Gardner, P.R.; Gardner, A.M.; Martin, L.A.; Salzman, A.L. Nitric oxide dioxygenase: An enzymic function for flavohemoglobin. Proc. Natl. Acad. Sci. USA 1998, 95, 10378–10383. [Google Scholar] [CrossRef] [Green Version]
- Gardner, P.R.; Gardner, A.M.; Martin, L.A.; Dou, Y.; Li, T.; Olson, J.S.; Zhu, H.; Riggs, A.F. Nitric-oxide dioxygenase activity and function of flavohemoglobins. sensitivity to nitric oxide and carbon monoxide inhibition. J. Biol. Chem. 2000, 275, 31581–31587. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.M.; Gardner, P.R. Flavohemoglobin detoxifies nitric oxide in aerobic, but not anaerobic, Escherichia coli. Evidence for a novel inducible anaerobic nitric oxide-scavenging activity. J. Biol. Chem. 2002, 277, 8166–8171. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Dubchak, I.L.; Arkin, A.P.; Alm, E.J.; Gelfand, M.S. Dissimilatory metabolism of nitrogen oxides in bacteria: Comparative reconstruction of transcriptional networks. PLoS Comput. Biol. 2005, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Spiro, S. Regulators of bacterial responses to nitric oxide. FEMS Microbiol. Rev. 2007, 31, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Constantinidou, C.; Hobman, J.L.; Griffiths, L.; Patel, M.D.; Penn, C.W.; Cole, J.A.; Overton, T.W. A reassessment of the FNR regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J. Biol. Chem. 2006, 281, 4802–4815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overton, T.W.; Griffiths, L.; Patel, M.D.; Hobman, J.L.; Penn, C.W.; Cole, J.A.; Constantinidou, C. Microarray analysis of gene regulation by oxygen, nitrate, nitrite, FNR, NarL and NarP during anaerobic growth of Escherichia coli: New insights into microbial physiology. Biochem. Soc. Trans. 2006, 34, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, R.C.; Evans, M.R.; Porwollik, S.; Vazquez-Torres, A.; Jones-Carson, J.; Troxell, B.; Libby, S.J.; McClelland, M.; Hassan, H.M. FNR is a global regulator of virulence and anaerobic metabolism in Salmonella enterica serovar Typhimurium (ATCC 14028s). J. Bacteriol. 2007, 189, 2262–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullan, S.T.; Gidley, M.D.; Jones, R.A.; Barrett, J.; Stevanin, T.M.; Read, R.C.; Green, J.; Poole, R.K. Nitric oxide in chemostat-cultured Escherichia coli is sensed by Fnr and other global regulators: Unaltered methionine biosynthesis indicates lack of S nitrosation. J. Bacteriol. 2007, 189, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Iovine, N.M.; Pursnani, S.; Voldman, A.; Wasserman, G.; Blaser, M.J.; Weinrauch, Y. Reactive nitrogen species contribute to innate host defense against Campylobacter jejuni. Infect. Immun. 2008, 76, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Callahan, S.M.; Dolislager, C.G.; Johnson, J.G. The host cellular immune response to infection by Campylobacter spp. and its role in disease. Infect. Immun. 2021, 89, e0011621. [Google Scholar] [CrossRef]
- Wainwright, L.M.; Elvers, K.T.; Park, S.F.; Poole, R.K. A truncated haemoglobin implicated in oxygen metabolism by the microaerophilic food-borne pathogen Campylobacter jejuni. Microbiology 2005, 151, 4079–4091. [Google Scholar] [CrossRef] [Green Version]
- Nardini, M.; Pesce, A.; Labarre, M.; Richard, C.; Bolli, A.; Ascenzi, P.; Guertin, M.; Bolognesi, M. Structural determinants in the group III truncated hemoglobin from Campylobacter jejuni. J. Biol. Chem. 2006, 281, 37803–37812. [Google Scholar] [CrossRef]
- Wainwright, L.M.; Wang, Y.; Park, S.F.; Yeh, S.R.; Poole, R.K. Purification and spectroscopic characterization of Ctb, a group III truncated hemoglobin implicated in oxygen metabolism in the food-borne pathogen Campylobacter jejuni. Biochemistry 2006, 45, 6003–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinajero-Trejo, M.; Shepherd, M. The globins of Campylobacter jejuni. Adv. Microb. Physiol. 2013, 63, 97–145. [Google Scholar] [PubMed]
- Tinajero-Trejo, M.; Vreugdenhil, A.; Sedelnikova, S.E.; Davidge, K.S.; Poole, R.K. Nitric oxide reactivities of the two globins of the foodborne pathogen Campylobacter jejuni: Roles in protection from nitrosative stress and analysis of potential reductants. Nitric Oxide Biol. Chem. 2013, 34, 65–75. [Google Scholar] [CrossRef]
- Ascenzi, P.; di Masi, A.; Tundo, G.R.; Pesce, A.; Visca, P.; Coletta, M. Nitrosylation mechanisms of Mycobacterium tuberculosis and Campylobacter jejuni truncated hemoglobins N, O, and P. PLoS ONE 2014, 9, e102811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascenzi, P.; Pesce, A. Peroxynitrite scavenging by Campylobacter jejuni truncated hemoglobin P. J. Biol. Inorg. Chem. 2017, 22, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Elvers, K.T.; Wu, G.; Gilberthorpe, N.J.; Poole, R.K.; Park, S.F. Role of an inducible single-domain hemoglobin in mediating resistance to nitric oxide and nitrosative stress in Campylobacter jejuni and Campylobacter coli. J. Bacteriol. 2004, 186, 5332–5341. [Google Scholar] [CrossRef] [Green Version]
- Avila-Ramirez, C.; Tinajero-Trejo, M.; Davidge, K.S.; Monk, C.E.; Kelly, D.J.; Poole, R.K. Do globins in microaerophilic Campylobacter jejuni confer nitrosative stress tolerance under oxygen limitation? Antioxid. Redox Signal. 2013, 18, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Egawa, T.; Wainwright, L.M.; Poole, R.K.; Yeh, S.R. Structural and functional properties of a truncated hemoglobin from a food-borne pathogen Campylobacter jejuni. J. Biol. Chem. 2007, 282, 13627–13636. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T. Leprosy and tuberculosis: An insight-review. Crit. Rev. Microbiol. 2007, 33, 15–66. [Google Scholar] [CrossRef]
- Schalcher, T.R.; Vieira, J.L.; Salgado, C.G.; Borges Rdos, S.; Monteiro, M.C. Antioxidant factors, nitric oxide levels, and cellular damage in leprosy patients. Rev. Soc. Bras. Med. Trop. 2013, 46, 645–649. [Google Scholar] [CrossRef]
- Keragala, B.; Herath, H.; Janapriya, G.; Vanitha, S.; Balendran, T.; Janani, T.; Keragala, T.S.; Gunasekera, C.N. Coexistence of mycobacterial infections—Mycobacterium tuberculosis and Mycobacterium leprae—In Sri Lanka: A case series. J. Med. Case Rep. 2020, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- Schon, T.; Gebre, N.; Sundqvist, T.; Habetmariam, H.S.; Engeda, T.; Britton, S. Increased levels of nitric oxide metabolites in urine from leprosy patients in reversal reaction. Lepr. Rev. 1999, 70, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Schon, T.; Hernandez-Pando, R.H.; Negesse, Y.; Leekassa, R.; Sundqvist, T.; Britton, S. Expression of inducible nitric oxide synthase and nitrotyrosine in borderline leprosy lesions. Br. J. Dermatol. 2001, 145, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Couture, M.; Yeh, S.R.; Wittenberg, B.A.; Wittenberg, J.B.; Ouellet, Y.; Rousseau, D.L.; Guertin, M. A cooperative oxygen-binding hemoglobin from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 1999, 96, 11223–11228. [Google Scholar] [CrossRef] [Green Version]
- Ouellet, H.; Ouellet, Y.; Richard, C.; Labarre, M.; Wittenberg, B.; Wittenberg, J.; Guertin, M. Truncated hemoglobin HbN protects Mycobacterium bovis from nitric oxide. Proc. Natl. Acad. Sci. USA 2002, 99, 5902–5907. [Google Scholar] [CrossRef] [Green Version]
- Pathania, R.; Navani, N.K.; Gardner, A.M.; Gardner, P.R.; Dikshit, K.L. Nitric oxide scavenging and detoxification by the Mycobacterium tuberculosis haemoglobin, HbN in Escherichia coli. Mol. Microbiol. 2002, 45, 1303–1314. [Google Scholar] [CrossRef]
- Milani, M.; Pesce, A.; Ouellet, H.; Guertin, M.; Bolognesi, M. Truncated hemoglobins and nitric oxide action. IUBMB Life 2003, 55, 623–627. [Google Scholar] [CrossRef]
- Ascenzi, P.; Milani, M.; Visca, P. Peroxynitrite scavenging by ferrous truncated hemoglobin GlbO from Mycobacterium leprae. Biochem. Biophys. Res. Commun. 2006, 351, 528–533. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bolognesi, M.; Milani, M.; Guertin, M.; Visca, P. Mycobacterial truncated hemoglobins: From genes to functions. Gene 2007, 398, 42–51. [Google Scholar] [CrossRef]
- Lama, A.; Pawaria, S.; Dikshit, K.L. Oxygen binding and NO scavenging properties of truncated hemoglobin, HbN, of Mycobacterium smegmatis. FEBS Lett. 2006, 580, 4031–4041. [Google Scholar] [CrossRef]
- Davidge, K.S.; Dikshit, K.L. Haemoglobins of Mycobacteria: Structural features and biological functions. Adv. Microb. Physiol 2013, 63, 147–194. [Google Scholar] [PubMed]
- Nardini, M.; Pesce, A.; Milani, M.; Bolognesi, M. Protein fold and structure in the truncated (2/2) globin family. Gene 2007, 398, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Vuletich, D.A.; Lecomte, J.T. A phylogenetic and structural analysis of truncated hemoglobins. J. Mol. Evol. 2006, 62, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, J.B.; Bolognesi, M.; Wittenberg, B.A.; Guertin, M. Truncated hemoglobins: A new family of hemoglobins widely distributed in bacteria, unicellular eukaryotes, and plants. J. Biol. Chem. 2002, 277, 871–874. [Google Scholar] [CrossRef] [Green Version]
- Ascenzi, P.; De Marinis, E.; Visca, P.; Ciaccio, C.; Coletta, M. Peroxynitrite detoxification by ferryl Mycobacterium leprae truncated hemoglobin O. Biochem. Biophys. Res. Commun. 2009, 380, 392–396. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.P.; Bisht, S.S.; Ajay, A.; Sharma, A.; Misra, M.; Gupt, M.P. Developments in chemical approaches to treat tuberculosis in the last decade. Curr. Med. Chem. 2012, 19, 488–517. [Google Scholar] [CrossRef]
- Zumla, A.; Chakaya, J.; Centis, R.; D’Ambrosio, L.; Mwaba, P.; Bates, M.; Kapata, N.; Nyirenda, T.; Chanda, D.; Mfinanga, S.; et al. Tuberculosis treatment and management—An update on treatment regimens, trials, new drugs, and adjunct therapies. Lancet Respir Med. 2015, 3, 220–234. [Google Scholar] [CrossRef]
- Ascenzi, P.; Coletta, A.; Cao, Y.; Trezza, V.; Leboffe, L.; Fanali, G.; Fasano, M.; Pesce, A.; Ciaccio, C.; Marini, S.; et al. Isoniazid inhibits the heme-based reactivity of Mycobacterium tuberculosis truncated hemoglobin N. PLoS ONE 2013, 8, e69762. [Google Scholar] [CrossRef] [Green Version]
- De Simone, G.; Ascenzi, P.; Polticelli, F. Nitrobindin: An Ubiquitous Family of All beta-Barrel Heme-proteins. IUBMB Life 2016, 68, 423–428. [Google Scholar] [CrossRef] [Green Version]
- De Simone, G.; di Masi, A.; Polticelli, F.; Ascenzi, P. Human nitrobindin: The first example of an all-beta-barrel ferric heme-protein that catalyzes peroxynitrite detoxification. FEBS Open Bio 2018, 8, 2002–2010. [Google Scholar] [CrossRef]
- De Simone, G.; di Masi, A.; Ciaccio, C.; Coletta, M.; Ascenzi, P. NO Scavenging through reductive nitrosylation of ferric Mycobacterium tuberculosis and Homo sapiens Nitrobindins. Int. J. Mol. Sci. 2020, 21, 9395. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; di Masi, A.; Vita, G.M.; Polticelli, F.; Pesce, A.; Nardini, M.; Bolognesi, M.; Ciaccio, C.; Coletta, M.; Turilli, E.S.; et al. Mycobacterial and human nitrobindins: Structure and function. Antioxid. Redox Signal. 2020, 33, 229–246. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; di Masi, A.; Fattibene, P.; Ciaccio, C.; Platas-Iglesias, C.; Coletta, M.; Pesce, A.; Ascenzi, P. Oxygen-mediated oxidation of ferrous nitrosylated nitrobindins. J. Inorg. Biochem. 2021, 224, 111579. [Google Scholar] [CrossRef]
- Lecuit, M. Listeria monocytogenes, a model in infection biology. Cell. Microbiol. 2020, 22, e13186. [Google Scholar] [CrossRef] [Green Version]
- Cossart, P.; Vicente, M.F.; Mengaud, J.; Baquero, F.; Perez-Diaz, J.C.; Berche, P. Listeriolysin O is essential for virulence of Listeria monocytogenes: Direct evidence obtained by gene complementation. Infect. Immun. 1989, 57, 3629–3636. [Google Scholar] [CrossRef] [Green Version]
- Tojo, A.; Guzman, N.J.; Garg, L.C.; Tisher, C.C.; Madsen, K.M. Nitric oxide inhibits bafilomycin-sensitive H(+)-ATPase activity in rat cortical collecting duct. Am. J. Physiol. 1994, 267, F509–F515. [Google Scholar] [CrossRef]
- Forgac, M. The vacuolar H+-ATPase of clathrin-coated vesicles is reversibly inhibited by S-nitrosoglutathione. J. Biol. Chem. 1999, 274, 1301–1305. [Google Scholar] [CrossRef] [Green Version]
- Akaike, T.; Fujii, S.; Sawa, T.; Ihara, H. Cell signaling mediated by nitrated cyclic guanine nucleotide. Nitric Oxide Biol. Chem. 2010, 23, 166–174. [Google Scholar] [CrossRef]
- Ascenzi, P.; Salvati, L.; Brunori, M. Does myoglobin protect Trypanosoma cruzi from the antiparasitic effects of nitric oxide? FEBS Lett. 2001, 501, 103–105. [Google Scholar] [CrossRef] [Green Version]
- Ascenzi, P.; Fasano, M.; Gradoni, L. Do hemoglobin and hemocyanin impair schistosoma killing by no? IUBMB Life 2002, 53, 287–288. [Google Scholar] [CrossRef]
- Bath, P.M.; Coleman, C.M.; Gordon, A.L.; Lim, W.S.; Webb, A.J. Nitric oxide for the prevention and treatment of viral, bacterial, protozoal and fungal infections. F1000Research 2021, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Gilles, H.M. Protozoal Diseases; Arnold: London, UK, 1999; Volume 94, pp. 113–127. [Google Scholar]
- Perez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Burleigh, B.A.; Andrews, N.W. Signaling and host cell invasion by Trypanosoma cruzi. Curr. Opin. Microbiol. 1998, 1, 461–465. [Google Scholar] [CrossRef]
- Soeiro Mde, N.; Paiva, M.M.; Barbosa, H.S.; Meirelles Mde, N.; Araujo-Jorge, T.C. A cardiomyocyte mannose receptor system is involved in Trypanosoma cruzi invasion and is down-modulated after infection. Cell Struct. Funct. 1999, 24, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalta, F.; Zhang, Y.; Bibb, K.E.; Kappes, J.C.; Lima, M.F. The cysteine-cysteine family of chemokines RANTES, MIP-1α, and MIP-1β induce trypanocidal activity in human macrophages via nitric oxide. Infect. Immun. 1998, 66, 4690–4695. [Google Scholar] [CrossRef] [Green Version]
- Aliberti, J.C.; Machado, F.S.; Souto, J.T.; Campanelli, A.P.; Teixeira, M.M.; Gazzinelli, R.T.; Silva, J.S. Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infect. Immun. 1999, 67, 4819–4826. [Google Scholar] [CrossRef] [Green Version]
- Thomson, L.; Gadelha, F.R.; Peluffo, G.; Vercesi, A.E.; Radi, R. Peroxynitrite affects Ca2+ transport in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1999, 98, 81–91. [Google Scholar] [CrossRef]
- Eu, J.P.; Xu, L.; Stamler, J.S.; Meissner, G. Regulation of ryanodine receptors by reactive nitrogen species. Biochem. Pharmacol. 1999, 57, 1079–1084. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Melby, P.C.; Troyer, D.A.; Freeman, G.L. Differential regulation of nitric oxide synthase isoforms in experimental acute chagasic cardiomyopathy. Clin. Exp. Immunol. 2000, 121, 112–119. [Google Scholar] [CrossRef]
- Stamler, J.S.; Meissner, G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev. 2001, 81, 209–237. [Google Scholar] [CrossRef]
- Holscher, C.; Kohler, G.; Muller, U.; Mossmann, H.; Schaub, G.A.; Brombacher, F. Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infect. Immun. 1998, 66, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vespa, G.N.; Cunha, F.Q.; Silva, J.S. Nitric oxide is involved in control of Trypanosoma cruzi-induced parasitemia and directly kills the parasite in vitro. Infect. Immun. 1994, 62, 5177–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petray, P.; Castanos-Velez, E.; Grinstein, S.; Orn, A.; Rottenberg, M.E. Role of nitric oxide in resistance and histopathology during experimental infection with Trypanosoma cruzi. Immunol. Lett. 1995, 47, 121–126. [Google Scholar] [CrossRef]
- Martins, G.A.; Cardoso, M.A.; Aliberti, J.C.; Silva, J.S. Nitric oxide-induced apoptotic cell death in the acute phase of Trypanosoma cruzi infection in mice. Immunol. Lett. 1998, 63, 113–120. [Google Scholar] [CrossRef]
- Silva, J.S.; Machado, F.S.; Martins, G.A. The role of nitric oxide in the pathogenesis of Chagas disease. Front. Biosci. 2003, 8, s314–s325. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chan, J.; Wittner, M.; Jelicks, L.A.; Morris, S.A.; Factor, S.M.; Weiss, L.M.; Braunstein, V.L.; Bacchi, C.J.; Yarlett, N.; et al. Expression of cardiac cytokines and inducible form of nitric oxide synthase (NOS2) in Trypanosoma cruzi-infected mice. J. Mol. Cell Cardiol. 1999, 31, 75–88. [Google Scholar] [CrossRef]
- Machado, F.S.; Martins, G.A.; Aliberti, J.C.; Mestriner, F.L.; Cunha, F.Q.; Silva, J.S. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 2000, 102, 3003–3008. [Google Scholar] [CrossRef] [Green Version]
- Santos, E.; Menezes Falcao, L. Chagas cardiomyopathy and heart failure: From epidemiology to treatment. Rev. Port. Cardiol. (Engl. Ed.) 2020, 39, 279–289. [Google Scholar] [CrossRef]
- Eich, R.F.; Li, T.; Lemon, D.D.; Doherty, D.H.; Curry, S.R.; Aitken, J.F.; Mathews, A.J.; Johnson, K.A.; Smith, R.D.; Phillips, G.N., Jr.; et al. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 1996, 35, 6976–6983. [Google Scholar] [CrossRef]
- Livingston, D.J.; McLachlan, S.J.; La Mar, G.N.; Brown, W.D. Myoglobin: Cytochrome b5 interactions and the kinetic mechanism of metmyoglobin reductase. J. Biol. Chem. 1985, 260, 15699–15707. [Google Scholar] [CrossRef]
- Ahmad, G.; Chami, B.; El Kazzi, M.; Wang, X.; Moreira, M.T.S.; Hamilton, N.; Maw, A.M.; Hambly, T.W.; Witting, P.K. Catalase-like antioxidant activity is unaltered in hypochlorous acid oxidized horse heart Myoglobin. Antioxidants 2019, 8, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmester, T.; Weich, B.; Reinhardt, S.; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Gradoni, L. Do neuroglobin and myoglobin protect Toxoplasma gondii from nitrosative stress? IUBMB Life 2005, 57, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Exner, M.; Nauser, T. Kinetic and mechanistic studies of the NO*-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry 2001, 40, 3385–3395. [Google Scholar] [CrossRef]
- Herold, S.; Fago, A.; Weber, R.E.; Dewilde, S.; Moens, L. Reactivity studies of the Fe(III) and Fe(II)NO forms of human neuroglobin reveal a potential role against oxidative stress. J. Biol. Chem. 2004, 279, 22841–22847. [Google Scholar] [CrossRef] [Green Version]
- Brunori, M.; Giuffre, A.; Nienhaus, K.; Nienhaus, G.U.; Scandurra, F.M.; Vallone, B. Neuroglobin, nitric oxide, and oxygen: Functional pathways and conformational changes. Proc. Natl. Acad. Sci. USA 2005, 102, 8483–8488. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Wainwright, L.M.; Poole, R.K. Microbial globins. Adv. Microb. Physiol. 2003, 47, 255–310. [Google Scholar]
- Szewczyk-Golec, K.; Pawlowska, M.; Wesolowski, R.; Wroblewski, M.; Mila-Kierzenkowska, C. Oxidative stress as a possible target in the treatment of toxoplasmosis: Perspectives and ambiguities. Int. J. Mol. Sci. 2021, 22, 5705. [Google Scholar] [CrossRef]
- Taylor-Robinson, A.W. Nitric oxide can be released as well as scavenged by haemoglobin: Relevance to its antimalarial activity. Parasite Immunol. 1998, 20, 49–50. [Google Scholar] [CrossRef]
- Milner, D.A., Jr. Malaria Pathogenesis. Cold Spring Harb. Perspect. Med. 2018, 8, a025569. [Google Scholar] [CrossRef] [Green Version]
- Jones, I.W.; Thomsen, L.L.; Knowles, R.; Gutteridge, W.E.; Butcher, G.A.; Sinden, R.E. Nitric oxide synthase activity in malaria-infected mice. Parasite Immunol. 1996, 18, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, J.R., Jr. A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide Biol. Chem. 1997, 1, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Seguin, M.C.; Klotz, F.W.; Schneider, I.; Weir, J.P.; Goodbary, M.; Slayter, M.; Raney, J.J.; Aniagolu, J.U.; Green, S.J. Induction of nitric oxide synthase protects against malaria in mice exposed to irradiated Plasmodium berghei infected mosquitoes: Involvement of interferon gamma and CD8+ T cells. J. Exp. Med. 1994, 180, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrevanian, H. Immune effector mechanisms of the nitric oxide pathway in malaria: Cytotoxicity versus cytoprotection. Braz. J. Infect. Dis. 2006, 10, 283–292. [Google Scholar] [CrossRef]
- LoVerde, P.T. Schistosomiasis. Adv. Exp. Med. Biol. 2019, 1154, 45–70. [Google Scholar]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- James, S.L.; Glaven, J. Macrophage cytotoxicity against schistosomula of Schistosoma mansoni involves arginine-dependent production of reactive nitrogen intermediates. J. Immunol. 1989, 143, 4208–4212. [Google Scholar]
- Hahn, U.K.; Bender, R.C.; Bayne, C.J. Involvement of nitric oxide in killing of Schistosoma mansoni sporocysts by hemocytes from resistant Biomphalaria glabrata. J. Parasitol. 2001, 87, 778–785. [Google Scholar] [CrossRef]
- Oswald, I.P.; Eltoum, I.; Wynn, T.A.; Schwartz, B.; Caspar, P.; Paulin, D.; Sher, A.; James, S.L. Endothelial cells are activated by cytokine treatment to kill an intravascular parasite, Schistosoma mansoni, through the production of nitric oxide. Proc. Natl. Acad. Sci. USA 1994, 91, 999–1003. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Oswald, I.P.; Eltoum, I.A.; Caspar, P.; Lowenstein, C.J.; Lewis, F.A.; James, S.L.; Sher, A. Elevated expression of Th1 cytokines and nitric oxide synthase in the lungs of vaccinated mice after challenge infection with Schistosoma mansoni. J. Immunol. 1994, 153, 5200–5209. [Google Scholar]
- Liu, J.; Pan, T.; You, X.; Xu, Y.; Liang, J.; Limpanont, Y.; Sun, X.; Okanurak, K.; Zheng, H.; Wu, Z.; et al. SjCa8, a calcium-binding protein from Schistosoma japonicum, inhibits cell migration and suppresses nitric oxide release of RAW264.7 macrophages. Parasit. Vectors 2015, 8, 513. [Google Scholar] [CrossRef] [Green Version]
- Gow, A.J.; Luchsinger, B.P.; Pawloski, J.R.; Singel, D.J.; Stamler, J.S. The oxyhemoglobin reaction of nitric oxide. Proc. Natl. Acad. Sci. USA 1999, 96, 9027–9032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K. The haemoglobin enzyme. Nature 1999, 401, 437–439. [Google Scholar] [CrossRef] [PubMed]
- Minning, D.M.; Gow, A.J.; Bonaventura, J.; Braun, R.; Dewhirst, M.; Goldberg, D.E.; Stamler, J.S. Ascaris haemoglobin is a nitric oxide-activated ‘deoxygenase’. Nature 1999, 401, 497–502. [Google Scholar] [CrossRef]
- Dold, C.; Holland, C.V. Ascaris and ascariasis. Microbes Infect. 2011, 13, 632–637. [Google Scholar] [CrossRef]
- Singel, D.J.; Stamler, J.S. Chemical physiology of blood flow regulation by red blood cells: The role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. 2005, 67, 99–145. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.W.; Stamler, J.S.; Piantadosi, C.A. Hemoglobin, nitric oxide and molecular mechanisms of hypoxic vasodilation. Trends Mol. Med. 2009, 15, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Palmer, L.A.; Doctor, A.; Gaston, B. SNO-hemoglobin and hypoxic vasodilation. Nat. Med. 2008, 14, 1009, author reply 1009–1010. [Google Scholar] [CrossRef]
- Gaston, B.; May, W.J.; Sullivan, S.; Yemen, S.; Marozkina, N.V.; Palmer, L.A.; Bates, J.N.; Lewis, S.J. Essential role of hemoglobin beta-93-cysteine in posthypoxia facilitation of breathing in conscious mice. J. Appl. Physiol. 2014, 116, 1290–1299. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.J.; Haynes, K.; Quinn, J. Nitrosative and oxidative stress responses in fungal pathogenicity. Curr. Opin. Microbiol. 2009, 12, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Missall, T.A.; Lodge, J.K.; McEwen, J.E. Mechanisms of resistance to oxidative and nitrosative stress: Implications for fungal survival in mammalian hosts. Eukaryot. Cell 2004, 3, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, G.K.K.; Padmavathi, A.R.; Nancharaiah, Y.V. Fungal infections: Pathogenesis, antifungals and alternate treatment approaches. Curr. Res. Microbial Sci. 2022, 3, 100137. [Google Scholar] [CrossRef] [PubMed]
- Gulati, M.; Nobile, C.J. Candida albicans biofilms: Development, regulation, and molecular mechanisms. Microbes Infect. 2016, 18, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hromatka, B.S.; Noble, S.M.; Johnson, A.D. Transcriptional response of Candida albicans to nitric oxide and the role of the YHB1 gene in nitrosative stress and virulence. Mol. Biol. Cell 2005, 16, 4814–4826. [Google Scholar] [CrossRef] [PubMed]
- Chiranand, W.; McLeod, I.; Zhou, H.; Lynn, J.J.; Vega, L.A.; Myers, H.; Yates, J.R., 3rd; Lorenz, M.C.; Gustin, M.C. CTA4 transcription factor mediates induction of nitrosative stress response in Candida albicans. Eukaryot. Cell 2008, 7, 268–278. [Google Scholar] [CrossRef] [Green Version]
- De Jesus-Berrios, M.; Liu, L.; Nussbaum, J.C.; Cox, G.M.; Stamler, J.S.; Heitman, J. Enzymes that counteract nitrosative stress promote fungal virulence. Curr. Biol. 2003, 13, 1963–1968. [Google Scholar] [CrossRef] [Green Version]
- Ullmann, B.D.; Myers, H.; Chiranand, W.; Lazzell, A.L.; Zhao, Q.; Vega, L.A.; Lopez-Ribot, J.L.; Gardner, P.R.; Gustin, M.C. Inducible defense mechanism against nitric oxide in Candida albicans. Eukaryot. Cell. 2004, 3, 715–723. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Simone, G.; di Masi, A.; Ascenzi, P. Strategies of Pathogens to Escape from NO-Based Host Defense. Antioxidants 2022, 11, 2176. https://doi.org/10.3390/antiox11112176
De Simone G, di Masi A, Ascenzi P. Strategies of Pathogens to Escape from NO-Based Host Defense. Antioxidants. 2022; 11(11):2176. https://doi.org/10.3390/antiox11112176
Chicago/Turabian StyleDe Simone, Giovanna, Alessandra di Masi, and Paolo Ascenzi. 2022. "Strategies of Pathogens to Escape from NO-Based Host Defense" Antioxidants 11, no. 11: 2176. https://doi.org/10.3390/antiox11112176
APA StyleDe Simone, G., di Masi, A., & Ascenzi, P. (2022). Strategies of Pathogens to Escape from NO-Based Host Defense. Antioxidants, 11(11), 2176. https://doi.org/10.3390/antiox11112176