
Uric Acid Reacts with Peroxidasin, Decreases Collagen IV Crosslink, Impairs Human Endothelial Cell Migration and Adhesion

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Procedures
2.1. Reagents and Antibodies
2.2. Cell Culture and Transfection
2.3. siRNA Transfection
2.4. Isolation of Secretome and Extracellular Matrix
2.5. Amplex Red Assay
2.6. Quantification of Bromo-Tyrosine by HPLC
2.7. Detection of Urate Oxidation Products by LC-MS/MS
2.8. Collagen IV Crosslink Assay
2.9. Adhesion Assay
2.10. Wound Healing Assay
2.11. SDS-PAGE Analysis
2.12. Protein Identification by Proteomics
2.12.1. In-Gel Digestion
2.12.2. In-Solution Digestion
2.12.3. Extracellular Matrix (ECM) Digestion
2.12.4. Data Dependent Acquisition (DDA) Proteomics
2.12.5. Protein Identification
2.13. Statistical Analysis
3. Results
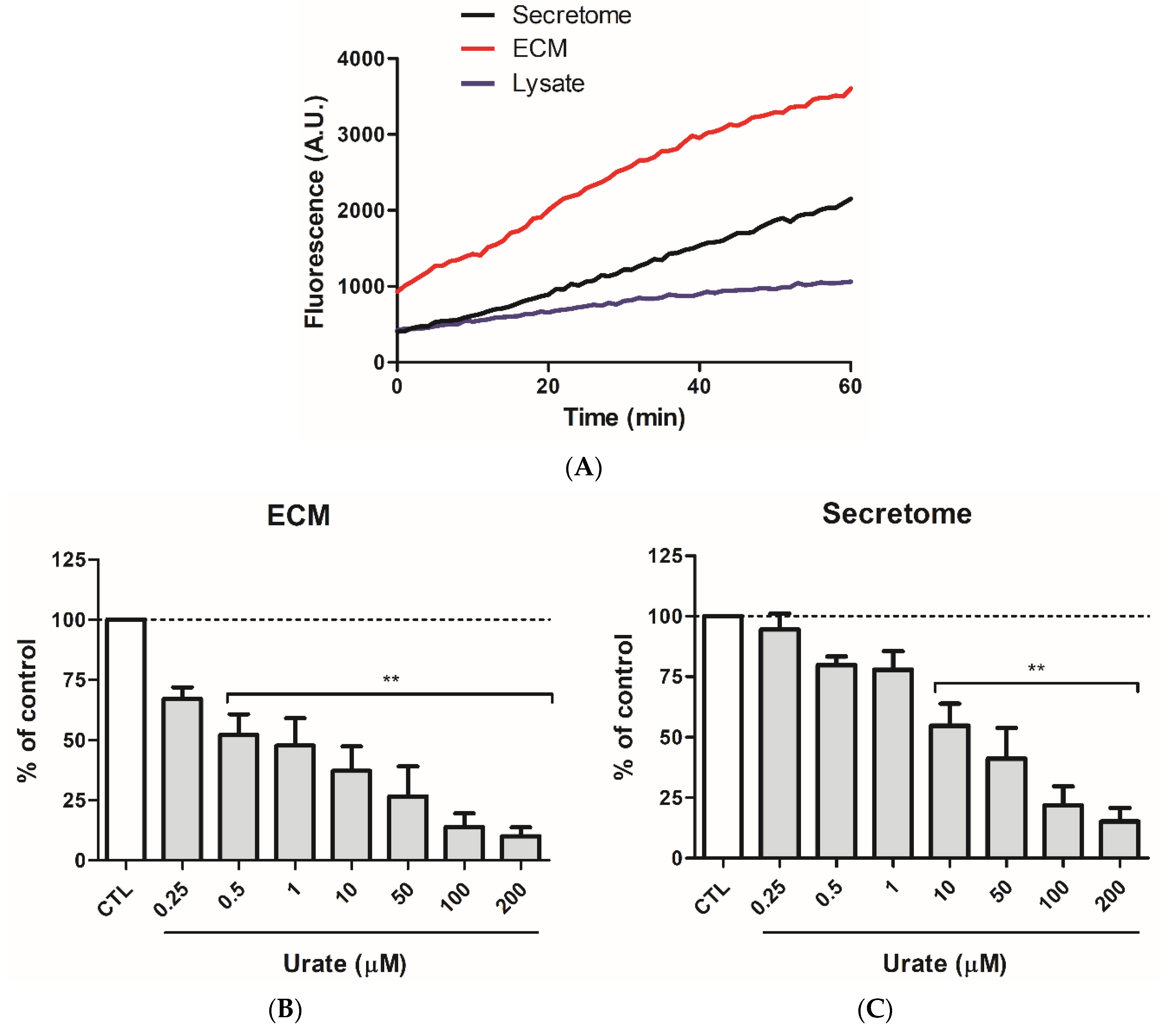
3.1. Urate Inhibits PXDN Released from HUVECs
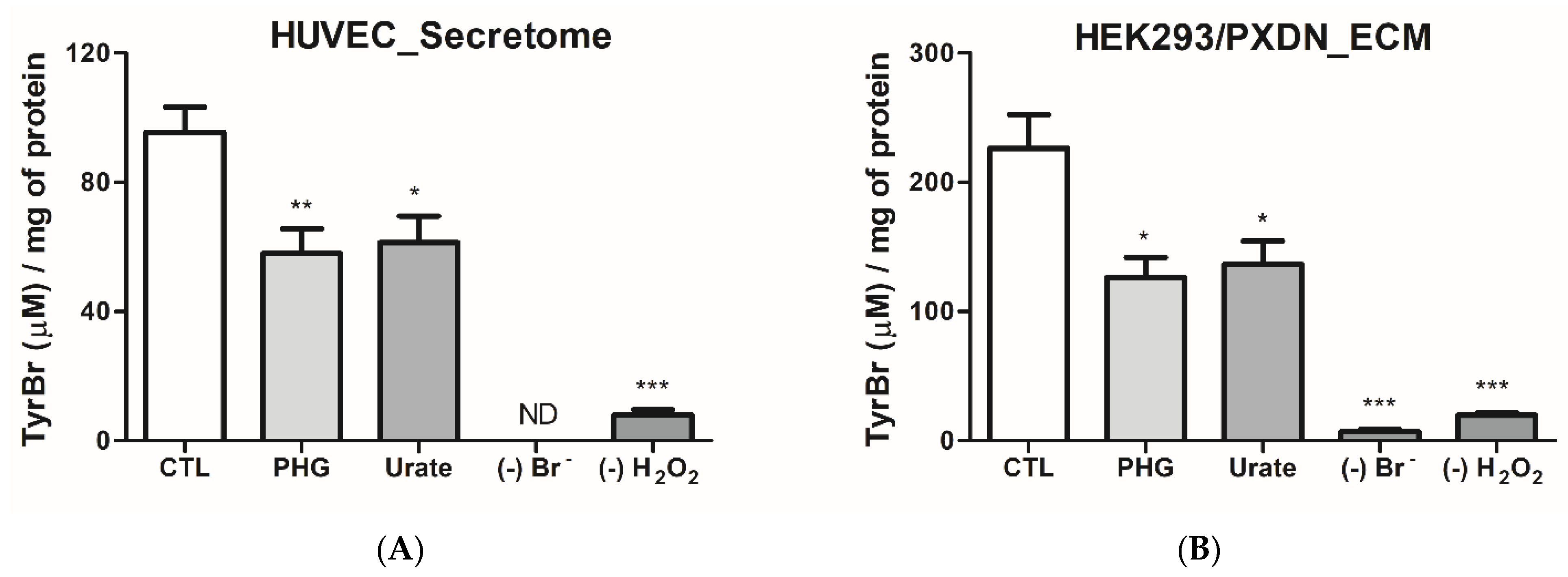
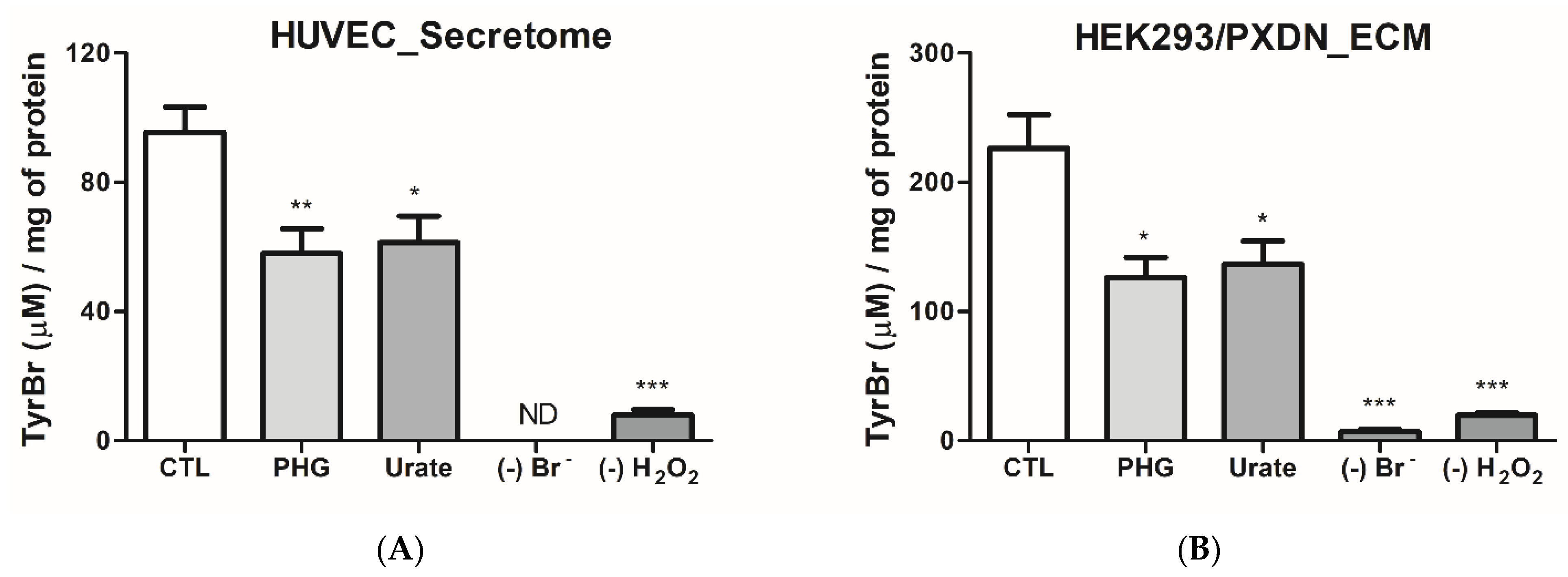
3.2. Urate Decreases PXDN-Bromination Activity
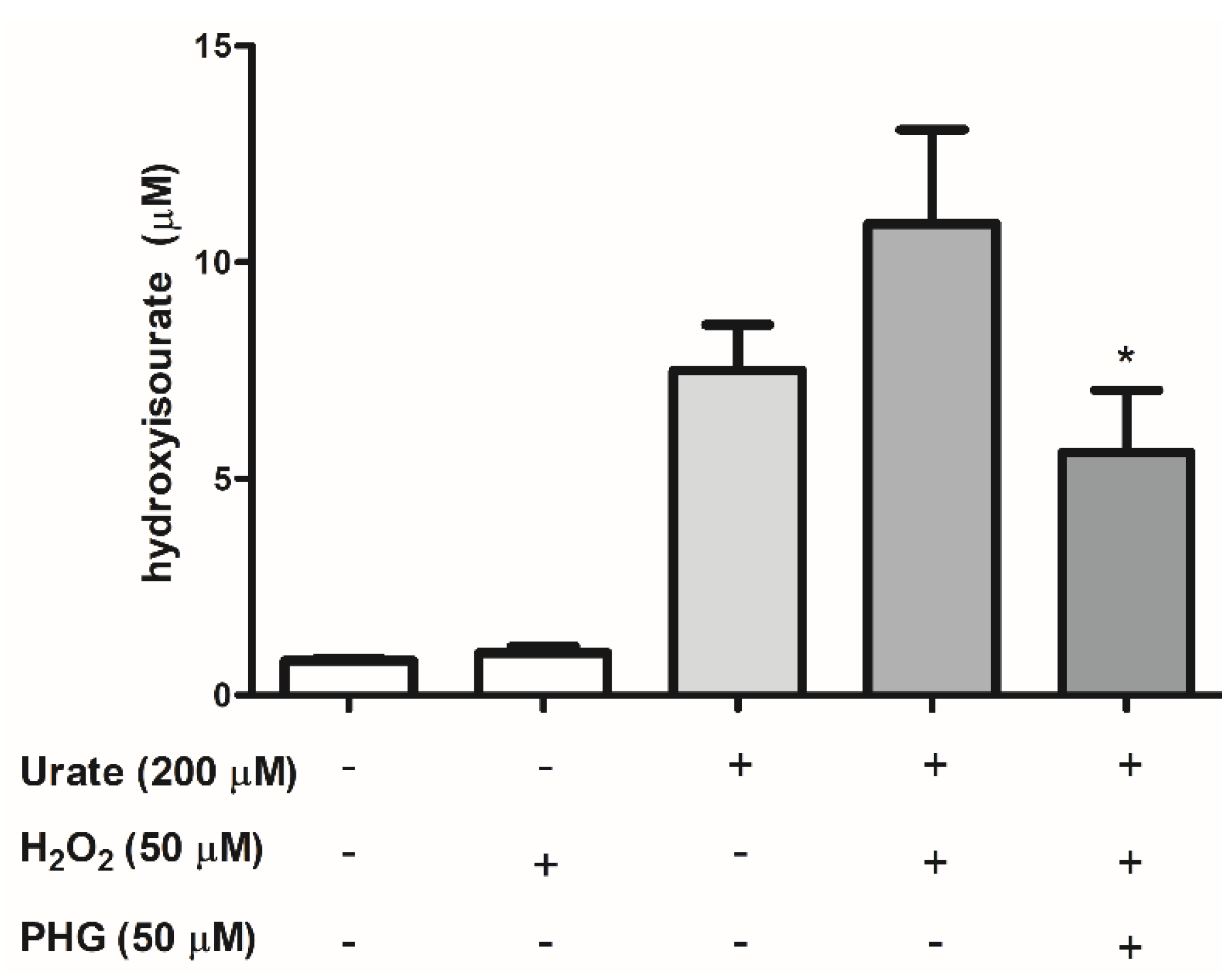
3.3. Identification of Urate Oxidation Products in HUVEC Secretome
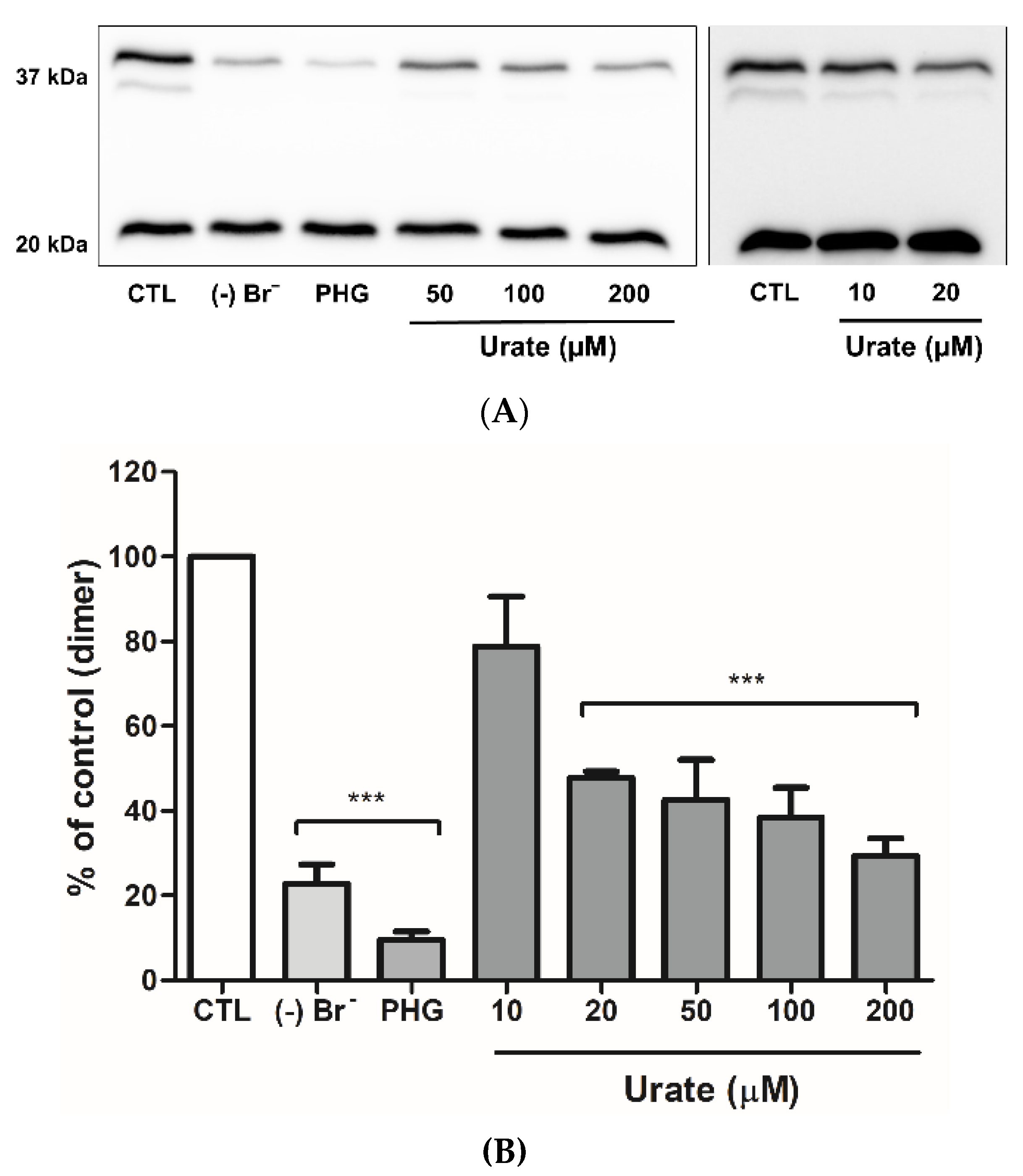
3.4. Urate Reduces Collagen IV Crosslinking
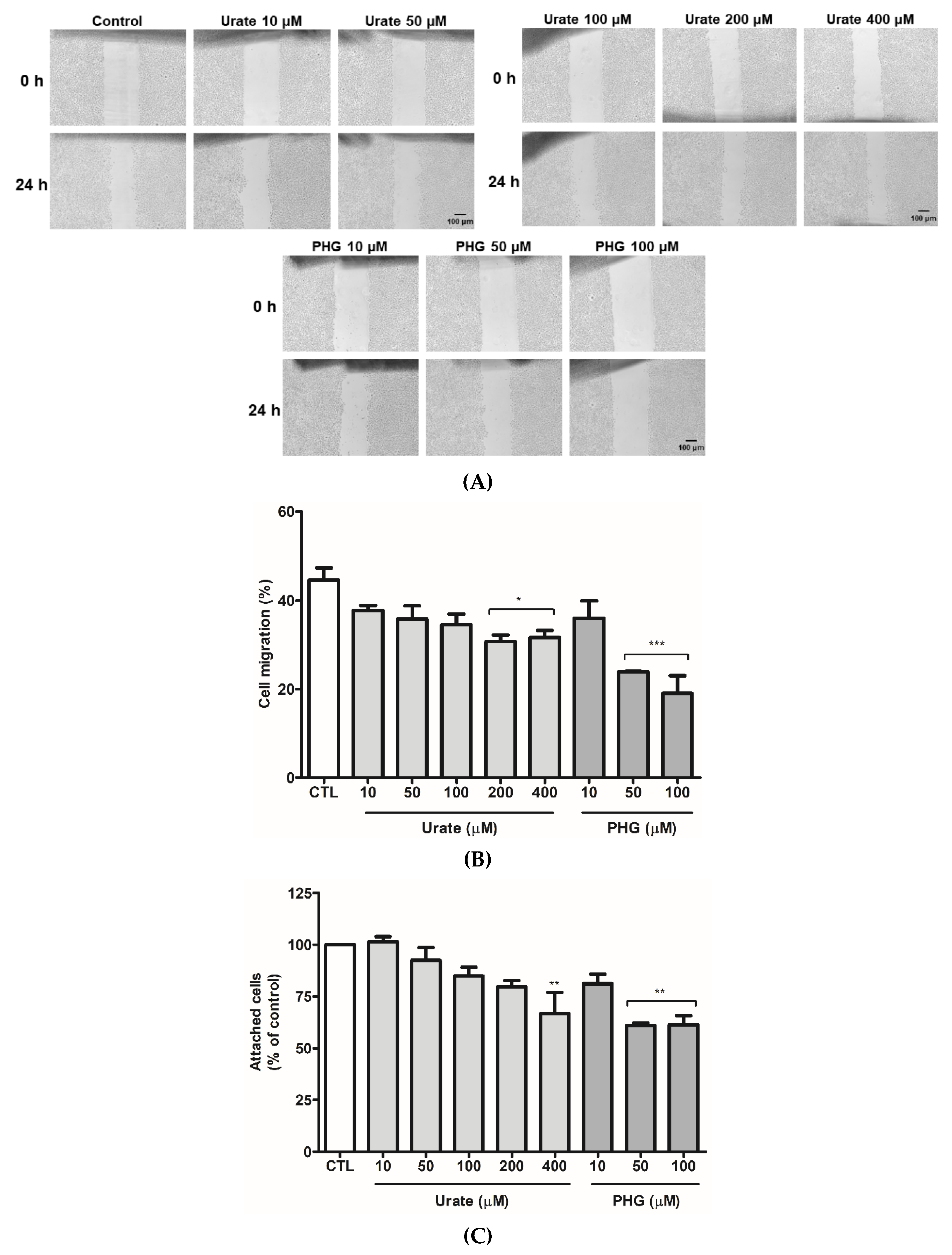
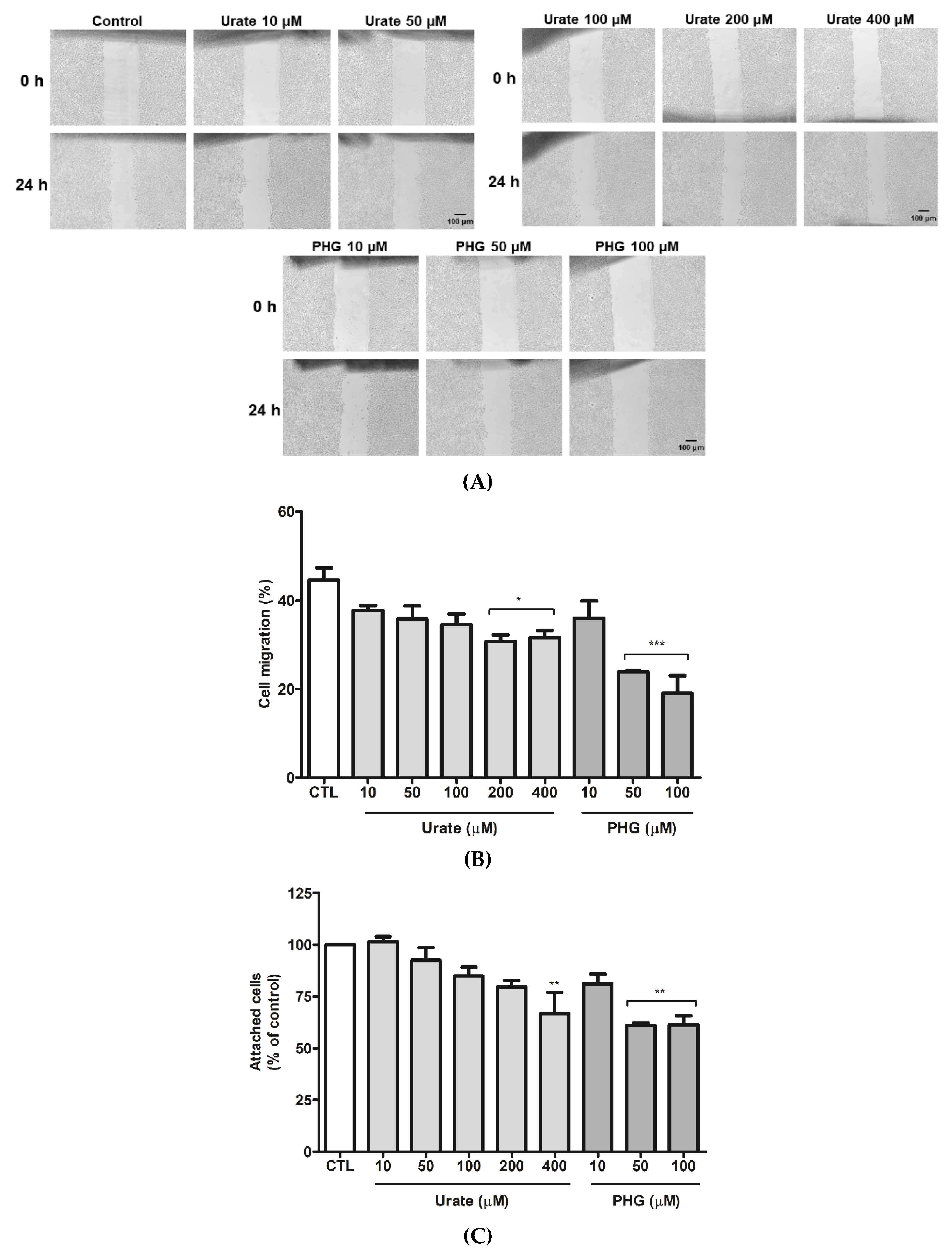
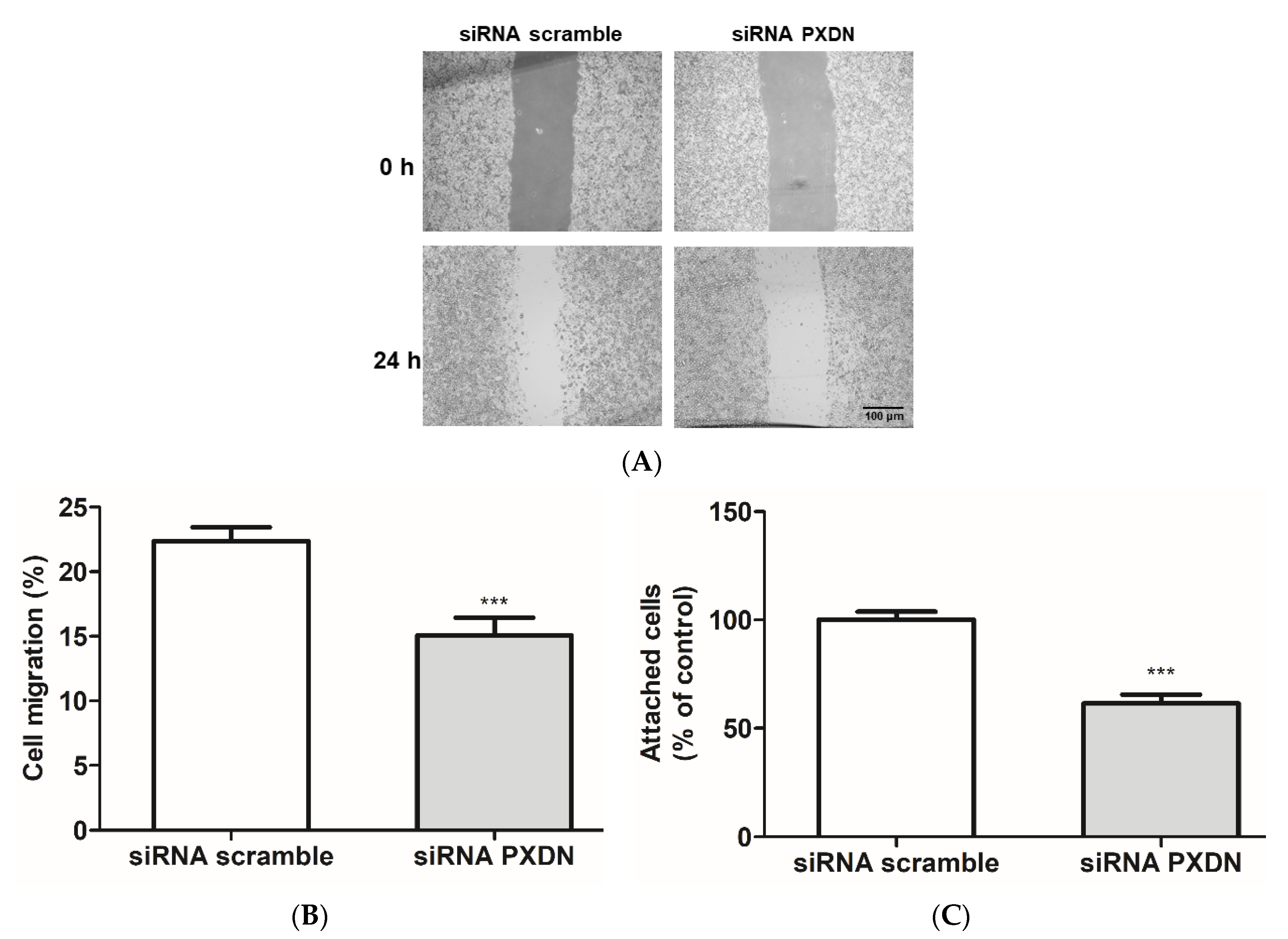
3.5. PXDN Inhibition Decreases HUVEC Migration and Adhesion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, R.J.; Rideout, B.A. Uric Acid and Diet—Insights into the Epidemic of Cardiovascular Disease. N. Engl. J. Med. 2004, 350, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.F. Towards the Physiological Function of Uric Acid. Free. Radic. Biol. Med. 1993, 14, 615–631. [Google Scholar] [CrossRef]
- Simic, M.G.; Jovanovic, S.V. Antioxidation Mechanisms of Uric Acid. J. Am. Chem. Soc. 1989, 111, 5778–5782. [Google Scholar] [CrossRef]
- Filipe, P.; Haigle, J.; Freitas, J.; Fernandes, A.; Mazière, J.C.; Mazière, C.; Santus, R.; Morlière, P. Anti- and pro-Oxidant Effects of Urate in Copper-Induced Low-Density Lipoprotein Oxidation. Eur. J. Biochem. 2002, 269, 5474–5483. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi-Oskouie, F.; Shi, Y. The Role of Uric Acid as an Endogenous Danger Signal in Immunity and Inflammation. Curr. Rheumatol. Rep. 2011, 13, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F. Mechanisms of Uric Acid Crystal-Mediated Autoinflammation. Immunol. Rev. 2010, 233, 218–232. [Google Scholar] [CrossRef]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Feig, D.I.; Kang, D.-H.; Johnson, R.J. Uric Acid and Cardiovascular Risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef]
- Hare, J.M.; Johnson, R.J. Uric Acid Predicts Clinical Outcomes in Heart Failure: Insights Regarding the Role of Xanthine Oxidase and Uric Acid in Disease Pathophysiology. Circulation 2003, 107, 1951–1953. [Google Scholar] [CrossRef] [Green Version]
- Santana, M.S.; Nascimento, K.P.; Lotufo, P.A.; Benseñor, I.M.; Meotti, F.C. Allantoin as an Independent Marker Associated with Carotid Intima-Media Thickness in Subclinical Atherosclerosis. Braz. J. Med. Biol. Res. 2018, 51, e7543. [Google Scholar] [CrossRef] [PubMed]
- Lotufo, P.A.; Baena, C.P.; Santos, I.S.; Bensenor, I.M. Serum Uric Acid and Prehypertension among Adults Free of Cardiovascular Diseases and Diabetes: Baseline of the Brazilian Longitudinal Study of Adult Health (ELSA-Brasil). Angiology 2016, 67, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Ndrepepa, G. Uric Acid and Cardiovascular Disease. Clin. Chim. Acta 2018, 484, 150–163. [Google Scholar] [CrossRef]
- Zhen, H.; Gui, F. The Role of Hyperuricemia on Vascular Endothelium Dysfunction. Biomed. Rep. 2017, 7, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G. Myeloperoxidase–A Bridge Linking Inflammation and Oxidative Stress with Cardiovascular Disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Watanabe, S.; Li, J.H.; Kang, D.H.; Li, P.; Nakagawa, T.; Wamsley, A.; Sheikh-Hamad, D.; Lan, H.Y.; Feng, L.; et al. Uric Acid Stimulates Monocyte Chemoattractant Protein-1 Production in Vascular Smooth Muscle Cells via Mitogen-Activated Protein Kinase and Cyclooxygenase-2. Hypertension 2003, 41, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Corry, D.B.; Eslami, P.; Yamamoto, K.; Nyby, M.D.; Makino, H.; Tuck, M.L. Uric Acid Stimulates Vascular Smooth Muscle Cell Proliferation and Oxidative Stress via the Vascular Renin-Angiotensin System. J. Hypertens. 2008, 26, 269–275. [Google Scholar] [CrossRef]
- Silva, R.P.; Carvalho, L.A.C.; Patricio, E.S.; Bonifacio, J.P.P.; Chaves-Filho, A.B.; Miyamoto, S.; Meotti, F.C. Identification of Urate Hydroperoxide in Neutrophils: A Novel pro-Oxidant Generated in Inflammatory Conditions. Free Radic. Biol. Med. 2018, 126, 177–186. [Google Scholar] [CrossRef]
- Meotti, F.C.; Jameson, G.N.L.; Turner, R.; Harwood, D.T.; Stockwell, S.; Rees, M.D.; Thomas, S.R.; Kettle, A.J. Urate as a Physiological Substrate for Myeloperoxidase. J. Biol. Chem. 2011, 286, 12901–12911. [Google Scholar] [CrossRef] [Green Version]
- Seidel, A.; Parker, H.; Turner, R.; Dickerhof, N.; Khalilova, I.S.; Wilbanks, S.M.; Kettle, A.J.; Jameson, G.N.L. Uric Acid and Thiocyanate as Competing Substrates of Lactoperoxidase. J. Biol. Chem. 2014, 289, 21937–21949. [Google Scholar] [CrossRef] [Green Version]
- Mineiro, M.F.; de Souza Patricio, E.; Peixoto, Á.S.; Araujo, T.L.S.; da Silva, R.P.; Moretti, A.I.S.; Lima, F.S.; Laurindo, F.R.M.; Meotti, F.C. Urate Hydroperoxide Oxidizes Endothelial Cell Surface Protein Disulfide Isomerase-A1 and Impairs Adherence. Biochim. Biophys. Acta 2020, 1864, 129481. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.A.C.C.; Truzzi, D.R.; Fallani, T.S.; Alves, S.V.; Toledo, J.C.; Augusto, O.; Netto, L.E.S.S.; Meotti, F.C. Urate Hydroperoxide Oxidizes Human Peroxiredoxin 1 and Peroxiredoxin 2. J. Biol. Chem. 2017, 292, 8705–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padiglia, A.; Medda, R.; Longu, S.; Pedersen, J.Z.; Floris, G. Uric Acid Is a Main Electron Donor to Peroxidases in Human Blood Plasma. Med. Sci. Monit. 2002, 8, BR454-9. [Google Scholar] [PubMed]
- Péterfi, Z.; Donkó, Á.; Orient, A.; Sum, A.; Prókai, Á.; Molnár, B.; Veréb, Z.; Rajnavölgyi, É.; Kovács, K.J.; Müller, V.; et al. Peroxidasin Is Secreted and Incorporated into the Extracellular Matrix of Myofibroblasts and Fibrotic Kidney. Am. J. Pathol. 2009, 175, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtmüller, P.G.; Zederbauer, M.; Jantschko, W.; Helm, J.; Bogner, M.; Jakopitsch, C.; Obinger, C. Active Site Structure and Catalytic Mechanisms of Human Peroxidases. Arch. Biochem. Biophys. 2006, 445, 199–213. [Google Scholar] [CrossRef]
- Soudi, M.; Paumann-Page, M.; Delporte, C.; Pirker, K.F.; Bellei, M.; Edenhofer, E.; Stadlmayr, G.; Battistuzzi, G.; Boudjeltia, K.Z.; Furtmüller, P.G.; et al. Multidomain Human Peroxidasin 1 Is a Highly Glycosylated and Stable Homotrimeric High Spin Ferric Peroxidase. J. Biol. Chem. 2015, 290, 10876–10890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebanoff, S.J. Myeloperoxidase: Friend and Foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Paumann-Page, M.; Katz, R.-S.; Bellei, M.; Schwartz, I.; Edenhofer, E.; Sevcnikar, B.; Soudi, M.; Hofbauer, S.; Battistuzzi, G.; Furtmüller, P.G.; et al. Pre-Steady-State Kinetics Reveal the Substrate Specificity and Mechanism of Halide Oxidation of Truncated Human Peroxidasin 1. J. Biol. Chem. 2017, 292, 4583–4592. [Google Scholar] [CrossRef] [Green Version]
- Gerson, C.; Sabater, J.; Scuri, M.; Torbati, A.; Coffey, R.; Abraham, J.W.; Lauredo, I.; Forteza, R.; Wanner, A.; Salathe, M.; et al. The Lactoperoxidase System Functions in Bacterial Clearance of Airways. Am. J. Respir. Cell Mol. Biol. 2000, 22, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Salerno, J.C.; Cao, Z.; Pagano, P.J.; Lambeth, J.D. Identification and Characterization of VPO1, a New Animal Heme-Containing Peroxidase. Free Radic. Biol. Med. 2008, 45, 1682–1694. [Google Scholar] [CrossRef] [Green Version]
- Lázár, E.E.; Péterfi, Z.; Sirokmány, G.; Kovács, H.A.; Klement, E.; Medzihradszky, K.F.; Geiszt, M. Structure-Function Analysis of Peroxidasin Provides Insight into the Mechanism of Collagen IV Crosslinking. Free Radic. Biol. Med. 2015, 83, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhave, G.; Cummings, C.F.; Vanacore, R.M.; Kumagai-Cresse, C.; Ero-Tolliver, I.A.; Rafi, M.; Kang, J.-S.S.; Pedchenko, V.; Fessler, L.I.; Fessler, J.H.; et al. Peroxidasin Forms Sulfilimine Chemical Bonds Using Hypohalous Acids in Tissue Genesis. Nat. Chem. Biol. 2012, 8, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCall, A.S.; Cummings, C.F.; Bhave, G.; Vanacore, R.; Page-McCaw, A.; Hudson, B.G. Bromine Is an Essential Trace Element for Assembly of Collagen IV Scaffolds in Tissue Development and Architecture. Cell 2014, 157, 1380–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevcnikar, B.; Paumann-Page, M.; Hofbauer, S.; Pfanzagl, V.; Furtmüller, P.G.; Obinger, C. Reaction of Human Peroxidasin 1 Compound I and Compound II with One-Electron Donors. Arch. Biochem. Biophys. 2020, 681, 108267. [Google Scholar] [CrossRef] [PubMed]
- Olszowy, H.A.; Rossiter, J.; Hegarty, J.; Geoghegan, P.; Haswell-Elkins, M. Background Levels of Bromide in Human Blood. J. Anal. Toxicol. 1998, 22, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.; Brennan, S.O.; Ashby, L.V.; Dickerhof, N.; Hamzah, M.R.; Pearson, J.F.; Stamp, L.K.; Kettle, A.J. Conjugation of Urate-Derived Electrophiles to Proteins during Normal Metabolism and Inflammation. J. Biol. Chem. 2018, 293, 19886–19898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliesiu, A.; Campeanu, A.; Dusceac, D. Serum Uric Acid and Cardiovascular Disease. Maedica 2010, 5, 186–192. [Google Scholar]
- Johnson, R.J.; Bakris, G.L.; Borghi, C.; Chonchol, M.B.; Feldman, D.; Lanaspa, M.A.; Merriman, T.R.; Moe, O.W.; Mount, D.B.; Sanchez Lozada, L.G.; et al. Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. Am. J. Kidney Dis. 2018, 71, 851–865. [Google Scholar] [CrossRef]
- Bathish, B.; Turner, R.; Paumann-Page, M.; Kettle, A.J.; Winterbourn, C.C. Characterisation of Peroxidasin Activity in Isolated Extracellular Matrix and Direct Detection of Hypobromous Acid Formation. Arch. Biochem. Biophys. 2018, 646, 120–127. [Google Scholar] [CrossRef]
- Wu, W.; Chen, Y.; D’Avignon, A.; Hazen, S.L. 3-Bromotyrosine and 3,5-Dibromotyrosine Are Major Products of Protein Oxidation by Eosinophil Peroxidase: Potential Markers for Eosinophii- Dependent Tissue Injury In Vivo. Biochemistry 1999, 38, 3538–3548. [Google Scholar] [CrossRef]
- Bathish, B.; Paumann-Page, M.; Paton, L.N.; Kettle, A.J.; Winterbourn, C.C. Peroxidasin Mediates Bromination of Tyrosine Residues in the Extracellular Matrix. J. Biol. Chem. 2020, 295, 12697–12705. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In Vitro Scratch Assay: A Convenient and Inexpensive Method for Analysis of Cell Migration In Vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.C.; Schmitt, L.R.; Hill, R.C.; Dzieciatkowska, M.; Maslanka, M.; Daamen, W.F.; van Kuppevelt, T.H.; Hof, D.J.; Hansen, K.C. Evaluation and Refinement of Sample Preparation Methods for Extracellular Matrix Proteome Coverage. Mol. Cell Proteom. 2021, 20, 100079. [Google Scholar] [CrossRef] [PubMed]
- Sevcnikar, B.; Schaffner, I.; Chuang, C.Y.; Gamon, L.; Paumann-Page, M.; Hofbauer, S.; Davies, M.J.; Furtmüller, P.G.; Obinger, C. The Leucine-Rich Repeat Domain of Human Peroxidasin 1 Promotes Binding to Laminin in Basement Membranes. Arch. Biochem. Biophys. 2020, 689, 108443. [Google Scholar] [CrossRef] [PubMed]
- Domogatskaya, A.; Rodin, S.; Tryggvason, K. Functional Diversity of Laminins. Annu. Rev. Cell Dev. Biol. 2012, 28, 523–553. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, H.K.; Naidansuren, P.; Ham, K.A.; Choi, H.S.; Ahn, H.Y.; Kim, M.; Kang, D.H.; Kang, S.W.; Joe, Y.A. Peroxidasin Is Essential for Endothelial Cell Survival and Growth Signaling by Sulfilimine Crosslink-Dependent Matrix Assembly. FASEB J. 2020, 34, 10228–10241. [Google Scholar] [CrossRef]
- da Silva, R.P. Investigation of Oxidative Metabolism of Uric Acid and Its Role in Redox Processes in Inflammation. Ph.D. Thesis, Universidade de São Paulo, Sao Paulo, Brazil, 2020. [Google Scholar]
- Shi, R.; Hu, C.; Yuan, Q.; Yang, T.; Peng, J.; Li, Y.; Bai, Y.; Cao, Z.; Cheng, G.; Zhang, G. Involvement of Vascular Peroxidase 1 in Angiotensin II-Induced Vascular Smooth Muscle Cell Proliferation. Cardiovasc. Res. 2011, 91, 27–36. [Google Scholar] [CrossRef]
- Medfai, H.; Khalil, A.; Rousseau, A.; Nuyens, V.; Paumann-Page, M.; Sevcnikar, B.; Furtmüller, P.; Obinger, C.; Moguilevsky, N.; Peulen, O.; et al. Human Peroxidasin 1 Promotes Angiogenesis through ERK1/2, Akt and FAK Pathways. Cardiovasc. Res. 2018, 115, 463–475. [Google Scholar] [CrossRef]
- Dougan, J.; Hawsawi, O.; Burton, L.J.; Edwards, G.; Jones, K.; Zou, J.; Nagappan, P.; Wang, G.; Zhang, Q.; Danaher, A.; et al. Proteomics-Metabolomics Combined Approach Identifies Peroxidasin as a Protector against Metabolic and Oxidative Stress in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3046. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.-Z.Z.; Liang, L. High Expression of PXDN Is Associated with Poor Prognosis and Promotes Proliferation, Invasion as Well as Migration in Ovarian Cancer. Ann. Diagn. Pathol. 2018, 34, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Prithviraj, P.; Lo, P.H.; Walkiewicz, M.; Anaka, M.; Woods, B.L.; Tan, B.S.; Behren, A.; Cebon, J.; McKeown, S.J. Identifying and Targeting Determinants of Melanoma Cellular Invasion. Oncotarget 2016, 7, 41186–41202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colon, S.; Luan, H.; Liu, Y.; Meyer, C.; Gewin, L.; Bhave, G. Peroxidasin and Eosinophil Peroxidase, but Not Myeloperoxidase, Contribute to Renal Fibrosis in the Murine Unilateral Ureteral Obstruction Model. Am. J. Physiol.-Ren. Physiol. 2019, 316, F360–F371. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bai, Y.; Li, N.; Hu, C.; Peng, J.; Cheng, G.; Zhang, G.; Shi, R. Vascular VPO1 Expression Is Related to the Endothelial Dysfunction in Spontaneously Hypertensive Rats. Biochem. Biophys. Res. Commun. 2013, 439, 511–516. [Google Scholar] [CrossRef]
- Peng, H.; Chen, L.; Huang, X.; Yang, T.; Yu, Z.; Cheng, G.; Zhang, G.; Shi, R. Vascular Peroxidase 1 up Regulation by Angiotensin II Attenuates Nitric Oxide Production through Increasing Asymmetrical Dimethylarginine in HUVECs. J. Am. Soc. Hypertens. 2016, 10, 741–751.e3. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Yuan, Q.; Li, X.H.; Hu, C.P.; Hu, R.; Zhang, G.G.; Li, D.; Li, Y.J. Role of Vascular Peroxidase 1 in Senescence of Endothelial Cells in Diabetes Rats. Int. J. Cardiol. 2015, 197, 182–191. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, R.; Cao, Z.; Zhang, G.; Cheng, G. VPO1 Mediates Oxidation of LDL and Formation of Foam Cells. Oncotarget 2016, 7, 35500–35511. [Google Scholar] [CrossRef] [Green Version]
- Paumann-Page, M.; Kienzl, N.F.; Motwani, J.; Bathish, B.; Paton, L.N.; Magon, N.J.; Sevcnikar, B.; Furtmüller, P.G.; Traxlmayr, M.W.; Obinger, C.; et al. Peroxidasin Protein Expression and Enzymatic Activity in Metastatic Melanoma Cell Lines Are Associated with Invasive Potential. Redox Biol. 2021, 46, 102090. [Google Scholar] [CrossRef]
- Cheng, G.; Li, H.; Cao, Z.; Qiu, X.; McCormick, S.; Thannickal, V.J.; Nauseef, W.M. Vascular Peroxidase-1 Is Rapidly Secreted, Circulates in Plasma, and Supports Dityrosine Cross-Linking Reactions. Free Radic. Biol. Med. 2011, 51, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Patrício, E.S.; Prado, F.M.; da Silva, R.P.; Carvalho, L.A.C.; Prates, M.V.C.; Dadamos, T.; Bertotti, M.; di Mascio, P.; Kettle, A.J.; Meotti, F.C. Chemical Characterization of Urate Hydroperoxide, A Pro-Oxidant Intermediate Generated by Urate Oxidation in Inflammatory and Photoinduced Processes. Chem. Res. Toxicol. 2015, 28, 1556–1566. [Google Scholar] [CrossRef]
- San Martín, A.; Griendling, K.K. Redox Control of Vascular Smooth Muscle Migration. Antioxid. Redox Signal. 2010, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Lauffenburger, D.A.; Horwitz, A.F. Cell Migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.Y.; Zhu, X.Y.; Zhang, J.W.; Feng, X.R.; Wang, Y.C.; Liu, M.L. Uric Acid Promotes Chemokine and Adhesion Molecule Production in Vascular Endothelium via Nuclear Factor-Kappa B Signaling. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yuan, Y.; Zhou, Y.; Zhao, M.; Chen, Y.; Cheng, J.; Lu, Y.; Liu, J. Phloretin Attenuates Hyperuricemia-Induced Endothelial Dysfunction through Co-Inhibiting Inflammation and GLUT9-Mediated Uric Acid Uptake. J. Cell. Mol. Med. 2017, 21, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Duan, X.M.; Liu, Y.; Yu, J.; Tang, Y.L.; Liu, Z.L.; Jiang, S.; Zhang, C.P.; Liu, J.Y.; Xu, J.X. Uric Acid Induces Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling Pathway. BioMed Res. Int. 2017, 2017, 4391920. [Google Scholar] [CrossRef]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia Induces Endothelial Dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.-A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.-H. Oxidative Stress with an Activation of the Renin-Angiotensin System in Human Vascular Endothelial Cells as a Novel Mechanism of Uric Acid-Induced Endothelial Dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Qi, W. Uric Acid, as a Double-Edged Sword, Affects the Activity of Epidermal Growth Factor (EGF) on Human Umbilical Vein Endothelial Cells by Regulating Aging Process. Bioengineered 2022, 13, 3877–3895. [Google Scholar] [CrossRef]
- Hong, Q.; Yu, S.; Geng, X.; Duan, L.; Zheng, W.; Fan, M.; Chen, X.; Wu, D. High Concentrations of Uric Acid Inhibit Endothelial Cell Migration via MiR-663 Which Regulates Phosphatase and Tensin Homolog by Targeting Transforming Growth Factor-Β1. Microcirculation 2015, 22, 306–314. [Google Scholar] [CrossRef]
- Ouyang, R.; Zhao, X.; Zhang, R.; Yang, J.; Li, S.; Deng, D. FGF21 Attenuates High Uric Acid-induced Endoplasmic Reticulum Stress, Inflammation and Vascular Endothelial Cell Dysfunction by Activating Sirt1. Mol. Med. Rep. 2022, 25, 1–9. [Google Scholar] [CrossRef]
- Li, P.; Zhang, L.; Zhang, M.; Zhou, C.; Lin, N. Uric Acid Enhances PKC-Dependent ENOS Phosphorylationand Mediates Cellular ER Stress: A Mechanism for Uric Acid-Induced Endothelial Dysfunction. Int. J. Mol. Med. 2016, 37, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dempsey, B.; Cruz, L.C.; Mineiro, M.F.; da Silva, R.P.; Meotti, F.C. Uric Acid Reacts with Peroxidasin, Decreases Collagen IV Crosslink, Impairs Human Endothelial Cell Migration and Adhesion. Antioxidants 2022, 11, 1117. https://doi.org/10.3390/antiox11061117
Dempsey B, Cruz LC, Mineiro MF, da Silva RP, Meotti FC. Uric Acid Reacts with Peroxidasin, Decreases Collagen IV Crosslink, Impairs Human Endothelial Cell Migration and Adhesion. Antioxidants. 2022; 11(6):1117. https://doi.org/10.3390/antiox11061117
Chicago/Turabian StyleDempsey, Bianca, Litiele Cezar Cruz, Marcela Franco Mineiro, Railmara Pereira da Silva, and Flavia Carla Meotti. 2022. "Uric Acid Reacts with Peroxidasin, Decreases Collagen IV Crosslink, Impairs Human Endothelial Cell Migration and Adhesion" Antioxidants 11, no. 6: 1117. https://doi.org/10.3390/antiox11061117
APA StyleDempsey, B., Cruz, L. C., Mineiro, M. F., da Silva, R. P., & Meotti, F. C. (2022). Uric Acid Reacts with Peroxidasin, Decreases Collagen IV Crosslink, Impairs Human Endothelial Cell Migration and Adhesion. Antioxidants, 11(6), 1117. https://doi.org/10.3390/antiox11061117