
P2X7 Receptor Augments LPS-Induced Nitrosative Stress by Regulating Nrf2 and GSH Levels in the Mouse Hippocampus

Abstract
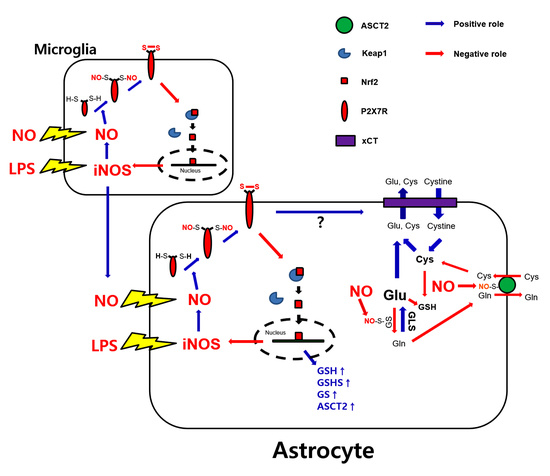
:
1. Introduction
2. Materials and Methods
2.1. Experimental Animals, Chemicals and LPS Treatment
2.2. GSH Assay
2.3. Immunohistochemistry
2.4. Western Blot
2.5. Measurement of S-Nitrosylation on GS and ASCT2
2.6. Data Analysis
3. Results
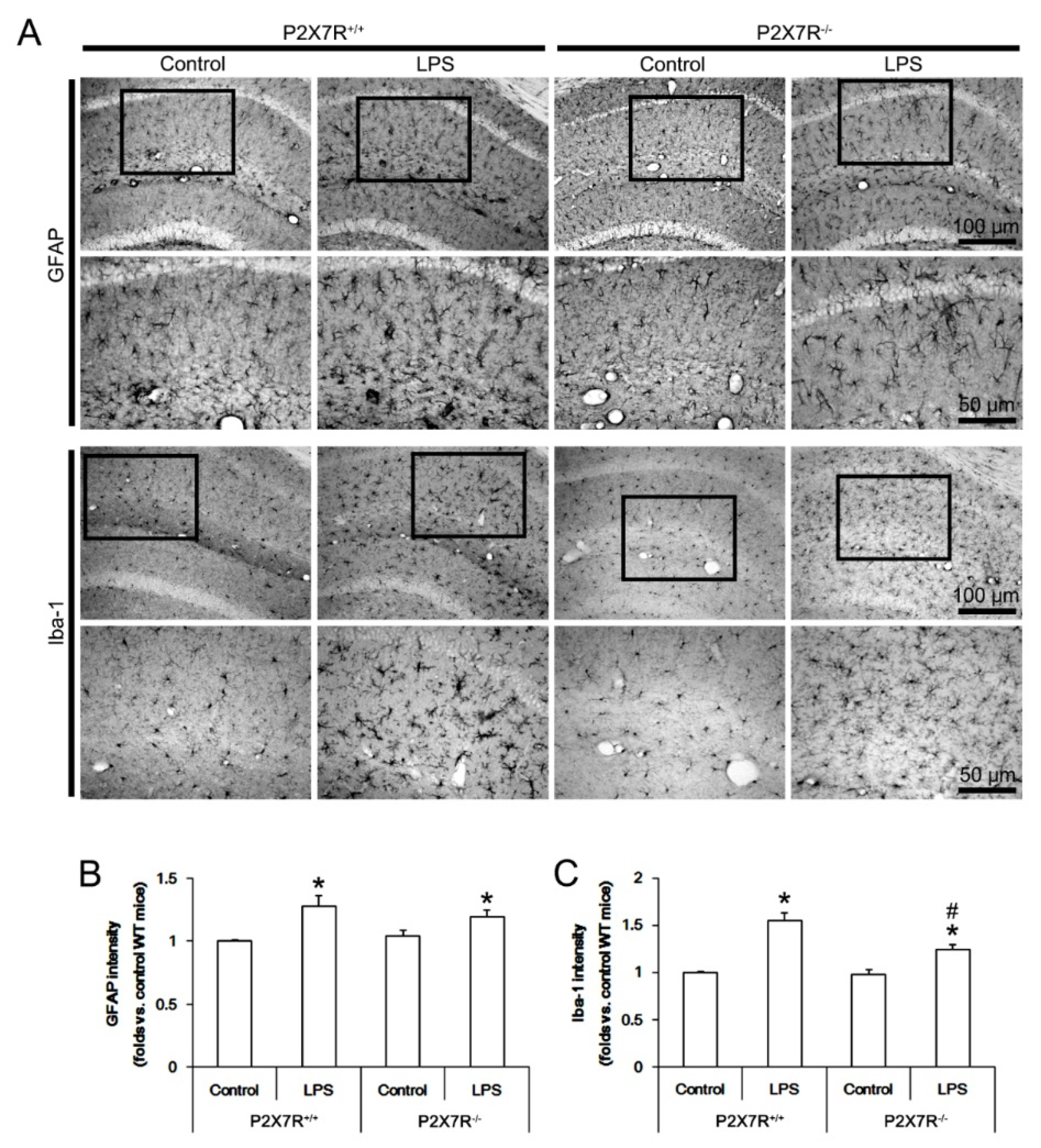
3.1. P2X7R Deletion Ameliorates Microglial Activation, but Not Reactive Astrogliosis in Response to LPS
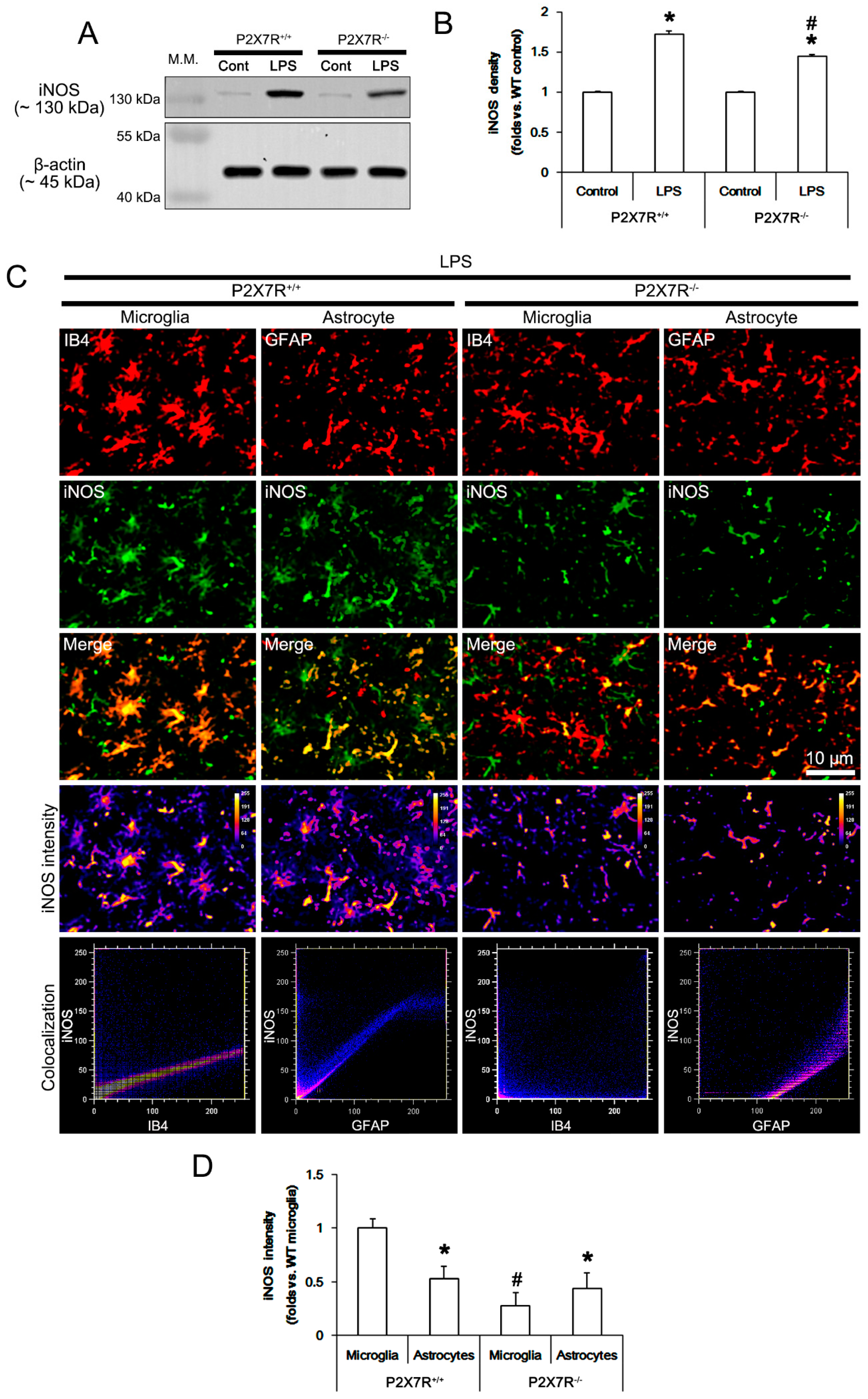
3.2. P2X7R Deletion Attenuates LPS-Induced iNOS Induction in Microglia Rather Than Astrocytes
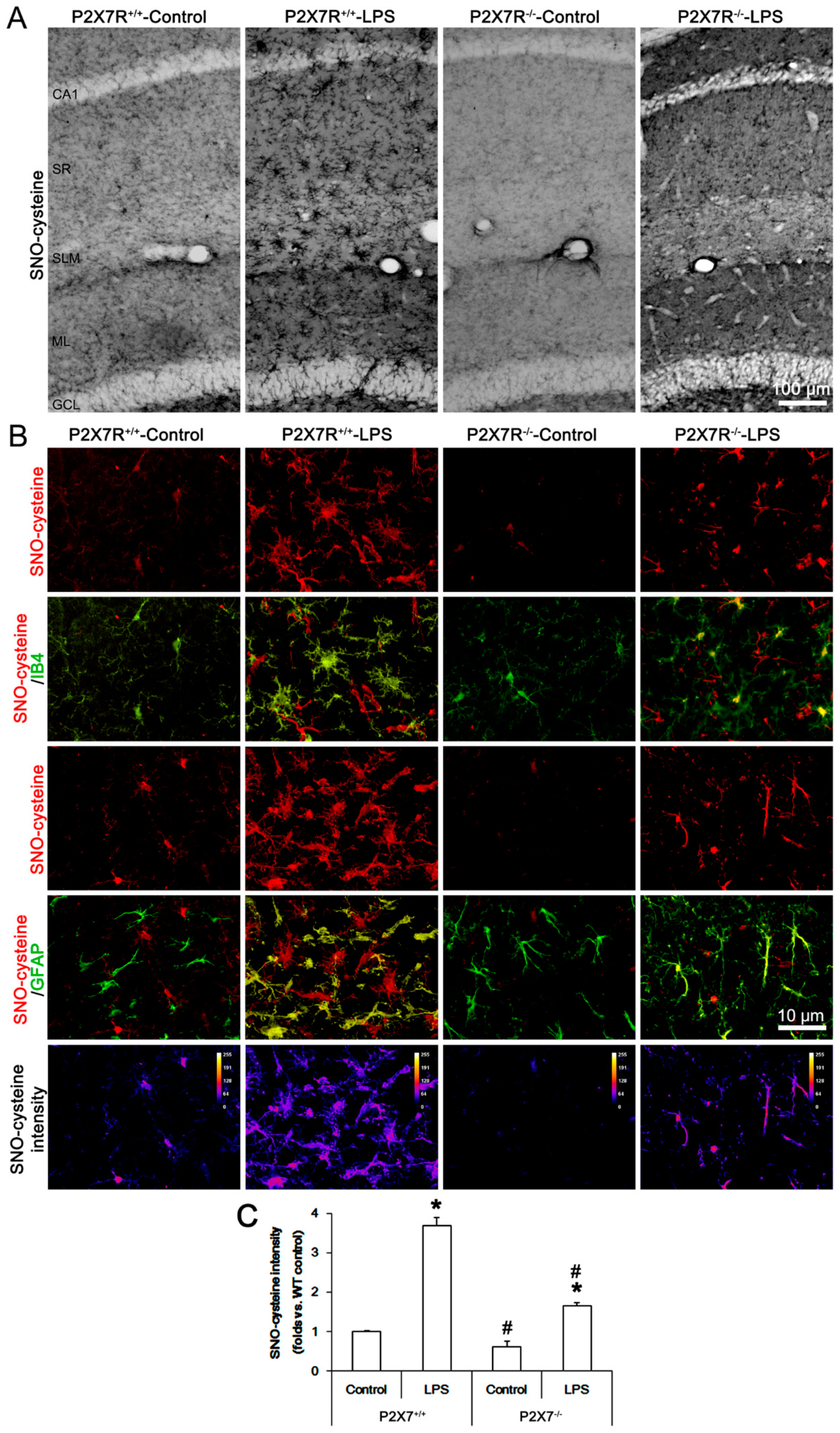
3.3. P2X7R Deletion Attenuates LPS-Induced SNO-Cysteine Production in Microglia and Astrocytes
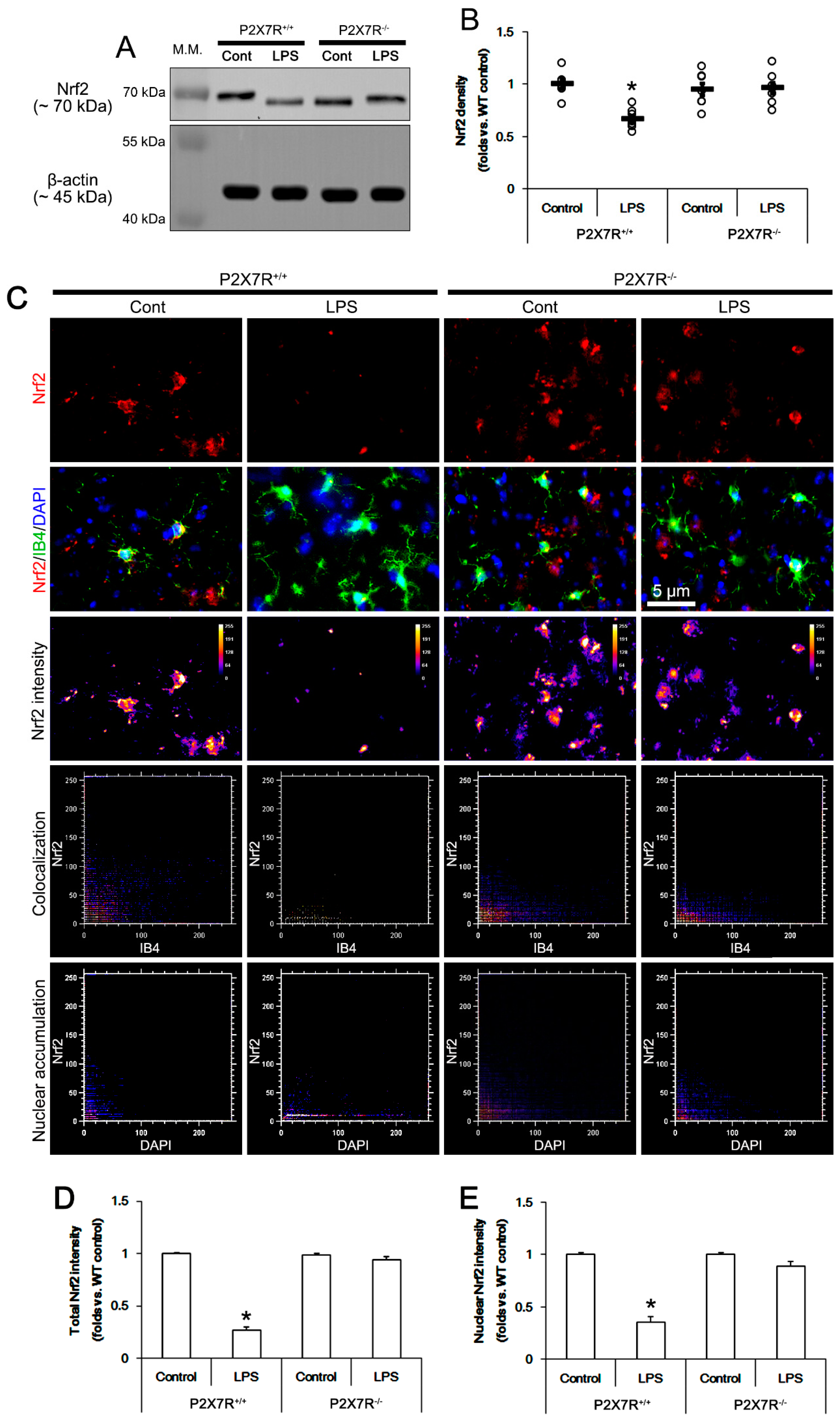
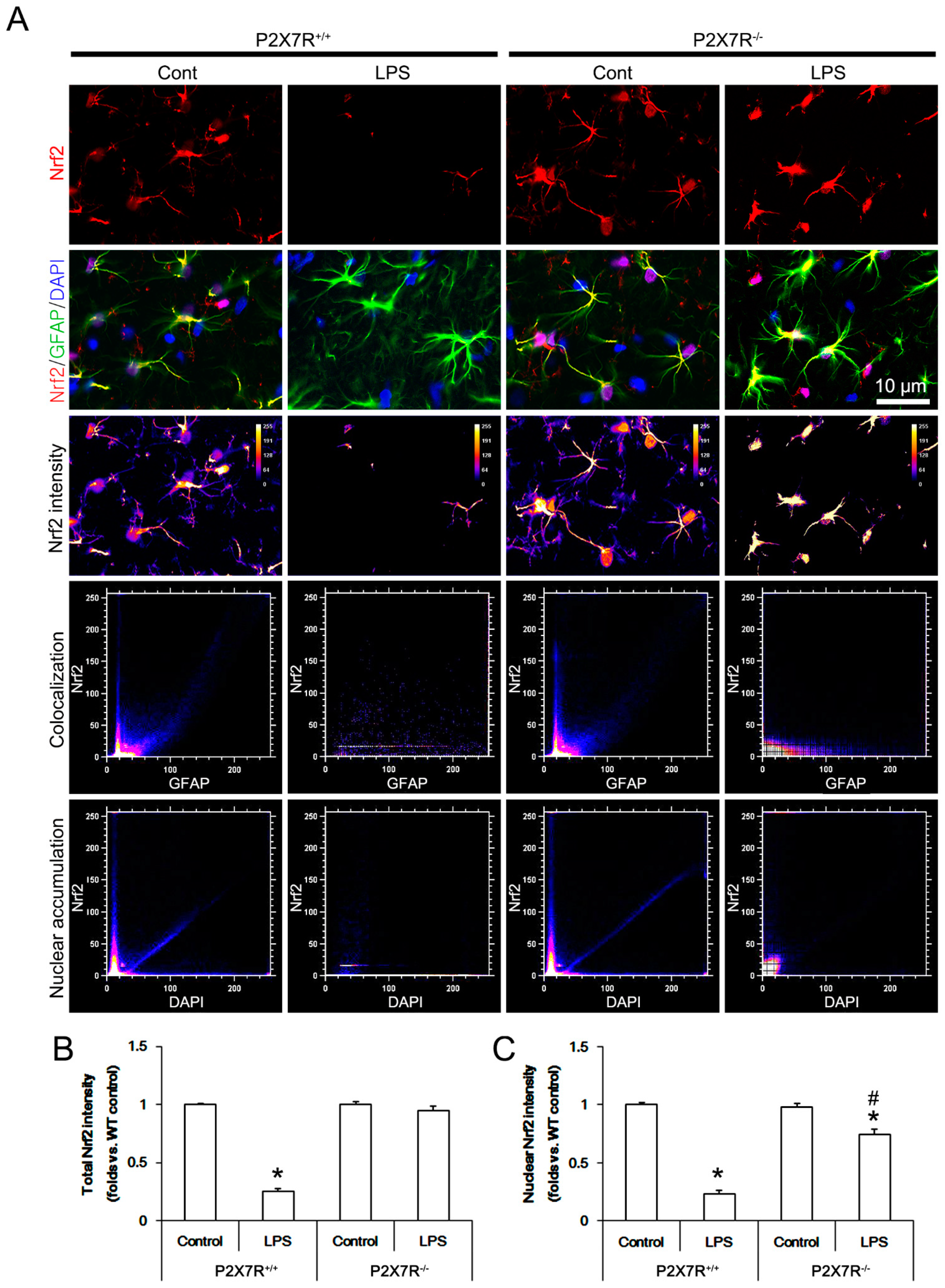
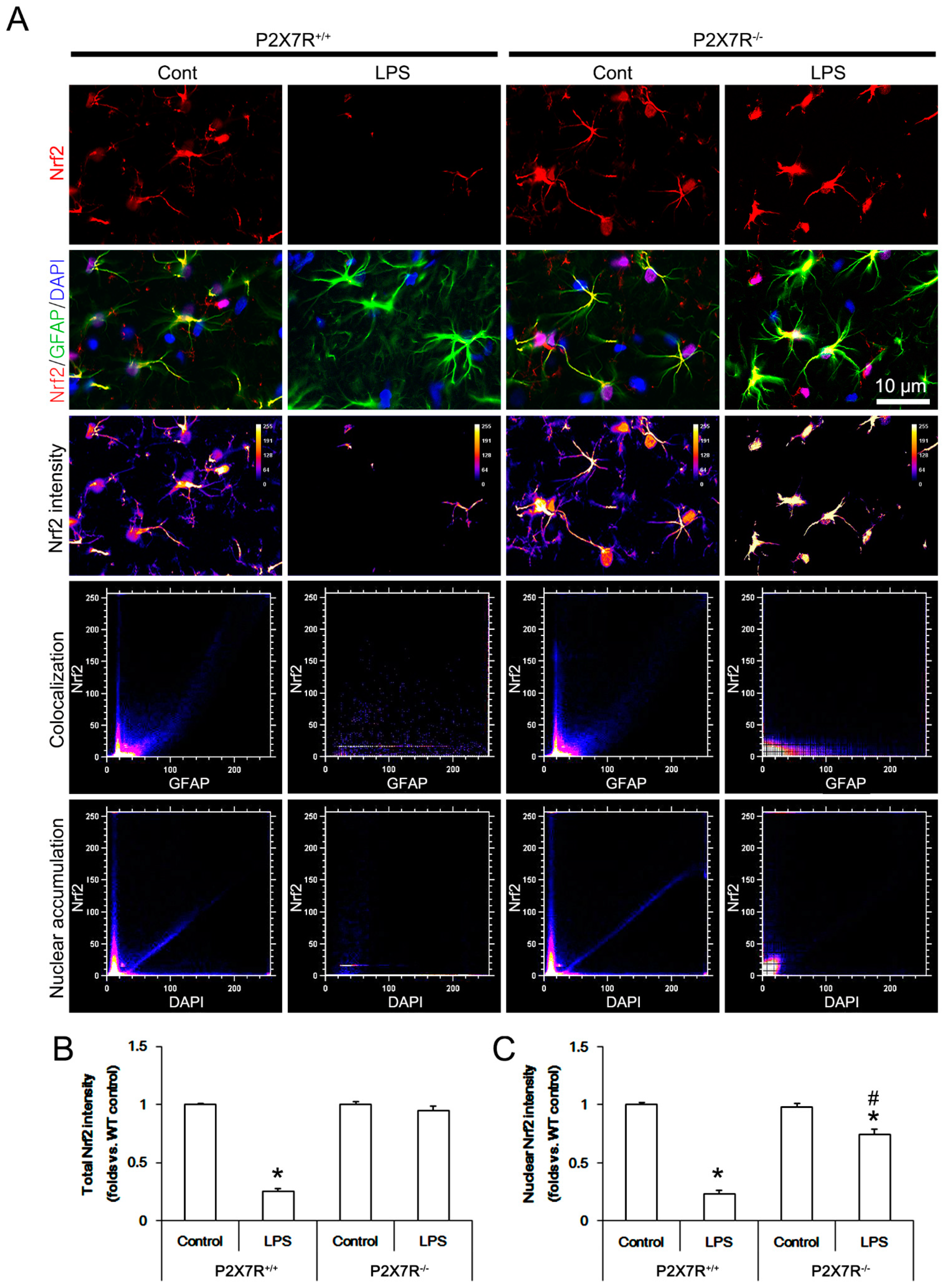
3.4. P2X7R Deletion Ameliorates LPS-Induced Nrf2 Downregulation
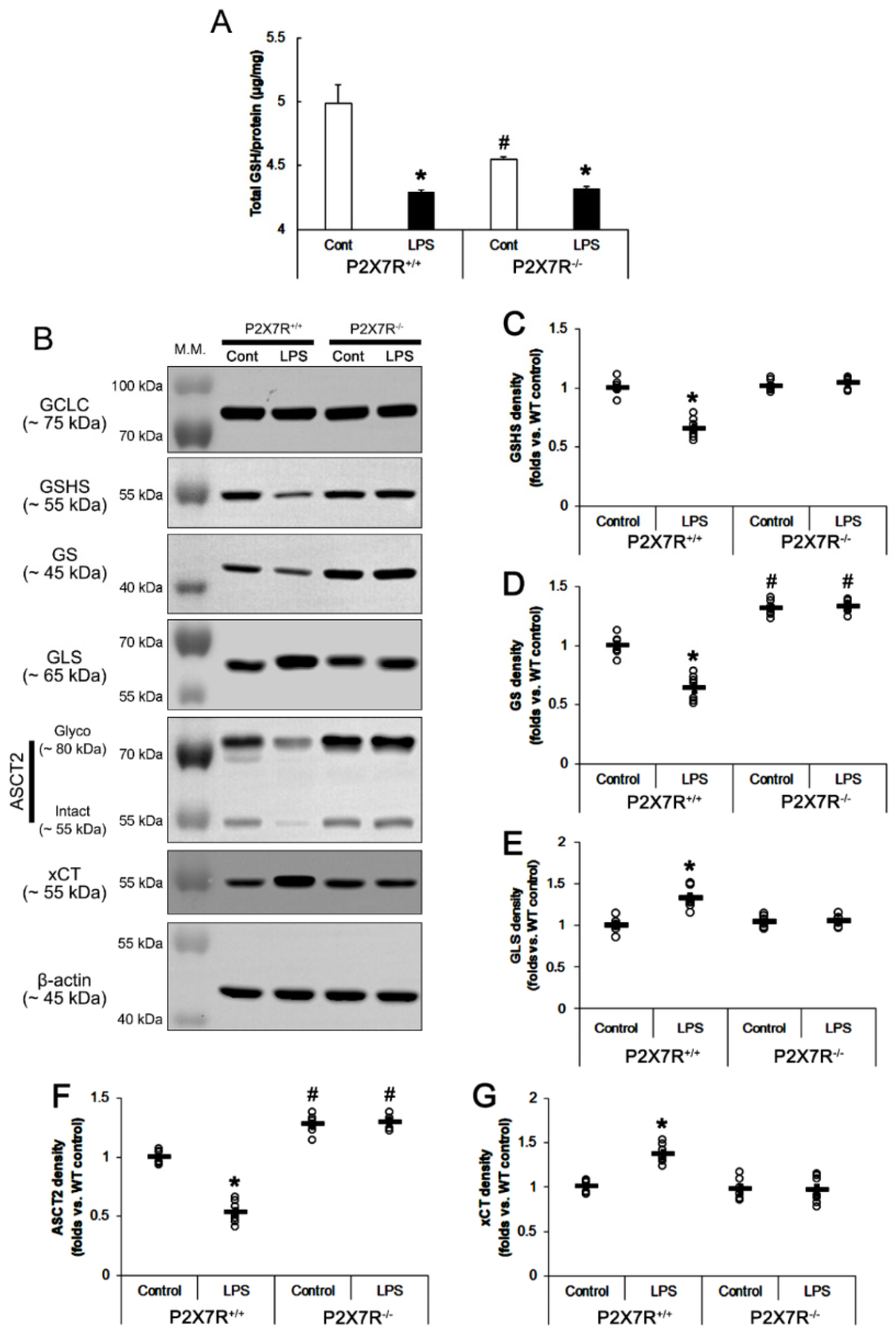
3.5. P2X7R Aggravates the Decreased GSH Concentration Induced by LPS
3.6. P2X7R Downregulates GSHS, but Not GCLC Expression following LPS Treatment
3.7. P2X7R Downregulates GS, but Increases GLS Expression following LPS Treatment
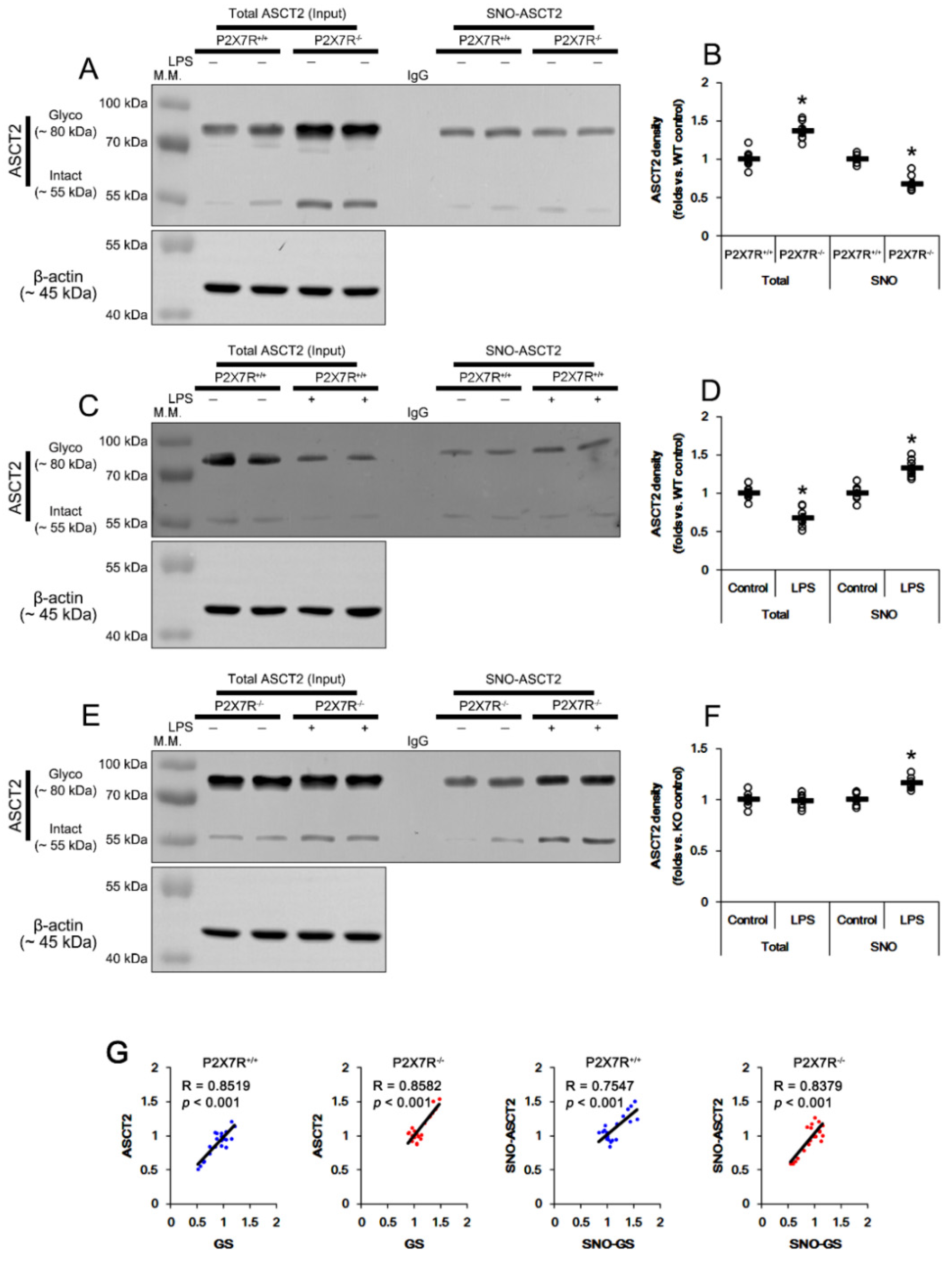
3.8. P2X7R Upregulates xCT, but Decreases ASCT2 Expression following LPS Treatment
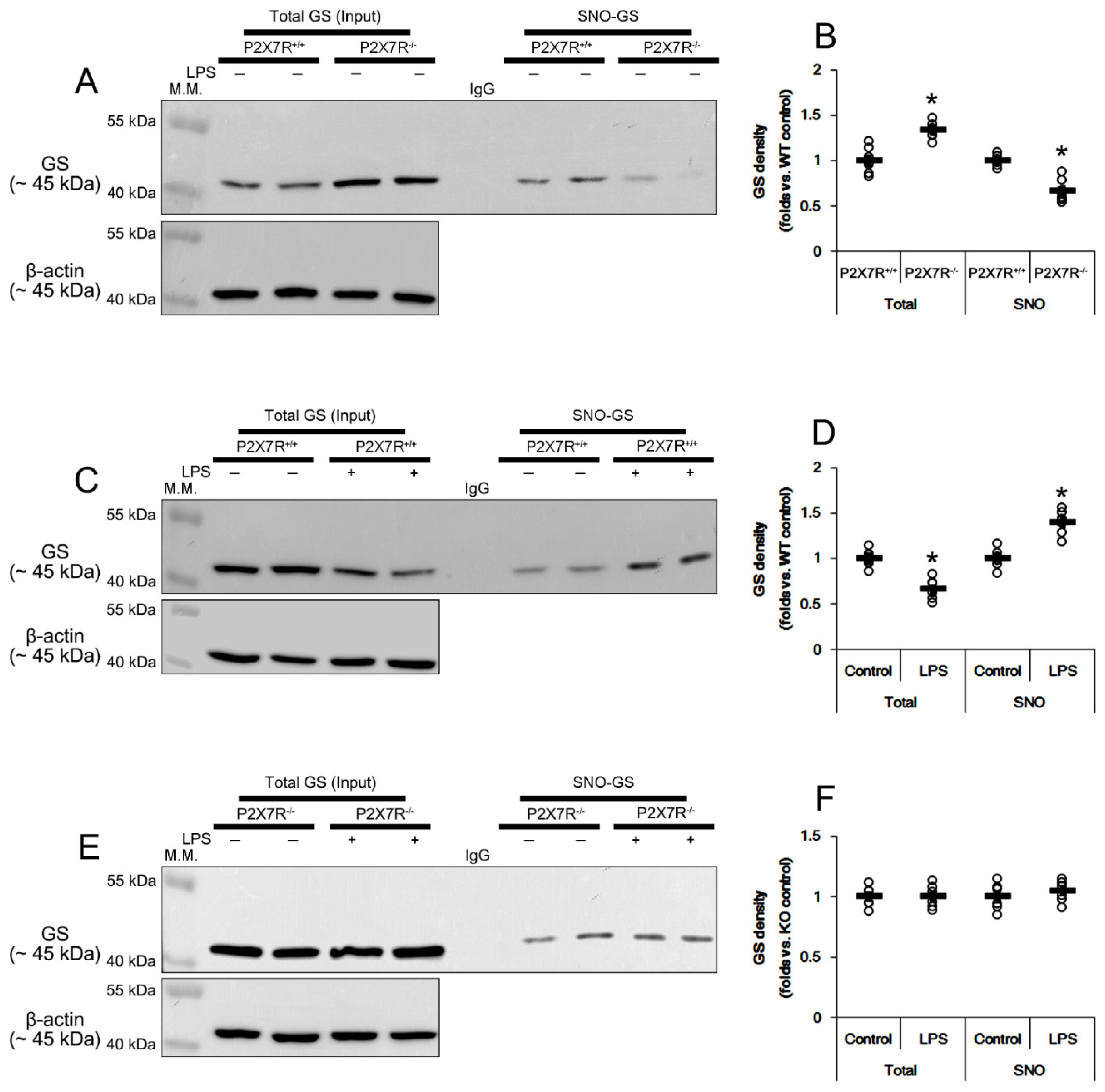
3.9. P2X7R Regulates S-Nitrosylation of GS and ASCT2 under Physiological and Post-LPS Treated Condition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kang, T.C. The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice. J. Clin. Investig. 2011, 121, 2037–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Kang, T.C. P2X7 receptor regulates leukocyte infiltrations in rat frontoparietal cortex following status epilepticus. J. Neuroinflamm. 2010, 7, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J.C.; Huang, W.C.; Chou, Y.C.; Tseng, C.H.; Lee, W.L.; Sun, S.H. Activation of P2X(7) receptors decreases glutamate uptake and glutamine synthetase activity in RBA-2 astrocytes via distinct mechanisms. J. Neurochem. 2008, 105, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Codocedo, J.F.; Godoy, J.A.; Poblete, M.I.; Inestrosa, N.C.; Huidobro-Toro, J.P. ATP induces NO production in hippocampal neurons by P2X(7) receptor activation independent of glutamate signaling. PLoS ONE 2013, 8, e57626. [Google Scholar] [CrossRef] [Green Version]
- Ficker, C.; Rozmer, K.; Kató, E.; Andó, R.D.; Schumann, L.; Krügel, U.; Franke, H.; Sperlágh, B.; Riedel, T.; Illes, P. Astrocyte-neuron interaction in the substantia gelatinosa of the spinal cord dorsal horn via P2X7 receptor-mediated release of glutamate and reactive oxygen species. Glia 2014, 62, 1671–1686. [Google Scholar] [CrossRef] [Green Version]
- Sperlágh, B.; Haskó, G.; Németh, Z.; Vizi, E.S. ATP released by LPS increases nitric oxide production in raw 264.7 macrophage cell line via P2Z/P2X7 receptors. Neurochem. Int. 1998, 33, 209–215. [Google Scholar] [CrossRef]
- Hu, Y.; Fisette, P.L.; Denlinger, L.C.; Guadarrama, A.G.; Sommer, J.A.; Proctor, R.A.; Bertics, P.J. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J. Biol. Chem. 1998, 273, 27170–27175. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.B.; Ryu, J.K.; Kim, S.U.; McLarnon, J.G. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J. Neurosci. 2007, 27, 4957–4968. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Meng, F.; Zhou, H.; Li, J.; Wang, Q.; Wei, F.; Cheng, J.; Greenlief, M.C.; Lubahn, B.M.; Sun, Y.G.; et al. NitroDIGE analysis reveals inhibition of protein S-nitrosylation by epigallocatechin gallates in lipopolysaccharide-stimulated microglial cells. J. Neuroinflamm. 2014, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Kim, J.E. Protein disulfide isomerase-mediated S-nitrosylation facilitates surface expression of P2X7 receptor following status epilepticus. J. Neuroinflamm. 2021, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-dependent detoxification processes in astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya-Tate, N.; Endou, H.; Kanai, Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J. Biol. Chem. 1996, 271, 14883–14890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Van Liefferinge, J.; Bentea, E.; Demuyser, T.; Albertini, G.; Follin-Arbelet, V.; Holmseth, S.; Merckx, E.; Sato, H.; Aerts, J.L.; Smolders, I.; et al. Comparative analysis of antibodies to xCT (Slc7a11): Forewarned is forearmed. J. Comp. Neurol. 2016, 524, 1015–1032. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Ruangkittisakul, A.; MacTavish, D.; Baker, G.B.; Ballanyi, K.; Jhamandas, J.H. Activity and metabolism-related Ca2+ and mitochondrial dynamics in co-cultured human fetal cortical neurons and astrocytes. Neuroscience 2013, 250, 520–535. [Google Scholar] [CrossRef]
- Pan, H.C.; Chou, Y.C.; Sun, S.H. P2X7 R-mediated Ca(2+)-independent d-serine release via pannexin-1 of the P2X7 R-pannexin-1 complex in astrocytes. Glia 2015, 63, 877–893. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.E. Deletion of P2X7 receptor decreases basal glutathione level by changing glutamate-glutamine cycle and neutral amino acid transporters. Cells 2020, 9, 995. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar] [PubMed]
- Noh, H.; Jeon, J.; Seo, H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 2014, 69, 35–40. [Google Scholar] [CrossRef]
- Venoji, R.; Amirtharaj, G.J.; Kini, A.; Vanaparthi, S.; Venkatraman, A.; Ramachandran, A. Enteral glutamine differentially regulates Nrf 2 along the villus-crypt axis of the intestine to enhance glutathione levels. J. Gastroenterol. Hepatol. 2015, 30, 1740–1747. [Google Scholar] [CrossRef]
- Guerra, A.N.; Fisette, P.L.; Pfeiffer, Z.A.; Quinchia-Rios, B.H.; Prabhu, U.; Aga, M.; Denlinger, L.C.; Gaudarrama, A.G.; Abozeid, S.; Sommer, J.A.; et al. Purinergic receptor regulation of LPS-induced signaling and pathophysiology. J. Endotoxin Res. 2003, 9, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-kappaB signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [Green Version]
- Noworyta-Sokolowska, K.; Gorska, A.; Golembiowska, K. LPS-induced oxidative stress and inflammatory reaction in the rat striatum. Pharmacol. Rep. 2013, 65, 863–869. [Google Scholar] [CrossRef]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin stimulates the SIRT1/Nrf2 signaling pathway counteracting lipopolysaccharide (LPS)-induced oxidative stress to rescue postnatal rat brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef]
- Townsend, B.E.; Johnson, R.W. Sulforaphane induces Nrf2 target genes and attenuates inflammatory gene expression in microglia from brain of young adult and aged mice. Exp. Gerontol. 2016, 73, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Kwon, K.H.; Cheong, S.H. Taurine chloramine suppresses LPS-induced neuroinflammatory responses through Nrf2-mediated heme oxygenase-1 expression in mouse BV2 microglial cells. Adv. Exp. Med. Biol. 2017, 975, 131–143. [Google Scholar] [PubMed]
- Sun, X.; Erb, H.; Murphy, T.H. Coordinate regulation of glutathione metabolism in astrocytes by Nrf2. Biochem. Biophys. Res. Commun. 2005, 326, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.M.; Nevarea, Z.; Gipp, J.J.; Mulcahy, R.T. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. Revision of the ARE consensus sequence. J. Biol. Chem. 2002, 277, 30730–30737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, P.B.; Sri Hari, A.; Day, B.J.; Patel, M. Post-translational activation of glutamate cysteine ligase with dimercaprol: A novel mechanism of inhibiting neuroinflammation in vitro. J. Biol. Chem. 2017, 292, 5532–5545. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yin, W.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. L-F001, a novel multifunctional ROCK inhibitor, suppresses neuroinflammation in vitro and in vivo: Involvement of NF-κB inhibition and Nrf2 pathway activation. Eur. J. Pharmacol. 2017, 806, 1–9. [Google Scholar] [CrossRef]
- Wu, P.S.; Ding, H.Y.; Yen, J.H.; Chen, S.F.; Lee, K.H.; Wu, M.J. Anti-inflammatory activity of 8-hydroxydaidzein in LPS-stimulated BV2 microglial cells via activation of Nrf2-antioxidant and attenuation of Akt/NF-κB-inflammatory signaling pathways, as well as inhibition of COX-2 activity. J. Agric. Food Chem. 2018, 66, 5790–5801. [Google Scholar] [CrossRef]
- Ko, K.; Yang, H.; Noureddin, M.; Iglesia-Ara, A.; Xia, M.; Wagner, C.; Luka, Z.; Mato, J.M.; Lu, S.C. Changes in S-adenosylmethionine and GSH homeostasis during endotoxemia in mice. Lab. Investig. 2008, 88, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Häberle, J.; Görg, B.; Toutain, A.; Rutsch, F.; Benoist, J.F.; Gelot, A.; Suc, A.-L.; Koch, H.G.; Schliess, F.; Häussinger, D. Inborn error of amino acid synthesis: Human glutamine synthetase deficiency. J. Inherit. Metab. Dis. 2006, 29, 352–358. [Google Scholar] [CrossRef]
- Hayashi, M.K. Structure-function relationship of transporters in the glutamate-glutamine cycle of the central nervous system. Int. J. Mol. Sci. 2018, 19, 1177. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Zhao, S.; Xia, X.; Li, C.; Li, C.; Ji, C.; Sheng, S.; Tang, Y.; Zhu, J.; Wang, Y.; et al. Glutaminase C regulates microglial activation and pro-inflammatory exosome release: Relevance to the pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 2019, 13, 264. [Google Scholar] [CrossRef]
- Gegelashvili, M.; Rodriguez-Kern, A.; Pirozhkova, I.; Zhang, J.; Sung, L.; Gegelashvili, G. High-affinity glutamate transporter GLAST/EAAT1 regulates cell surface expression of glutamine/neutral amino acid transporter ASCT2 in human fetal astrocytes. Neurochem. Int. 2006, 48, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Brookes, N.; Ganapathy, V.; Dimmer, K.S.; Wagner, C.A.; Lang, F.; Bröer, S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 1999, 73, 2184–2194. [Google Scholar] [PubMed]
- Asahi, M.; Fujii, J.; Suzuki, K.; Seo, H.G.; Kuzuya, T.; Hori, M.; Tada, M.; Fujii, S.; Taniguchi, N. Inactivation of glutathione peroxidase by nitric oxide. Implication for cytotoxicity. J. Biol. Chem. 1995, 270, 21035–21039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, K.; Doulias, P.T.; Evans, P.; Krizman, E.N.; Jackson, J.G.; Horyn, O.; Daikhin, Y.; Nissim, I.; Yudkoff, M.; Nissim, I.; et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal. 2015, 8, ra68. [Google Scholar] [CrossRef] [Green Version]
- Miñana, M.D.; Kosenko, E.; Marcaida, G.; Hermenegildo, C.; Montoliu, C.; Grisolía, S.; Felipo, V. Modulation of glutamine synthesis in cultured astrocytes by nitric oxide. Cell. Mol. Neurobiol. 1997, 17, 433–445. [Google Scholar] [CrossRef]
- Kosenko, E.; Llansola, M.; Montoliu, C.; Monfort, P.; Rodrigo, R.; Hernandez-Viadel, M.; Erceg, S.; Sánchez-Perez, A.M.; Felipo, V. Glutamine synthetase activity and glutamine content in brain: Modulation by NMDA receptors and nitric oxide. Neurochem. Int. 2003, 43, 493–499. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Pappacoda, G.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Cys site-directed mutagenesis of the human SLC1A5 (ASCT2) transporter: Structure/function relationships and crucial role of Cys467 for redox sensing and glutamine transport. Int. J. Mol. Sci. 2018, 19, 648. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, T.; Matsuda, Y.; Wada, M.; Takahashi, S.; Fujita, T. Functional regulation of Na+-dependent neutral amino acid transporter ASCT2 by S-nitrosothiols and nitric oxide in Caco-2 cells. FEBS Lett. 2005, 579, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Barbierato, M.; Facci, L.; Argentini, C.; Marinelli, C.; Skaper, S.D.; Giusti, P. Astrocyte-microglia cooperation in the expression of a pro-inflammatory phenotype. CNS Neurol. Disord. Drug Targets 2013, 12, 608–618. [Google Scholar] [CrossRef]
- Aga, M.; Watters, J.J.; Pfeiffer, Z.A.; Wiepz, G.J.; Sommer, J.A.; Bertics, P.J. Evidence for nucleotide receptor modulation of cross talk between MAP kinase and NF-kappa B signaling pathways in murine RAW 264.7 macrophages. Am. J. Physiol. Cell Physiol. 2004, 286, 923–930. [Google Scholar] [CrossRef]
- Mann, G.E. Nrf2-mediated redox signalling in vascular health and disease. Free Radic. Biol. Med. 2014, 75, S1. [Google Scholar] [CrossRef] [PubMed]
- Owuor, E.D.; Kong, A.N. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002, 64, 765–770. [Google Scholar] [CrossRef]
- Leite, J.A.; Isaksen, T.J.; Heuck, A.; Scavone, C.; Lykke-Hartmann, K. The α2 Na+/K+-ATPase isoform mediates LPS-induced neuroinflammation. Sci. Rep. 2020, 10, 14180. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Rahman, S.U.; Hao, Q.; Li, W.; Liu, Z.; Ali Shah, F.; Murtaza, I.; Zhang, Z.; Yang, X.; Liu, G.; et al. Melatonin prevents neuroinflammation and relieves depression by attenuating autophagy impairment through FOXO3a regulation. J. Pineal Res. 2020, 69, e12667. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.L.; Fuller, S.; Patel, M.; Kersaitis, C.; Ooi, L.; Münch, G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol. 2013, 1, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Mann, G.E. Redox status in mammalian cells and stem cells during culture in vitro: Critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox Biol. 2014, 2, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Valdovinos-Flores, C.; Limón-Pacheco, J.H.; León-Rodríguez, R.; Petrosyan, P.; Garza-Lombó, C.; Gonsebatt, M.E. Systemic L-buthionine-S-R-sulfoximine treatment increases plasma NGF and upregulates L-cys/L-cys2 transporter and γ-glutamylcysteine ligase mRNAs through the NGF/TrkA/Akt/Nrf2 pathway in the striatum. Front. Cell. Neurosci. 2019, 13, 325. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Tamba, M.; Bannai, S.; Sato, H. Induction of cystine/glutamate transporter in bacterial lipopolysaccharide induced endotoxemia in mice. J. Inflamm. 2007, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Pacchioni, A.M.; Vallone, J.; Melendez, R.I.; Shih, A.; Murphy, T.H.; Kalivas, P.W. Nrf2 gene deletion fails to alter psychostimulant-induced behavior or neurotoxicity. Brain Res. 2007, 1127, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Saito, Y.; Nishio, K.; Yoshida, Y.; Ashida, H.; Niki, E. Gamma-tocopheryl quinone, not alpha-tocopheryl quinone, induces adaptive response through up-regulation of cellular glutathione and cysteine availability via activation of ATF4. Free Radic. Res. 2008, 42, 674–687. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the system x(C)-cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, S.; Marshall, Z.M.; Whorton, A.R. A cystine-cysteine shuttle mediated by xCT facilitates cellular responses to S-nitrosoalbumin. Am. J. Physiol. Cell. Physiol. 2008, 294, C1012–C1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollinger, K.R.; Zhu, X.; Khoury, E.S.; Thomas, A.G.; Liaw, K.; Tallon, C.; Ying, W.; Prchalova, E.; Atsushi, K.; Camilo, R.; et al. Glutamine antagonist JHU-083 normalizes aberrant hippocampal glutaminase activity and improves cognition in APOE4 mice. J. Alzheimers Dis. 2020, 77, 437–447. [Google Scholar] [CrossRef]
- Thomas, A.G.; O’Driscoll, C.M.; Bressler, J.; Kaufmann, W.; Rojas, C.J.; Slusher, B.S. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem. Biophys. Res. Commun. 2014, 443, 32–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of glutamine synthetase enhances inflammatory response in microglial cells. Antioxid. Redox Signal. 2017, 26, 351–363. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, H.; Geng, B.; Xie, Q.; Li, W.; Deng, Y.; Shi, W.; Pan, Y.; Kang, X.; Wang, J. 2-arachidonyl glycerol modulates astrocytic glutamine synthetase via p38 and ERK1/2 pathways. J. Neuroinflamm. 2018, 15, 220. [Google Scholar] [CrossRef]
- Görg, B.; Qvartskhava, N.; Voss, P.; Grune, T.; Häussinger, D.; Schliess, F. Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Lett. 2007, 581, 84–90. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| ASCT2 | Rabbit | Alomone labs (Jerusalem, Israel) (#ANT-082) | 1:500 (WB) |
| GCLC | Rabbit | Abcam (Waltham, MA, USA) (#ab190685) | 1:2000 (WB) |
| GFAP | Mouse | Millipore ( Burlington, MS, USA) (#MAB3402) | 1:2000 (IH) |
| GLS | Rabbit | Abcam (#ab93434) | 1:1000 (WB) |
| GS | Mouse | Millipore (#MAB302) | 1:1000 (WB) |
| GSHS | Rabbit | Abcam (#ab133592) | 1:2000 (WB) |
| IB4 | Vector (Los Altos, CA, USA) (#B-1205) | 1:200 (histochemistry) | |
| SNO-cysteine | Rabbit | Abcam (#ab94930) | 1:1000 (IH) |
| iNOS | Rabbit | Novus Biologicals (Centennial, CO, USA) (#NB300-605) | 1:100 (IH) 1:500 (WB) |
| Iba-1 | Rabbit | Biocare Medical (Pacheco, CA, USA) (#CP 290) | 1:500 (IH) |
| Nrf2 | Rabbit | Abcam (#ab137550) | 1:200 (IH) 1:1000 (IH) |
| xCT | Rabbit | Abcam (#ab175186) | 1:1000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-S.; Kim, J.-E. P2X7 Receptor Augments LPS-Induced Nitrosative Stress by Regulating Nrf2 and GSH Levels in the Mouse Hippocampus. Antioxidants 2022, 11, 778. https://doi.org/10.3390/antiox11040778
Lee D-S, Kim J-E. P2X7 Receptor Augments LPS-Induced Nitrosative Stress by Regulating Nrf2 and GSH Levels in the Mouse Hippocampus. Antioxidants. 2022; 11(4):778. https://doi.org/10.3390/antiox11040778
Chicago/Turabian StyleLee, Duk-Shin, and Ji-Eun Kim. 2022. "P2X7 Receptor Augments LPS-Induced Nitrosative Stress by Regulating Nrf2 and GSH Levels in the Mouse Hippocampus" Antioxidants 11, no. 4: 778. https://doi.org/10.3390/antiox11040778
APA StyleLee, D.-S., & Kim, J.-E. (2022). P2X7 Receptor Augments LPS-Induced Nitrosative Stress by Regulating Nrf2 and GSH Levels in the Mouse Hippocampus. Antioxidants, 11(4), 778. https://doi.org/10.3390/antiox11040778