
Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. SE Induction and Chronic Epilepsy Model
2.3. BSO and NAC Treatment
2.4. Western Blot
2.5. Immunohistochemistry
2.6. Data Analysis
3. Results
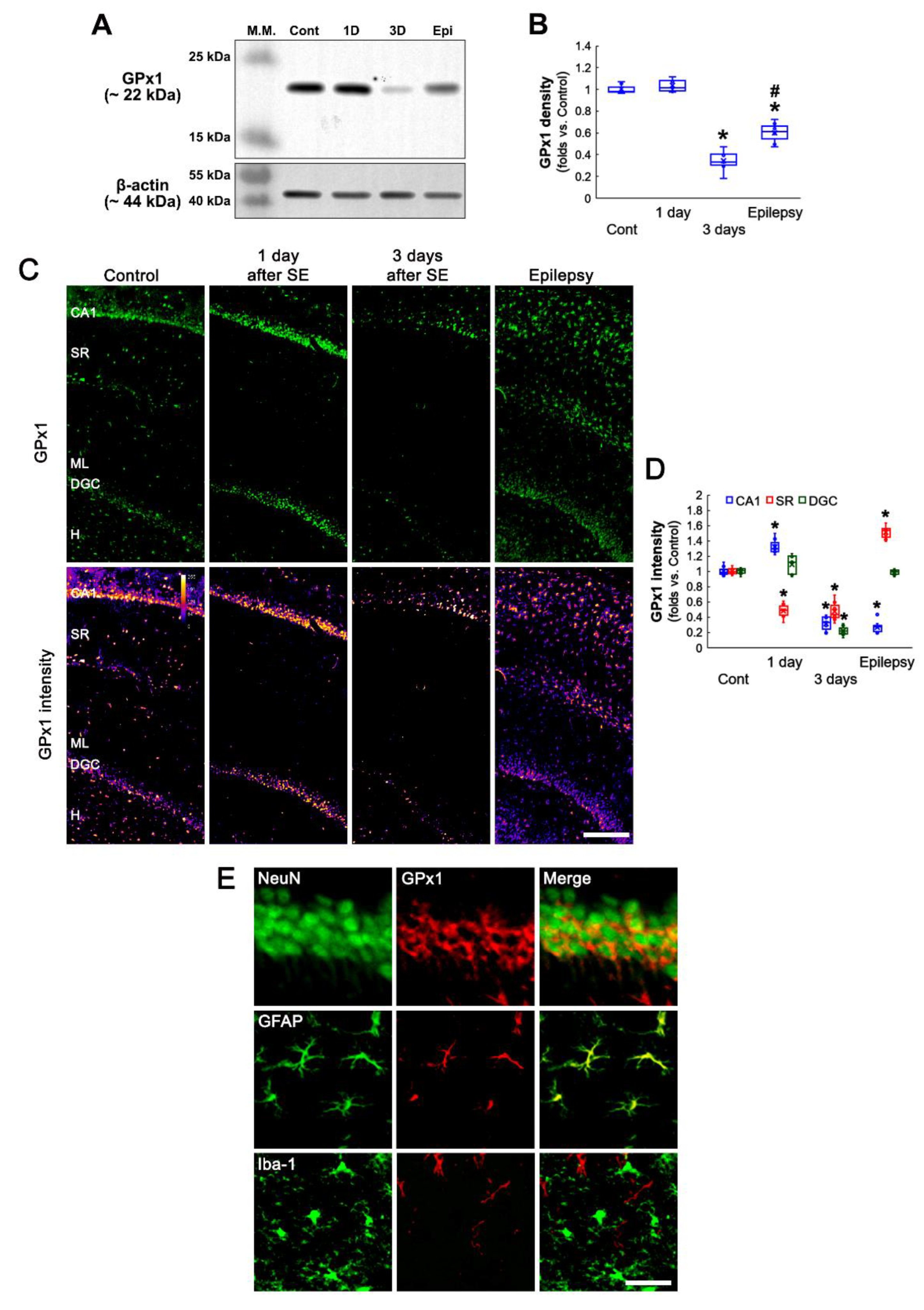
3.1. SE Changes Hippocampal GPx1 Expression in a Regional Specific Manner
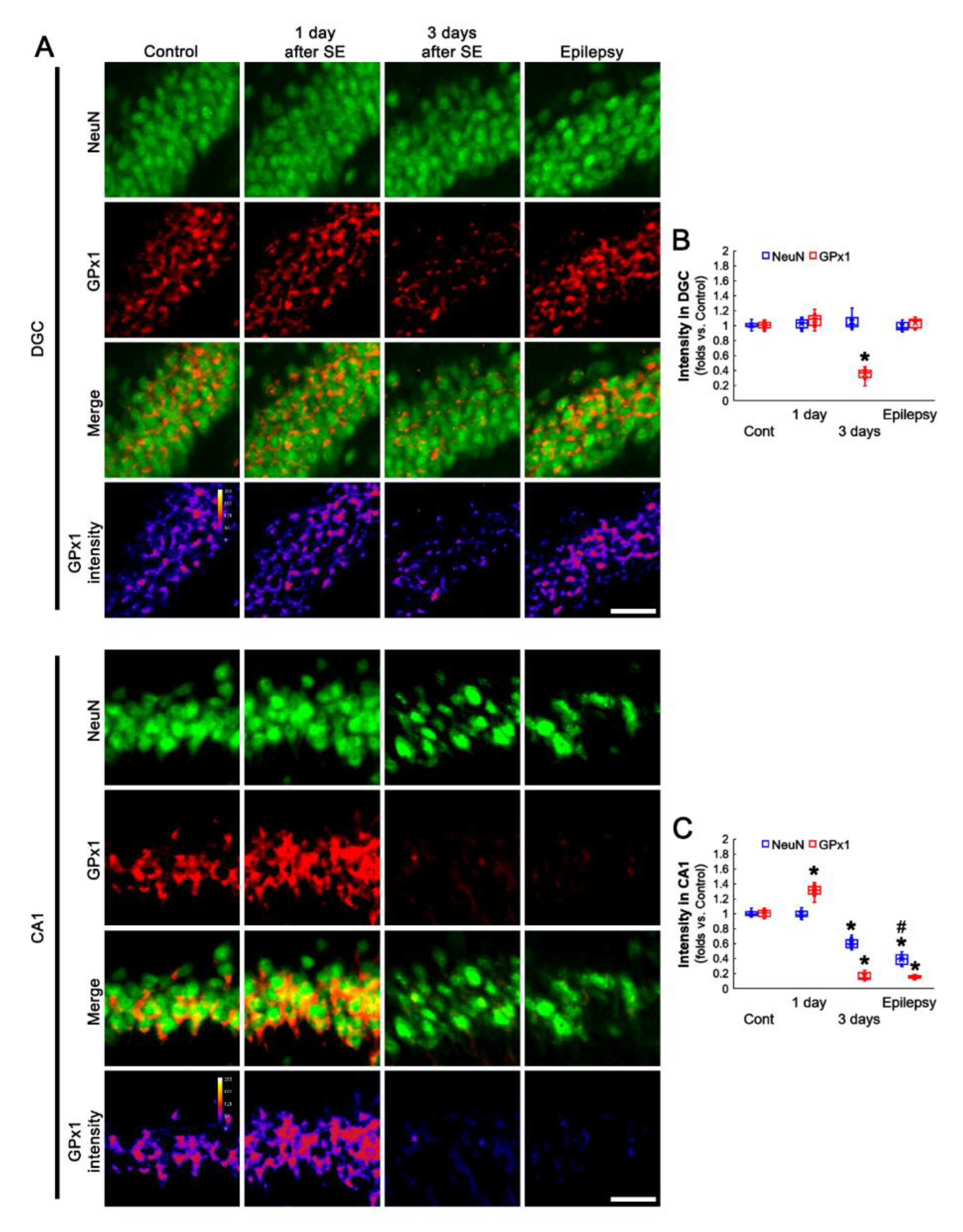
3.2. Altered GPx1 Expression Is Relevant to Neuronal Vulnerability to SE
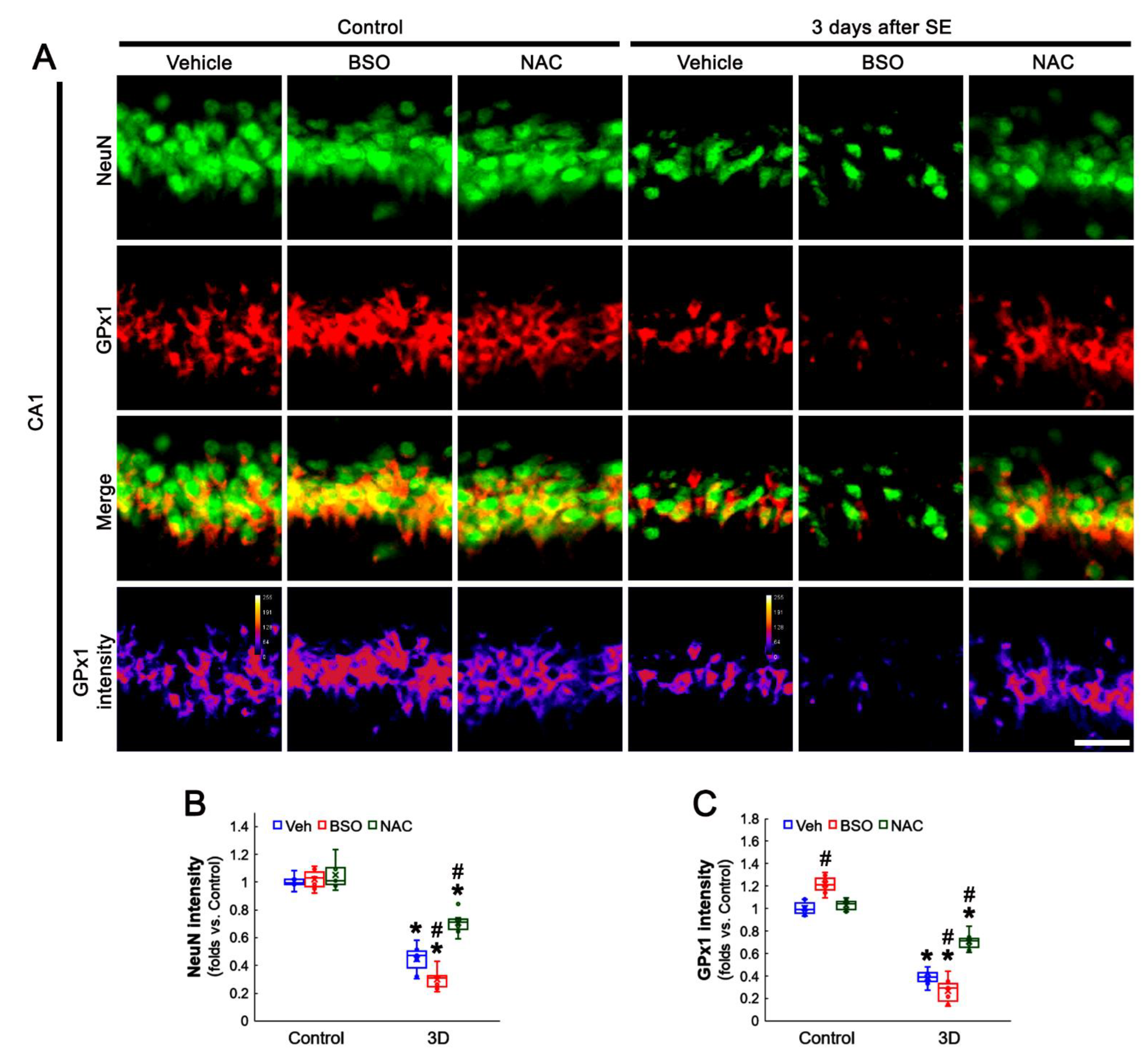
3.3. BSO and NAC Differently Affect GPx1 Expression in CA1 Neurons under Physiological and Post-SE Conditions
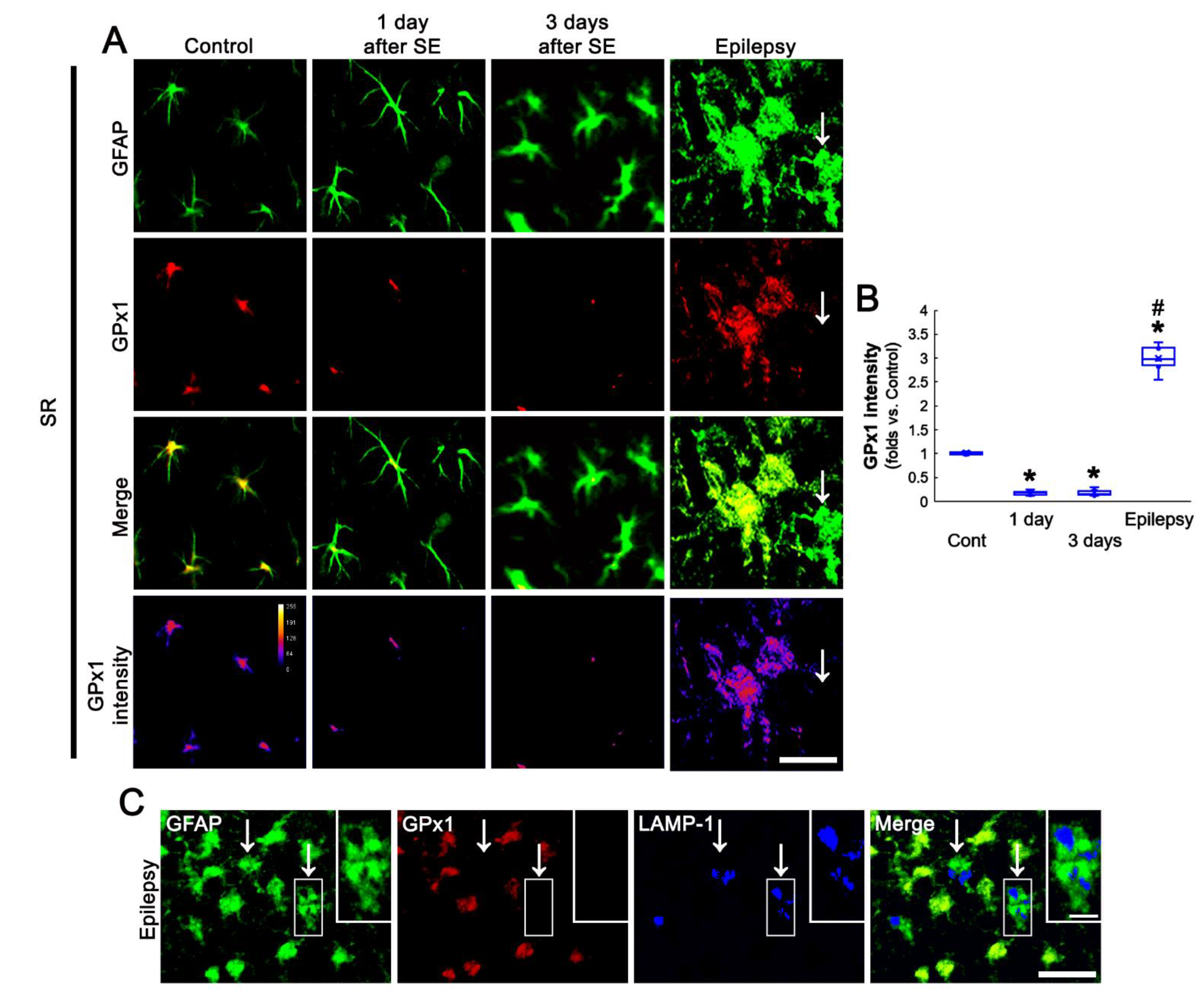
3.4. SE Leads to the Biphasic Alterations in GPx1 Expression in CA1 Astrocytes
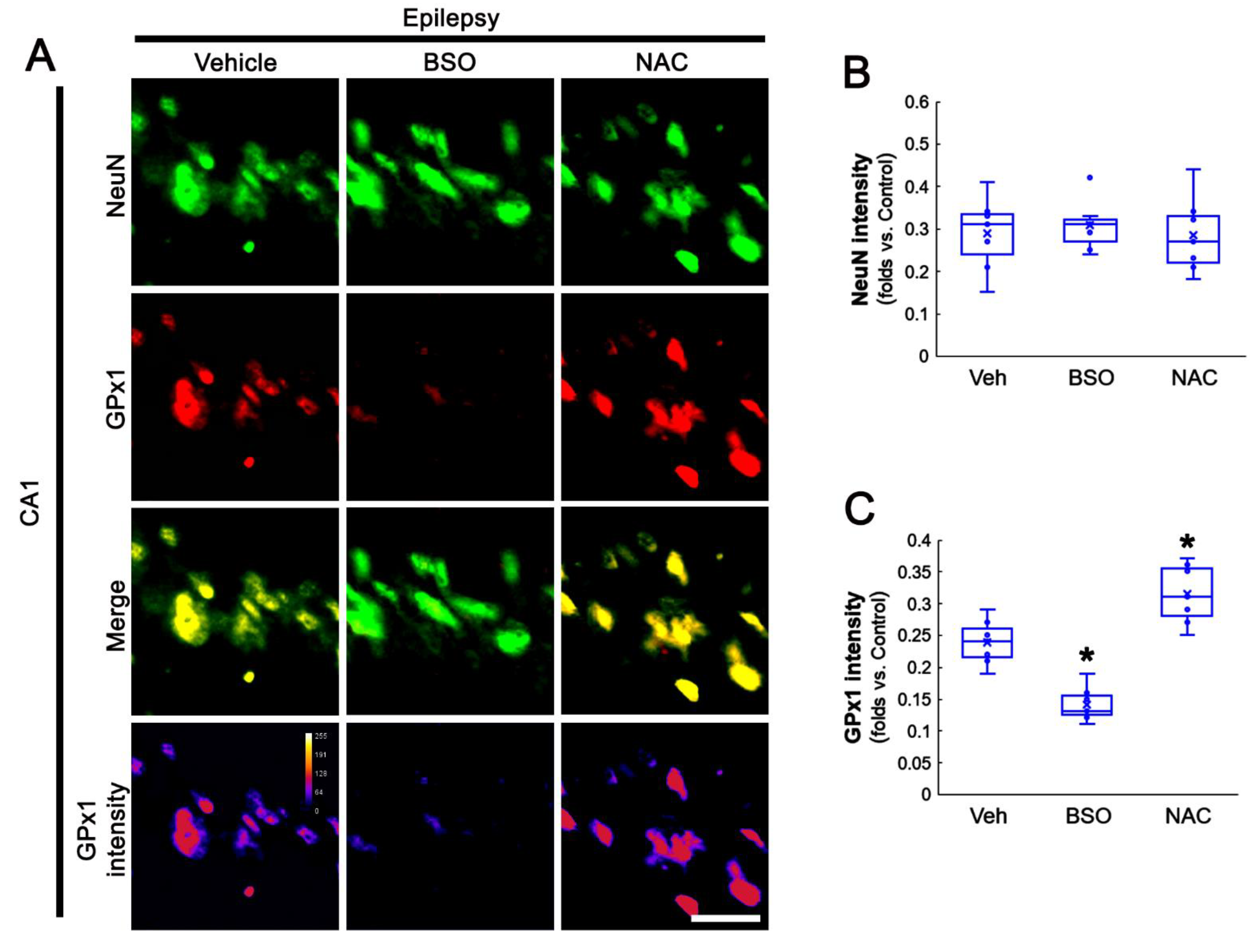
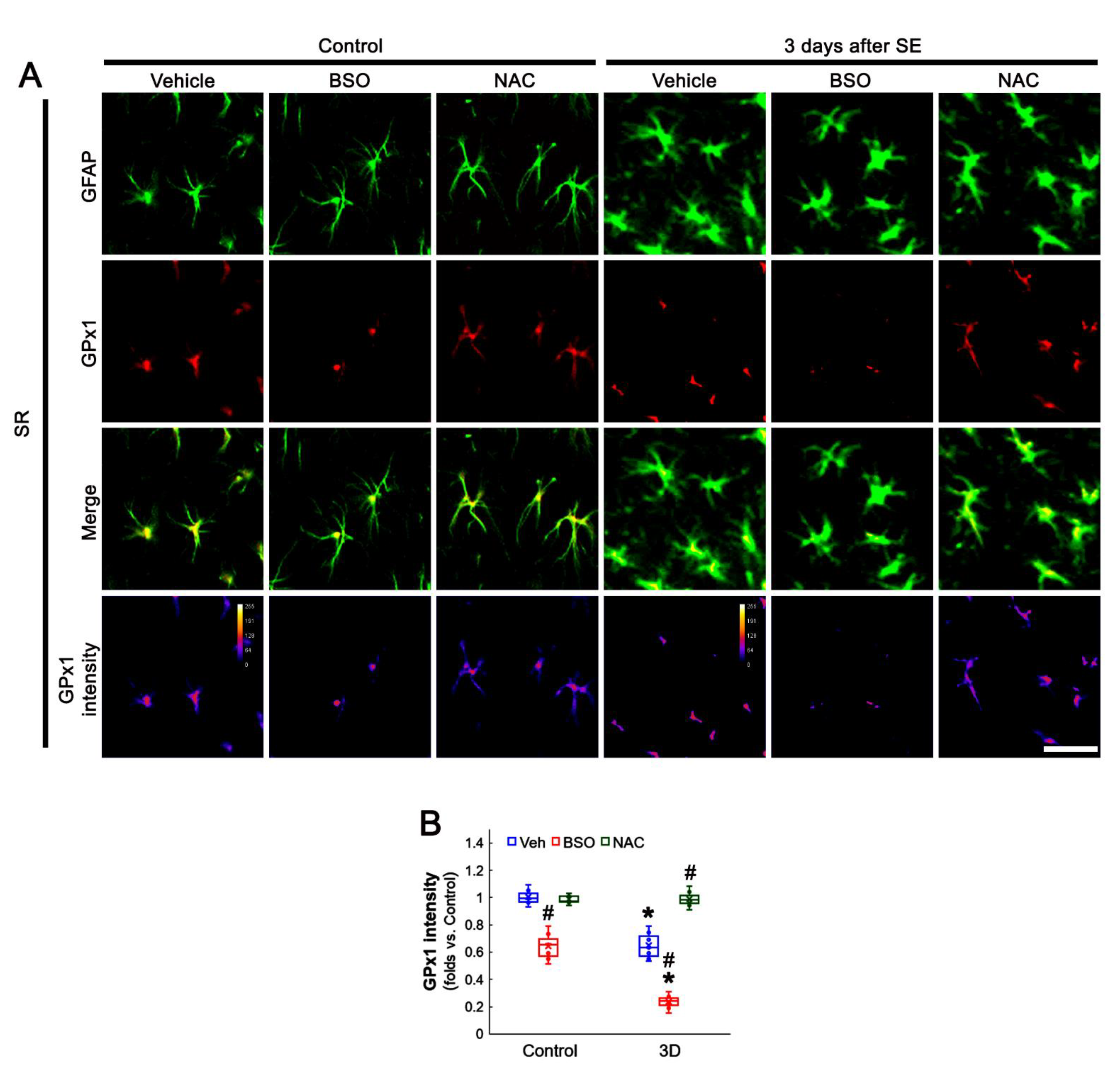
3.5. BSO Enhances, but NAC Abrogates SE-Induced GPx1 Downregulation in CA1 Astrocytes
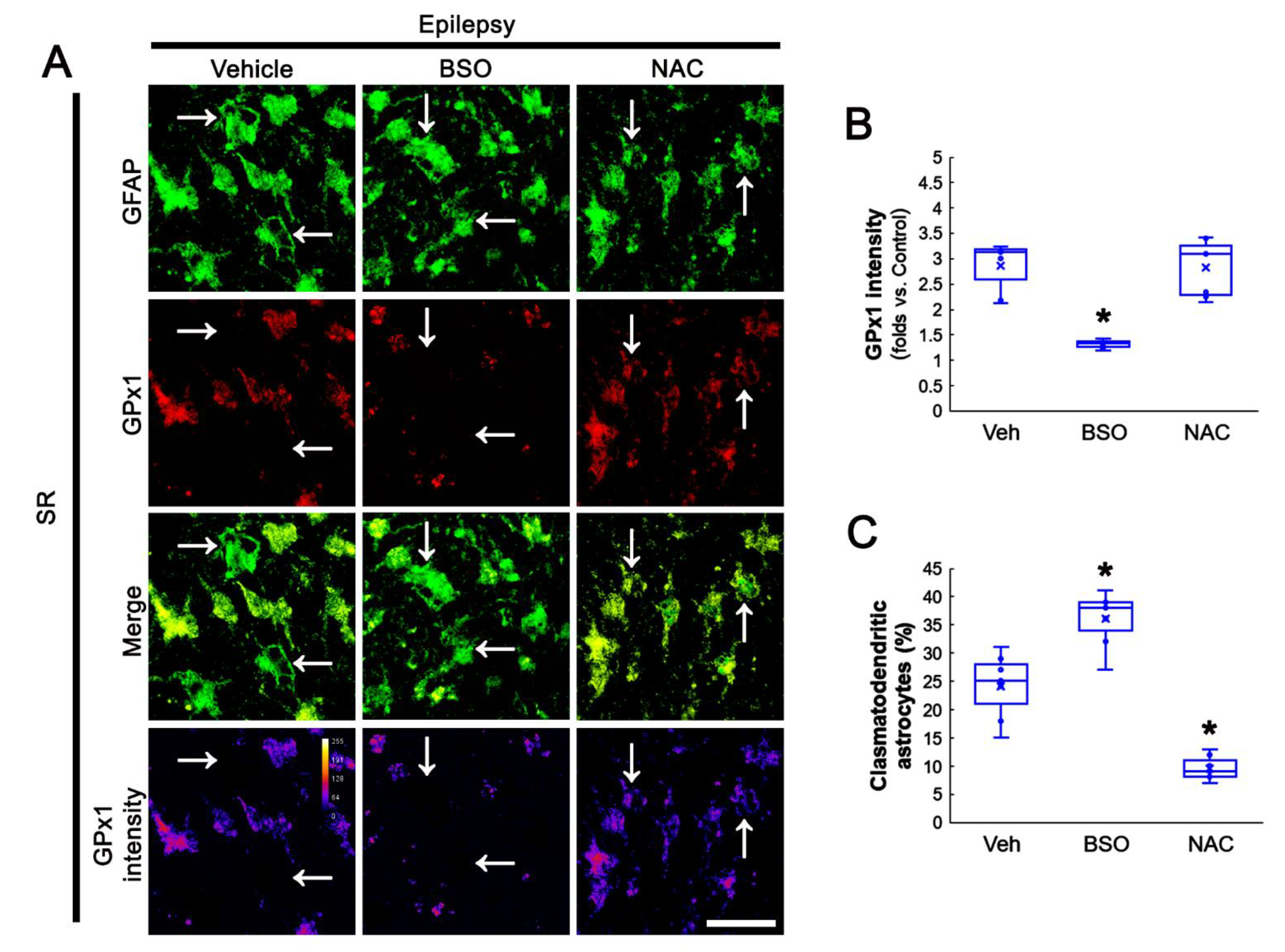
3.6. BSO Exacerbates, but NAC Attenuates Clasmatodendrosis in CA1 Astrocytes of Chronic Epilepsy Rats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 1999, 27, 951–965. [Google Scholar] [CrossRef]
- Marinho, H.S.; Antunes, F.; Pinto, R.E. Role of glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase in the reduction of lysophospholipid hydroperoxides. Free Radic. Biol. Med. 1997, 22, 871–883. [Google Scholar] [CrossRef]
- de Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Liddell, J.R.; Hoepken, H.H.; Crack, P.J.; Robinson, S.R.; Dringen, R. Glutathione peroxidase 1 and glutathione are required to protect mouse astrocytes from iron-mediated hydrogen peroxide toxicity. J. Neurosci. Res. 2006, 84, 578–586. [Google Scholar] [CrossRef]
- Shin, E.J.; Hwang, Y.G.; Pham, D.T.; Lee, J.W.; Lee, Y.J.; Pyo, D. Glutathione peroxidase-1 overexpressing transgenic mice are protected from neurotoxicity induced by microcystin-leucine-arginine. Environ. Toxicol. 2018, 33, 1019–1028. [Google Scholar] [CrossRef]
- Weisbrot-Lefkowitz, M.; Reuhl, K.; Perry, B.; Chan, P.H.; Inouye, M.; Mirochnitchenko, O. Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Brain Res. Mol. Brain Res. 1998, 53, 333–338. [Google Scholar] [CrossRef]
- Banerjee, P.N.; Filippi, D.; Allen Hauser, W. The descriptive epidemiology of epilepsy—A review. Epilepsy Res. 2009, 85, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Dichter, M.A. Emerging insights into mechanisms of epilepsy: Implications for new antiepileptic drug development. Epilepsia 1994, 35, S51–S57. [Google Scholar] [CrossRef]
- Dalby, N.O.; Mody, I. The process of epileptogenesis: A pathophysiological approach. Curr. Opin. Neurol. 2001, 14, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.P.; Garcia-cairasco, N.; Ca, E.A. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002, 50, 93–103. [Google Scholar] [CrossRef]
- Wahab, A.; Albus, K.; Gabriel, S.; Heinemann, U. In search of models of pharmacoresistant epilepsy. Epilepsia 2010, 51, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Trinka, E.; Cock, H.; Hesdorffer. D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus—Report of the ILAE task force on classification of status epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef]
- Schmoll, H.; Badan, I.; Grecksch, G.; Walker, L.; Kessler, C.; Popa-Wagner, A. Kindling status in sprague-dawley rats induced by pentylenetetrazole: Involvement of a critical development period. Am. J. Pathol. 2003, 162, 1027–1034. [Google Scholar] [CrossRef]
- Buga, A.-M.; Vintilescu, R.; Balseanu, A.T.; Pop, O.T.; Streba, C.; Toescu, E.; Popa-Wagner, A. Repeated PTZ treatment at 25-day intervals leads to a highly efficient accumulation of doublecortin in the dorsal hippocampus of rats. PLoS ONE 2012, 7, e39302. [Google Scholar] [CrossRef]
- Cavalheiro, E.A.; Leite, J.P.; Bortolotto, Z.A.; Turski, W.A.; Ikonomidou, C.; Turski, L. Long-term effects of pilocarpine in rats: Structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia 1991, 32, 778–782. [Google Scholar] [CrossRef]
- Goffin, K.; Nissinen, J.; Van Laere, K.; Pitkänen, A. Cyclicity of spontaneous recurrent seizures in pilocarpine model of temporal lobe epilepsy in rat. Exp. Neurol. 2007, 205, 501–505. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R.; Ingvar, M. Laminar damage of neurons and astrocytes in neocortex and hippocampus of rat after long-lasting status epilepticus induced by pilocarpine. Epilepsy Res. Suppl. 1996, 12, 309–316. [Google Scholar]
- Schmidt-Kastner, R.; Ingvar, M. Loss of immunoreactivity for glial fibrillary acidic protein (GFAP) in astrocytes as a marker for profound tissue damage in substantia nigra and basal cortical areas after status epilepticus induced by pilocarpine in rat. Glia 1994, 12, 165–172. [Google Scholar] [CrossRef]
- Kang, T.C.; Kim, D.S.; Kwak, S.E.; Kim, J.E.; Won, M.H.; Kim, D.W.; Choi, S.Y.; Kwon, O.S. Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 2006, 54, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. F-actin depolymerization accelerates clasmatodendrosis via activation of lysosome-derived autophagic astroglial death. Brain Res. Bull. 2011, 85, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Kang, T.C. P2X7 receptor differentially modulates astroglial apoptosis and clasmatodendrosis in the rat brain following status epilepticus. Hippocampus 2011, 21, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- González-Reyes, S.; Santillán-Cigales, J.J.; Jiménez-Osorio, A.S.; Pedraza-Chaverri, J.; Guevara-Guzmán, R. Glycyrrhizin ameliorates oxidative stress and inflammation in hippocampus and olfactory bulb in lithium/pilocarpine-induced status epilepticus in rats. Epilepsy Res. 2016, 126, 126–133. [Google Scholar] [CrossRef]
- Trépanier, G.; Furling, D.; Puymirat, J.; Mirault, M.E. Immunocytochemical localization of seleno-glutathione peroxidase in the adult mouse brain. Neuroscience 1996, 75, 231–243. [Google Scholar] [CrossRef]
- Power, J.H.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009, 117, 63–73. [Google Scholar] [CrossRef]
- Bellissimo, M.I.; Amado, D.; Abdalla, D.S.; Ferreira, E.C.; Cavalheiro, E.A.; Naffah-Mazzacoratti, M.G. Superoxide dismutase, glutathione peroxidase activities and the hydroperoxide concentration are modified in the hippocampus of epileptic rats. Epilepsy Res. 2001, 46, 121–128. [Google Scholar] [CrossRef]
- Eraković, V.; Zupan, G.; Varljen, J.; Laginja, J.; Simonić, A. Lithium plus pilocarpine induced status epilepticus-biochemical changes. Neurosci. Res. 2000, 36, 157–166. [Google Scholar] [CrossRef]
- Boonplueang, R.; Akopian, G.; Stevenson, F.F.; Kuhlenkamp, J.F.; Lu, S.C.; Walsh, J.P.; Andersen, J.K. Increased susceptibility of glutathione peroxidase-1 transgenic mice to kainic acid-related seizure activity and hippocampal neuronal cell death. Exp. Neurol. 2005, 192, 203–214. [Google Scholar] [CrossRef]
- Krifka, S.; Hiller, K.A.; Spagnuolo, G.; Jewett, A.; Schmalz, G.; Schweikl, H. The influence of glutathione on redox regulation by antioxidant proteins and apoptosis in macrophages exposed to 2-hydroxyethyl methacrylate (HEMA). Biomaterials 2012, 33, 5177–5186. [Google Scholar] [CrossRef]
- Gallorini, M.; Petzel, C.; Bolay, C.; Hiller, K.A.; Cataldi, A.; Buchalla, W.; Krifka, S.; Schweikl, H. Activation of the Nrf2-regulated antioxidant cell response inhibits HEMA-induced oxidative stress and supports cell viability. Biomaterials 2015, 56, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Cabrera, R.; Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Escobar, J.; Sastre, J.; Almeida, A.; Bolaños, J.P. γ-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat. Commun. 2012, 3, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Xu, J.; Liang, C.; Liu, J.; Hua, J.; Zhang, Y.; Ni, Q.; Shi, S.; Yu, X. GPx1 is involved in the induction of protective autophagy in pancreatic cancer cells in response to glucose deprivation. Cell Death Dis. 2018, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Hyun, H.W.; Min, S.J.; Kang, T.C. Sustained HSP25 expression induces clasmatodendrosis via ER stress in the rat hippocampus. Front. Cell. Neurosci. 2017, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Lee, D.S.; Park, H.; Kim, T.H.; Kang, T.C. Inhibition of AKT/GSK3β/CREB pathway improves the responsiveness to AMPA receptor antagonists by regulating GRIA1 surface expression in chronic epilepsy rats. Biomedicines 2021, 9, 425. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. CDDO-Me attenuates astroglial autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-Mediated signaling pathways in the hippocampus of chronic epilepsy rats. Antioxidants 2021, 10, 655. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.E. Deletion of P2X7 receptor decreases basal glutathione level by changing glutamate-glutamine cycle and neutral amino acid transporters. Cells 2020, 9, 995. [Google Scholar] [CrossRef]
- Reyes, R.C.; Cittolin-Santos, G.F.; Kim, J.E.; Won, S.J.; Brennan-Minnella, A.M.; Katz, M.; Glass, G.A.; Swanson, R.A. Neuronal glutathione content and antioxidant capacity can be normalized in situ by N-acetyl cysteine concentrations attained in human cerebrospinal fluid. Neurotherapeutics 2016, 13, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Mathern, G.W.; Babb, T.L.; Vickrey, B.G.; Melendez, M.; Pretorius, J.K. The clinical-pathogenic mechanisms of hippocampal neuron loss and surgical outcomes in temporal lobe epilepsy. Brain 1995, 118, 105–118. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kang, T.C. The role of TRPC6 in seizure susceptibility and seizure-related neuronal damage in the rat dentate gyrus. Neuroscience 2015, 307, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Vizuete, A.F.K.; Hansen, F.; Negri, E.; Leite, M.C.; de Oliveira, D.L.; Gonçalves, C.A. Effects of dexamethasone on the Li-pilocarpine model of epilepsy: Protection against hippocampal inflammation and astrogliosis. J. Neuroinflamm. 2018, 15, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishmanova-Doseva, M.; Peychev, L.; Yoanidu, L.; Uzunova, Y.; Atanasova, M.; Georgieva, K.; Tchekalarova, J. Anticonvulsant effects of topiramate and lacosamide on pilocarpine-induced status epilepticus in rats: A role of reactive oxygen species and inflammation. Int. J. Mol. Sci. 2021, 22, 2264. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Ordy, J.M.; Wengenack, T.M.; Bialobok, P.; Coleman, P.D.; Rodier, P.; Baggs, R.B.; Dunlap, W.P.; Kates, B. Selective vulnerability and early progression of hippocampal CA1 pyramidal cell degeneration and GFAP-positive astrocyte reactivity in the rat four-vessel occlusion model of transient global ischemia. Exp. Neurol. 1993, 119, 128–139. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. p47Phox/CDK5/DRP1-mediated mitochondrial fission evokes PV cell degeneration in the rat dentate gyrus following status epilepticus. Front. Cell. Neurosci. 2017, 11, 267. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me selectively attenuates CA1 neuronal death induced by status epilepticus via facilitating mitochondrial fission independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [Green Version]
- Waldbaum, S.; Liang, L.P.; Patel, M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J. Neurochem. 2010, 115, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Makino, N.; Sasaki, K.; Hashida, K.; Sakakura, Y. A metabolic model describing the H2O2 elimination by mammalian cells including H2O2 permeation through cytoplasmic and peroxisomal membranes: Comparison with experimental data. Biochim. Biophys. Acta 2004, 1673, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Ali, U.; Iannello, R.C.; Hertzog, P.; Crack, P.J. Diminished Akt phosphorylation in neurons lacking glutathione peroxidase-1 (Gpx1) leads to increased susceptibility to oxidative stress-induced cell death. J. Neurochem. 2005, 92, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Penfield, W. Neuroglia and microglia—The interstitial tissue of the central nervous system. In Special Cytology, the Form and Function of the Cell in Health and Disease; Cowdry, E.V., Ed.; Hoeber: New York, NY, USA, 1928; pp. 1033–1068. [Google Scholar]
- Duchen, L.W. General pathology of neurons and neuroglia. In Greenfield’s Neuropathology; Adams, J.H., Duchen, L.W., Eds.; Oxford: New York, NY, USA, 1992; pp. 1–68. [Google Scholar]
- Sakai, K.; Fukuda, T.; Iwadate, K. Beading of the astrocytic processes (clasmatodendrosis) following head trauma is associated with protein degradation pathways. Brain Inj. 2013, 27, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Bouchat, J.; Gilloteaux, J.; Suain, V.; Van Vlaender, D.; Brion, J.P.; Nicaise, C. Ultrastructural analysis of thalamus damages in a mouse model of osmotic-induced demyelination. Neurotox. Res. 2019, 36, 144–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Kim, J.E.; Kwak, S.E.; Choi, K.C.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. Spatiotemporal characteristics of astroglial death in the rat hippocampo-entorhinal complex following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2008, 511, 581–598. [Google Scholar] [CrossRef]
- Yüzbaşioğlu, A.; Karataş, H.; Gürsoy-Ozdemir, Y.; Saygi, S.; Akalan, N.; Söylemezoğlu, F.; Dalkara, T.; Kocaefe, Y.C.; Ozgüç, M. Changes in the expression of selenoproteins in mesial temporal lobe epilepsy patients. Cell. Mol. Neurobiol. 2009, 29, 1223–1231. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| GPx1 | Sheep | Biosensis (#S-072-100) | 1:10,000 (WB) 1:2000 (IH) |
| NeuN | Guinea pig | Millipore (#ABN90P) | 1:1000 (IH) |
| LAMP-1 | Rabbit | Abcam (#ab24170) | 1:100 (IH) |
| GFAP | Mouse | Millipore (#MAB3402) | 1:2000 (IH) |
| Iba-1 | Rabbit | Biocare Medical (#CP 290) | 1:500 (IH) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Lee, D.-S.; Kim, T.-H.; Kang, T.-C. Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus. Antioxidants 2022, 11, 756. https://doi.org/10.3390/antiox11040756
Kim J-E, Lee D-S, Kim T-H, Kang T-C. Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus. Antioxidants. 2022; 11(4):756. https://doi.org/10.3390/antiox11040756
Chicago/Turabian StyleKim, Ji-Eun, Duk-Shin Lee, Tae-Hyun Kim, and Tae-Cheon Kang. 2022. "Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus" Antioxidants 11, no. 4: 756. https://doi.org/10.3390/antiox11040756
APA StyleKim, J.-E., Lee, D.-S., Kim, T.-H., & Kang, T.-C. (2022). Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus. Antioxidants, 11(4), 756. https://doi.org/10.3390/antiox11040756