
Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies


Abstract
1. Introduction
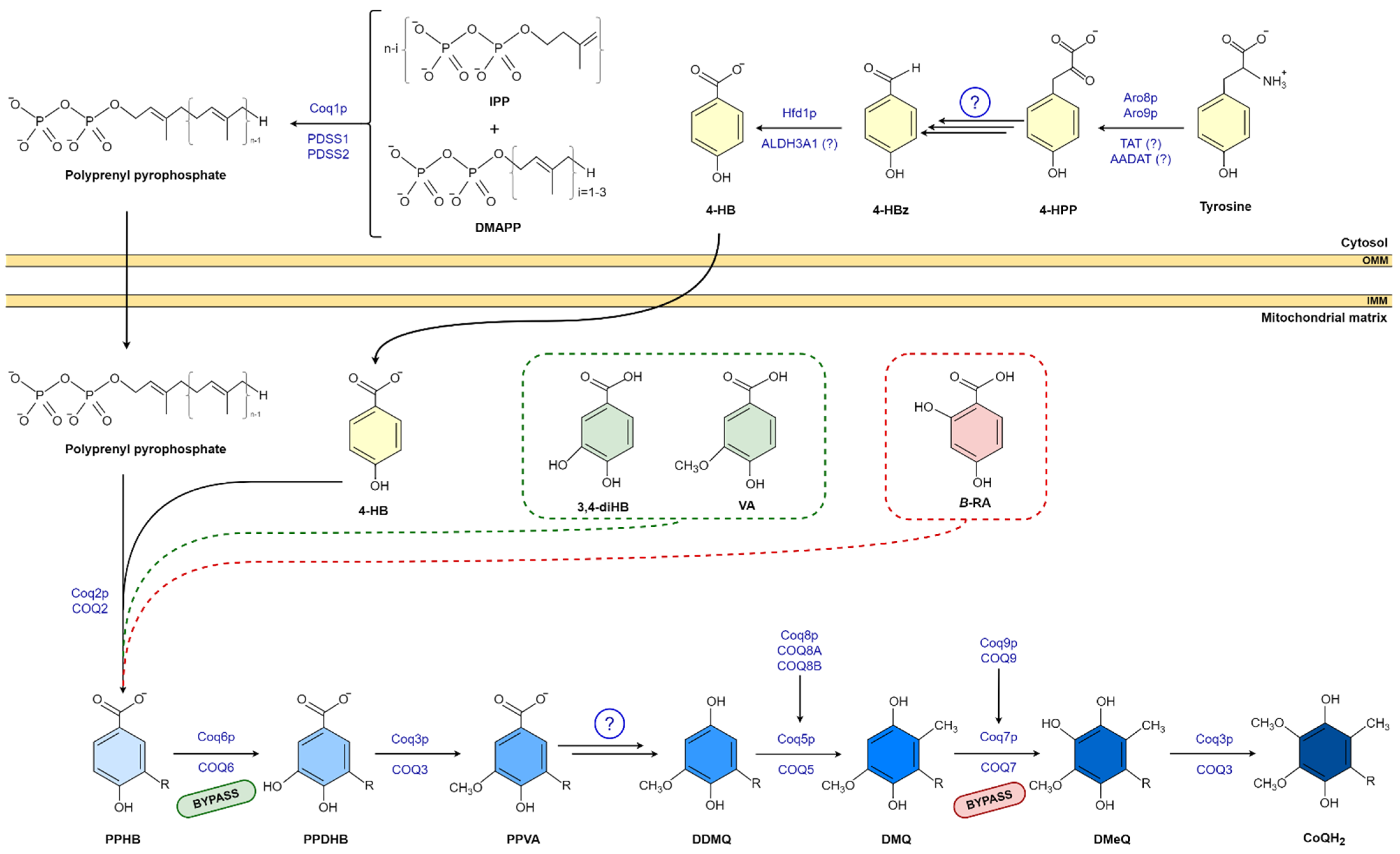
2. CoQ Biosynthesis
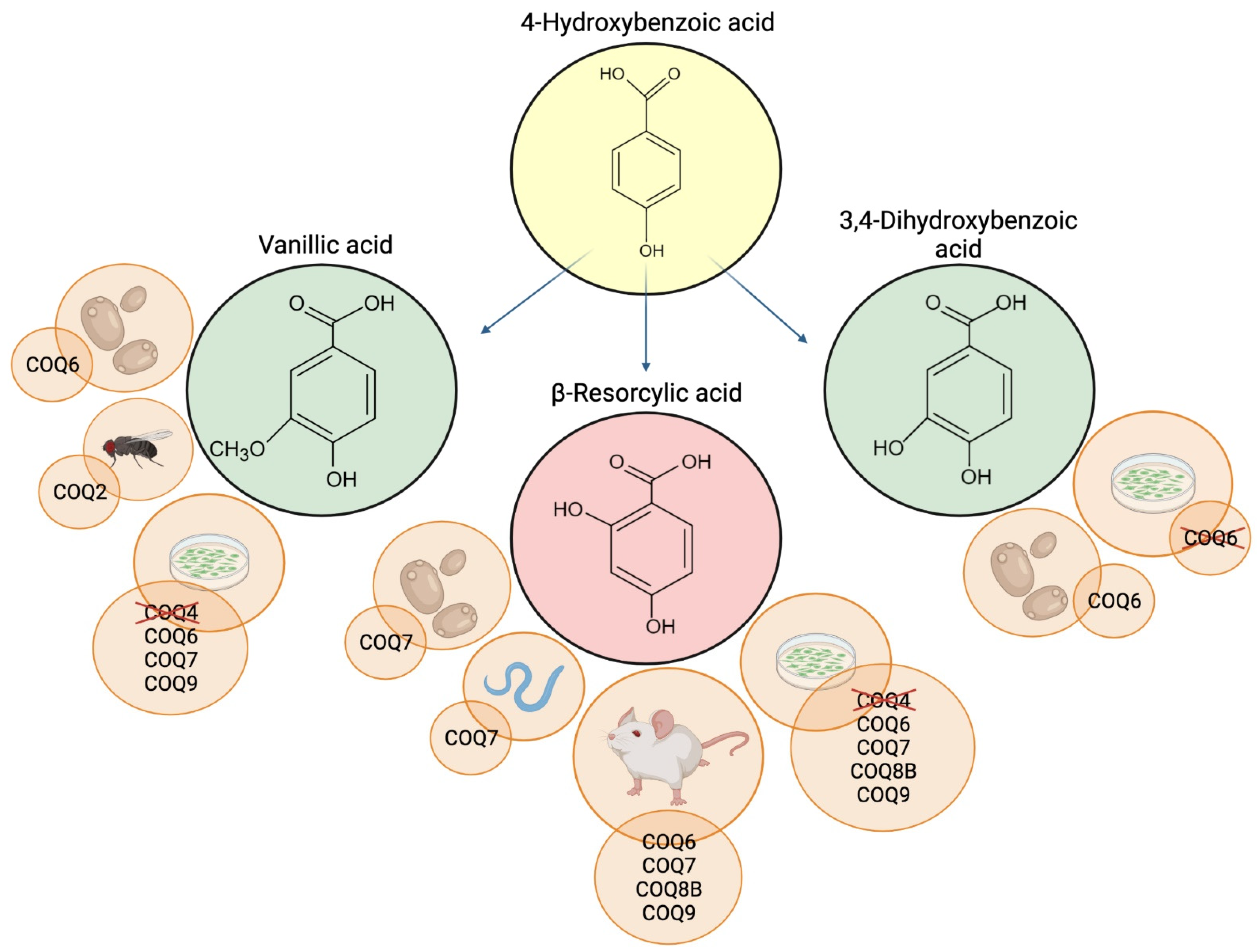
3. 4-Hydroxybenzoic Acid Analogs
3.1. 3,4-Dihydroxybenzoic Acid and Vanillic Acid
3.2. β-Resorcylic Acid
4. Molecular Mechanisms of 4-HB Analogs
5. Conclusions Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta (BBA)—Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef]
- Festenstein, G.N.; Heaton, F.W.; Lowe, J.S.; Morton, R.A. A constituent of the unsaponifiable portion of animal tissue lipids. Biochem. J. 1955, 59, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L.; Hatefi, Y.; Lester, R.L.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta 1957, 25, 220–221. [Google Scholar] [CrossRef]
- González-García, P.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; López-Herrador, S.; Hidalgo-Gutiérrez, A.; López, L.C. Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings. Antioxidants 2021, 10, 1687. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; González-García, P.; Chiozzi, R.Z.; Acuña-Castroviejo, D.; López, L.C. β-RA Targets Mitochondrial Metabolism and Adipogenesis, Leading to Therapeutic Benefits against CoQ Deficiency and Age-Related Overweight. Biomedicines 2021, 9, 1457. [Google Scholar] [CrossRef]
- Heaton, R.A.; Heales, S.; Rahman, K.; Sexton, D.W.; Hargreaves, I. The Effect of Cellular Coenzyme Q10 Deficiency on Lysosomal Acidification. J. Clin. Med. 2020, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef]
- Hirano, M.; Garone, C.; Quinzii, C.M. CoQ10 deficiencies and MNGIE: Two treatable mitochondrial disorders. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2012, 1820, 625–631. [Google Scholar] [CrossRef]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ10 Deficient Endothelial Cell Culture Model for the Investigation of CoQ10 Blood–Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q 10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 893–901. [Google Scholar] [CrossRef]
- Yang, H.Y.; Song, J.F. Inclusion of coenzyme Q10 with beta-cyclodextrin studied by polarography. Yao Xue Xue Bao 2006, 41, 671–674. [Google Scholar]
- Pastor-Maldonado, C.J.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10: Novel Formulations and Medical Trends. Int. J. Mol. Sci. 2020, 21, 8432. [Google Scholar] [CrossRef]
- Awad, A.M.; Bradley, M.C.; Fernández-Del-Río, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [CrossRef]
- Payet, L.-A.; Leroux, M.; Willison, J.C.; Kihara, A.; Pelosi, L.; Pierrel, F. Mechanistic Details of Early Steps in Coenzyme Q Biosynthesis Pathway in Yeast. Cell Chem. Biol. 2016, 23, 1241–1250. [Google Scholar] [CrossRef]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.P.; Medana, C.; Stainier, D.Y.; et al. Ubiad1 Is an Antioxidant Enzyme that Regulates eNOS Activity by CoQ10 Synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef]
- Teclebrhan, H.; Jakobsson-Borin, A.; Brunk, U.; Dallner, G. Relationship between the endoplasmic reticulum-Golgi membrane system and ubiquinone biosynthesis. Biochim. Biophys. Acta (BBA)—Lipids Lipid Metab. 1995, 1256, 157–165. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; del Rio, L.F.; Bradley, M.C.; Dunn, C.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact 2019, 2, 1–14. [Google Scholar] [CrossRef]
- Subramanian, K.; Jochem, A.; Le Vasseur, M.; Lewis, S.; Paulson, B.R.; Reddy, T.R.; Russell, J.D.; Coon, J.J.; Pagliarini, D.J.; Nunnari, J. Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER–mitochondria contacts. J. Cell Biol. 2019, 218, 1353–1369. [Google Scholar] [CrossRef]
- Fernández-Del-Río, L.; Clarke, C. Coenzyme Q Biosynthesis: An Update on the Origins of the Benzenoid Ring and Discovery of New Ring Precursors. Metabolites 2021, 11, 385. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. The Complexity of Making Ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.S.; Kazemzadeh, K.; Vragniau, C.; Pelosi, L.; Pierrel, F. Advances in bacterial pathways for the biosynthesis of ubiquinone. Biochim. Biophys. Acta 2020, 1861, 148259. [Google Scholar] [CrossRef]
- Hayashi, K.; Ogiyama, Y.; Yokomi, K.; Nakagawa, T.; Kaino, T.; Kawamukai, M. Functional Conservation of Coenzyme Q Biosynthetic Genes among Yeasts, Plants, and Humans. PLoS ONE 2014, 9, e99038. [Google Scholar] [CrossRef]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2009, 1791, 69–75. [Google Scholar] [CrossRef]
- Saiki, R.; Ogiyama, Y.; Kainou, T.; Nishi, T.; Matsuda, H.; Kawamukai, M. Pleiotropic phenotypes of fission yeast defective in ubiquinone-10 production. A study from theabc1Sp (coq8Sp)mutant. BioFactors 2003, 18, 229–235. [Google Scholar] [CrossRef]
- He, C.H.; Xie, L.X.; Allan, C.M.; Tran, U.C.; Clarke, C.F. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2014, 1841, 630–644. [Google Scholar] [CrossRef][Green Version]
- Hsieh, E.J.; Gin, P.; Gulmezian, M.; Tran, U.P.C.; Saiki, R.; Marbois, B.N.; Clarke, C.F. Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q biosynthetic complex. Arch. Biochem. Biophys. 2007, 463, 19–26. [Google Scholar] [CrossRef]
- Johnson, A.; Gin, P.; Marbois, B.N.; Hsieh, E.J.; Wu, M.H.; Barros, M.; Clarke, C.F.; Tzagoloff, A. COQ9, a New Gene Required for the Biosynthesis of Coenzyme Q in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 31397–31404. [Google Scholar] [CrossRef]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby, C.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Allan, C.M.; Hill, S.; Morvaridi, S.; Saiki, R.; Johnson, J.S.; Liau, W.-S.; Hirano, K.; Kawashima, T.; Ji, Z.; Loo, J.A.; et al. A conserved START domain coenzyme Q-binding polypeptide is required for efficient Q biosynthesis, respiratory electron transport, and antioxidant function in Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2012, 1831, 776–791. [Google Scholar] [CrossRef]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef]
- Nichols, B.P.; Green, J.M. Cloning and sequencing of Escherichia coli ubiC and purification of chorismate lyase. J. Bacteriol. 1992, 174, 5309–5316. [Google Scholar] [CrossRef][Green Version]
- Siebert, M.; Severin, K.; Heide, L. Formation of 4-hydroxybenzoate in Escherichia coli: Characterization of the ubiC gene and its encoded enzyme chorismate pyruvate-lyase. Microbiology 1994, 140, 897–904. [Google Scholar] [CrossRef]
- Lawrence, J.; Cox, G.B.; Gibson, F. Biosynthesis of Ubiquinone in Escherichia coli K-12: Biochemical and Genetic Characterization of a Mutant Unable to Convert Chorismate into 4-Hydroxybenzoate. J. Bacteriol. 1974, 118, 41–45. [Google Scholar] [CrossRef]
- Olson, R.E.; Dialamieh, G.H.; Bentley, R.; Springer, C.M.; Ramsey, V.G. Pattern Of Labeling In Coenzyme Q9 After Administration of Isotopic Acetate And Aromatic Amino Acids To Rats. J. Biol. Chem. 1965, 240, 514–523. [Google Scholar] [CrossRef]
- Olson, R.E. Biosynthesis of Ubiquinones in Animals. Klotho 1967, 24, 551–574. [Google Scholar] [CrossRef]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, J.-Y.; Wang, J.; Poplawsky, A.; Lin, S.; Zhu, B.; Chang, C.; Zhou, T.; Zhang, L.-H.; He, Y.-W. The diffusible factor synthase XanB2 is a bifunctional chorismatase that links the shikimate pathway to ubiquinone and xanthomonadins biosynthetic pathways. Mol. Microbiol. 2012, 87, 80–93. [Google Scholar] [CrossRef]
- Block, A.; Widhalm, J.R.; Fatihi, A.; Cahoon, R.E.; Wamboldt, Y.; Elowsky, C.; Mackenzie, S.A.; Cahoon, E.B.; Chapple, C.; Dudareva, N.; et al. The Origin and Biosynthesis of the Benzenoid Moiety of Ubiquinone (Coenzyme Q) in Arabidopsis. Plant Cell 2014, 26, 1938–1948. [Google Scholar] [CrossRef]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Bünning, G.; Freyer, C.; Prokisch, H.; Karall, D.; Wredenberg, A.; Wedell, A.; López, L.C.; et al. Detection of 6-demethoxyubiquinone in CoQ 10 deficiency disorders: Insights into enzyme interactions and identification of potential therapeutics. Mol. Genet. Metab. 2017, 121, 216–223. [Google Scholar] [CrossRef]
- Nambudiri, A.M.D.; Brockman, D.; Alam, S.S.; Rudney, H. Alternate routes for ubiquinone biosynthesis in rats. Biochem. Biophys. Res. Commun. 1977, 76, 282–288. [Google Scholar] [CrossRef]
- Goewert, R.R.; Sippel, C.; Grimm, M.F.; Olson, R.E. Identification of 3-methoxy-4-hydroxy-5-hexaprenylbenzoic acid as a new intermediate in ubiquinone biosynthesis by Saccharomyces cerevisiae. FEBS Lett. 1978, 87, 219–221. [Google Scholar] [CrossRef]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q Biosynthesis: Coq6 Is Required for the C5-Hydroxylation Reaction and Substrate Analogs Rescue Coq6 Deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef]
- Xie, L.X.; Ozeir, M.; Tang, J.Y.; Chen, J.Y.; Jaquinod, S.-K.; Fontecave, M.; Clarke, C.F.; Pierrel, F. Overexpression of the Coq8 Kinase in Saccharomyces cerevisiae coq Null Mutants Allows for Accumulation of Diagnostic Intermediates of the Coenzyme Q6 Biosynthetic Pathway. J. Biol. Chem. 2012, 287, 23571–23581. [Google Scholar] [CrossRef]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2013, 1842, 1–6. [Google Scholar] [CrossRef]
- Hermle, T.; Braun, D.A.; Helmstädter, M.; Huber, T.B.; Hildebrandt, F. Modeling Monogenic Human Nephrotic Syndrome in the Drosophila Garland Cell Nephrocyte. J. Am. Soc. Nephrol. 2016, 28, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.J.A.; Trevisson, E.; Canton, M.; Vazquez-Fonseca, L.; Morbidoni, V.; Baschiera, E.; Frasson, C.; Pelosi, L.; Rascalou, B.; Desbats, M.A.; et al. Vanillic Acid Restores Coenzyme Q Biosynthesis and ATP Production in Human Cells LackingCOQ6. Oxidative Med. Cell. Longev. 2019, 2019, 3904905. [Google Scholar] [CrossRef]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Muskiet, F.A.; Groen, A. Urinary excretion of conjugated homovanillic acid, 3,4-dihydroxyphenylacetic acid, p-hydroxyphenylacetic acid, and vanillic acid by persons on their usual diet and patients with neuroblastoma. Clin. Chem. 1979, 25, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Panoutsopoulos, G.; Beedham, C. Enzymatic Oxidation of Vanillin, Isovanillin and Protocatechuic Aldehyde with Freshly Prepared Guinea Pig Liver Slices. Cell. Physiol. Biochem. 2005, 15, 089–098. [Google Scholar] [CrossRef] [PubMed]
- Gitzinger, M.; Kemmer, C.; Fluri, D.A.; El-Baba, M.D.; Weber, W.; Fussenegger, M. The food additive vanillic acid controls transgene expression in mammalian cells and mice. Nucleic Acids Res. 2011, 40, e37. [Google Scholar] [CrossRef]
- Marbois, B.N.; Clarke, C.F. The COQ7 Gene Encodes a Protein in Saccharomyces cerevisiae Necessary for Ubiquinone Biosynthesis. J. Biol. Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef]
- Padilla, S.; Tran, U.C.; Jiménez-Hidalgo, M.; López-Martín, J.M.; Martín-Montalvo, A.; Clarke, C.F.; Navas, P.; Santos-Ocaña, C. Hydroxylation of demethoxy-Q6 constitutes a control point in yeast coenzyme Q6 biosynthesis. Cell. Mol. Life Sci. 2008, 66, 173–186. [Google Scholar] [CrossRef]
- Falk, M.J.; Kayser, E.-B.; Morgan, P.G.; Sedensky, M.M. Mitochondrial Complex I Function Modulates Volatile Anesthetic Sensitivity in C. elegans. Curr. Biol. 2006, 16, 1641–1645. [Google Scholar] [CrossRef]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novelCOQ7defect using 2,4–dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef]
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Gutiérrez, A.H.; Barriocanal-Casado, E.; Bakkali, M.; Casado, M.E.D.; Sánchez-Maldonado, L.; Romero, M.; Sayed, R.; Prehn, C.; Escames, G.; Duarte, J.; et al. β-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 R239X mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef]
- Liu, J.-L.; Yee, C.; Wang, Y.; Hekimi, S. A single biochemical activity underlies the pleiotropy of the aging-related protein CLK-1. Sci. Rep. 2017, 7, 859. [Google Scholar] [CrossRef]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sánchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q bio-synthesis. Proc. Natl. Acad. Sci. USA 2014, 111, 4697–4705. [Google Scholar] [CrossRef]
- Clarke, N.E.; Mosher, R.E.; Clarke, C.N. Phenolic Compounds in the Treatment of Rheumatic Fever. Circulation 1953, 7, 247–257. [Google Scholar] [CrossRef]
- Sahil, K.; Souravh, B. A Review on Protocatechuic Acid and Its Pharmacological Potential. ISRN Pharmacol. 2014, 2014, 952943. [Google Scholar] [CrossRef]
- Guan, S.; Bao, Y.-M.; Jiang, B.; An, L.-J. Protective effect of protocatechuic acid from Alpinia oxyphylla on hydrogen peroxide-induced oxidative PC12 cell death. Eur. J. Pharmacol. 2006, 538, 73–79. [Google Scholar] [CrossRef]
- Zhang, Q.; de Mejia, E.G.; Luna-Vital, D.A.; Tao, T.; Chandrasekaran, S.; Chatham, L.; Juvik, J.; Singh, V.; Kumar, D. Relationship of phenolic composition of selected purple maize (Zea mays L.) genotypes with their anti-inflammatory, anti-adipogenic and anti-diabetic potential. Food Chem. 2019, 289, 739–750. [Google Scholar] [CrossRef]
- Singh, B.; Kumar, A.; Singh, H.; Kaur, S.; Arora, S.; Singh, B. Protective effect of vanillic acid against diabetes and diabetic nephropathy by attenuating oxidative stress and upregulation of NF-κB, TN -α and COX-2 proteins in rats. Phytother. Res. 2022, 36, 1338–1352. [Google Scholar] [CrossRef]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef]
- Xie, L.X.; Williams, K.J.; He, C.H.; Weng, E.; Khong, S.; Rose, T.E.; Kwon, O.; Bensinger, S.J.; Marbois, B.N.; Clarke, C.F. Resveratrol and para-coumarate serve as ring precursors for coenzyme Q biosynthesis. J. Lipid Res. 2015, 56, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Diaz, B.G.; Barca, E.; Balreira, A.; Lopez, L.C.; Tadesse, S.; Krishna, S.; Naini, A.; Mariotti, C.; Castellotti, B.; Quinzii, C.M. Lack of aprataxin impairs mitochondrial functions via downregulation of the APE1/NRF1/NRF2 pathway. Hum. Mol. Genet. 2015, 24, 4516–4529. [Google Scholar] [CrossRef] [PubMed]
- Compagnoni, G.M.; Kleiner, G.; Bordoni, A.; Fortunato, F.; Ronchi, D.; Salani, S.; Guida, M.; Corti, C.; Pichler, I.; Bergamini, C.; et al. Mitochondrial dysfunction in fibroblasts of Multiple System Atrophy. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Barca, E.; Kleiner, G.; Tang, G.; Ziosi, M.; Tadesse, S.; Masliah, E.; Louis, E.D.; Faust, P.; Kang, U.J.; Torres, J.; et al. Decreased Coenzyme Q10 Levels in Multiple System Atrophy Cerebellum. J. Neuropathol. Exp. Neurol. 2016, 75, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Martín, M.Á.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Aguilera, J.C.R.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q 10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Analog | Model | Molecular Defect | CoQ | DMQ | Mitochondrial Respiration | Oxidative Stress | Growth/Morphology/LIFESPAN | Other | References |
|---|---|---|---|---|---|---|---|---|---|
| 3,4-diHB | S. cerevisiae | Coq6 | + | + | [46,48] | ||||
| Human cells | COQ6 | - | [51] | ||||||
| VA | D. melanogaster | Coq2 | + | + | [49] | ||||
| Human cells | COQ4 | - | [43] | ||||||
| S. cerevisiae | Coq6 | + | + | [46,48] | |||||
| Human cells | COQ6 | + | + | + | [50] | ||||
| Human cells | COQ6 | + | [51] | ||||||
| Human cells | COQ9 | + | + | [43] | |||||
| β-RA | Human cells | COQ4 | - | [43] | |||||
| Human cells | COQ6 | + | [51] | ||||||
| Mouse | Coq6 | + | improved kidney function | [51] | |||||
| S. cerevisiae | Coq7 | + | + | [47] | |||||
| C. elegans | Coq7 | + | + | + | [63] | ||||
| Human cells | COQ7 | + | + | increased COQ proteins levels | [43] | ||||
| Human cells | COQ7 | + | + | [60] | |||||
| Human cells | COQ7 | + | + | + | [21,57] | ||||
| Mouse | Coq7 | + | + | + | + | decreased lactate and TG levels | [21] | ||
| Human cells | COQ8B | + | + | [52] | |||||
| Mouse | Coq8B | + | + | improved kidney function | [52] | ||||
| Human cells | COQ9 | + | + | increased COQ proteine levels | [43] | ||||
| Human cells | COQ9 | + | [61] | ||||||
| Mouse | Coq9 | + | + | + | + | [61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesini, A.; Hidalgo-Gutierrez, A.; Quinzii, C.M. Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies. Antioxidants 2022, 11, 665. https://doi.org/10.3390/antiox11040665
Pesini A, Hidalgo-Gutierrez A, Quinzii CM. Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies. Antioxidants. 2022; 11(4):665. https://doi.org/10.3390/antiox11040665
Chicago/Turabian StylePesini, Alba, Agustin Hidalgo-Gutierrez, and Catarina M. Quinzii. 2022. "Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies" Antioxidants 11, no. 4: 665. https://doi.org/10.3390/antiox11040665
APA StylePesini, A., Hidalgo-Gutierrez, A., & Quinzii, C. M. (2022). Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies. Antioxidants, 11(4), 665. https://doi.org/10.3390/antiox11040665