

Identification of Heme Oxygenase-1 as a Putative DNA-Binding Protein

 , , ,
, , , 
Abstract
1. Introduction
2. Material and Methods
2.1. MLE-12 Cell Culture
2.2. CRISPR/Cas9 Deficient of HO-1 in MLE-12 Cells
2.3. Transcriptional Analysis
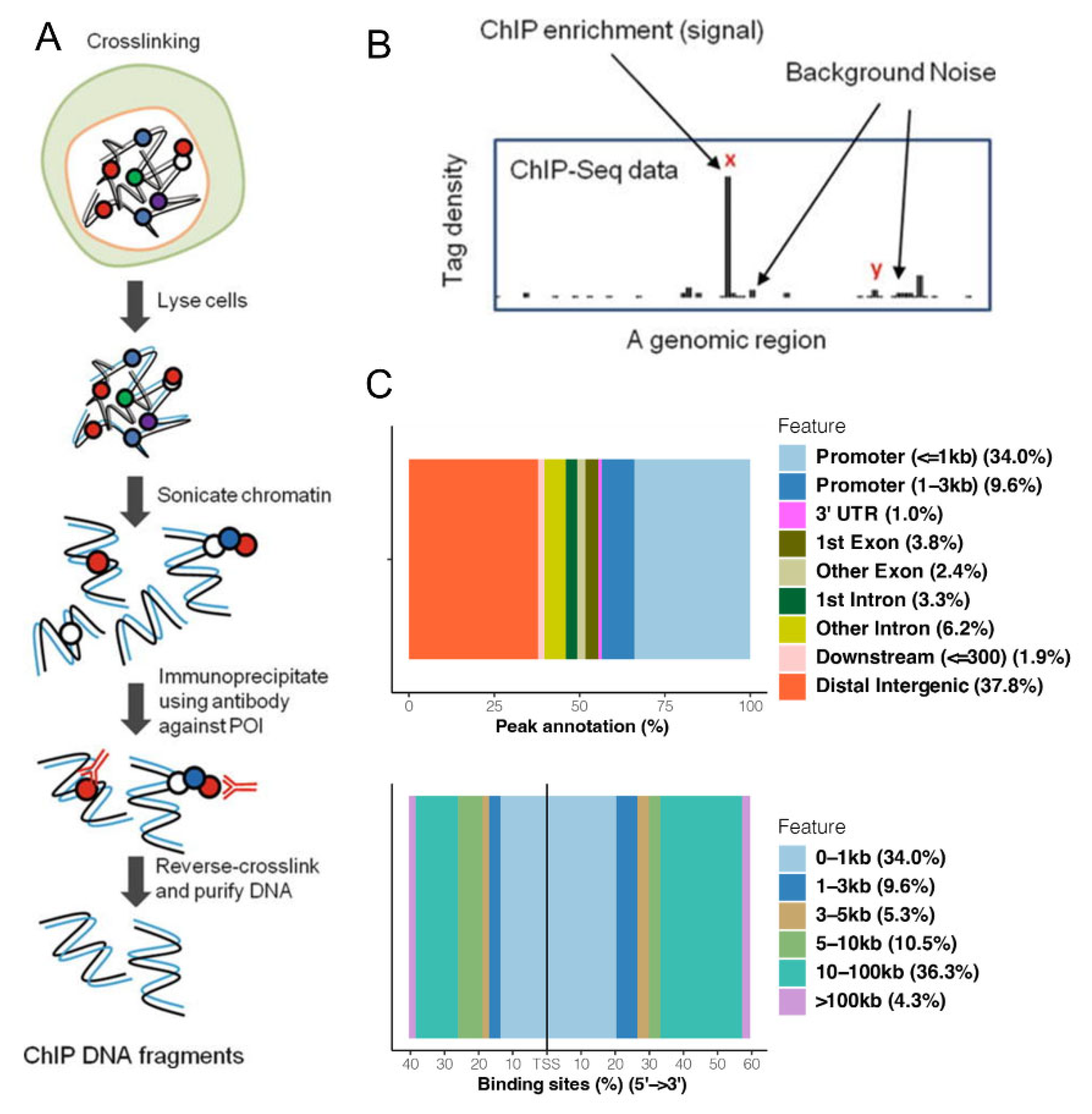
2.4. ChIP-Seq Assay
2.5. Computational Analysis of ChIP-Seq and Motif Binding
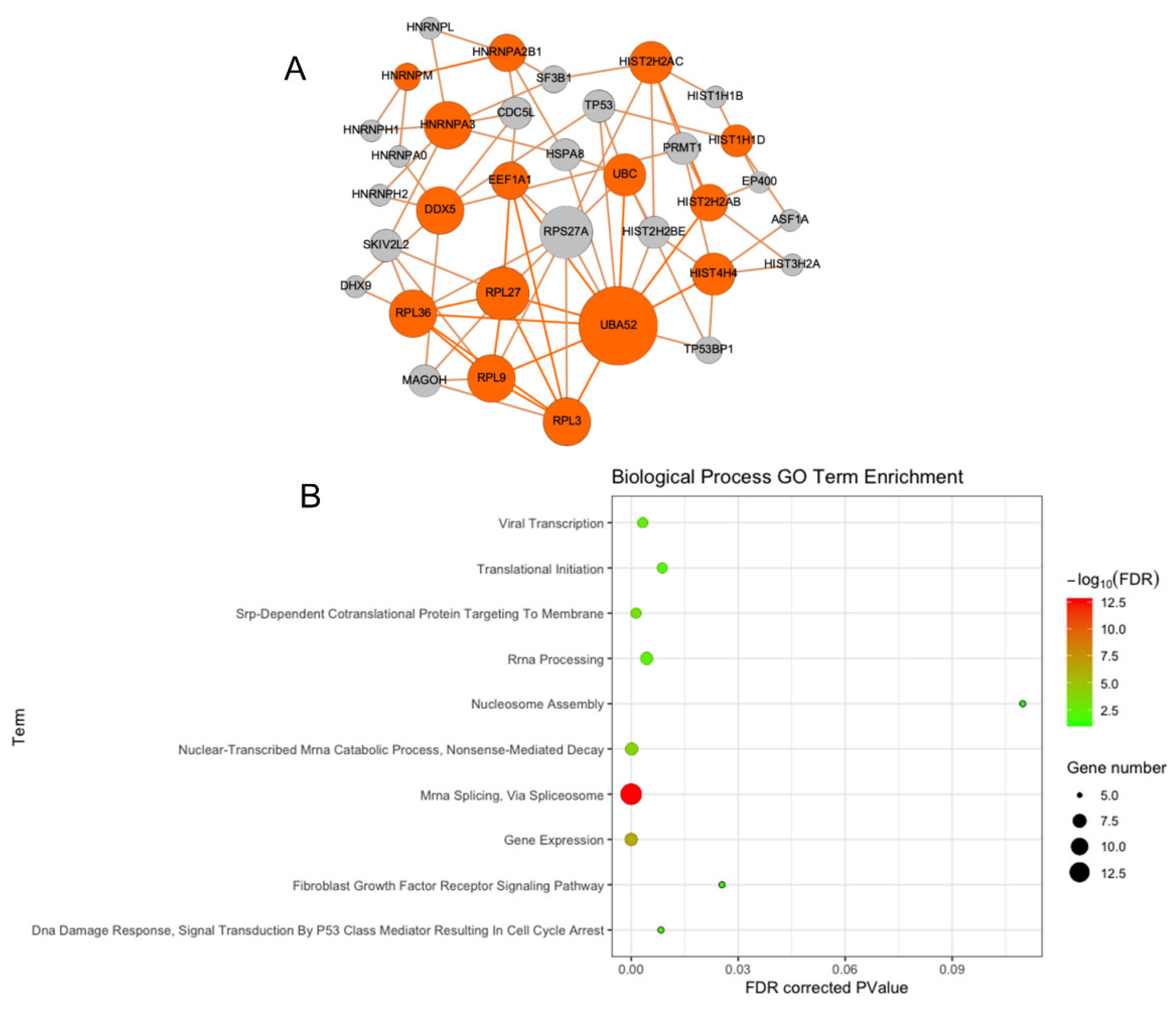
2.6. Performing Protein–Protein Interaction (PPI) Networks (Proteinarium)
2.7. Functional Enrichment
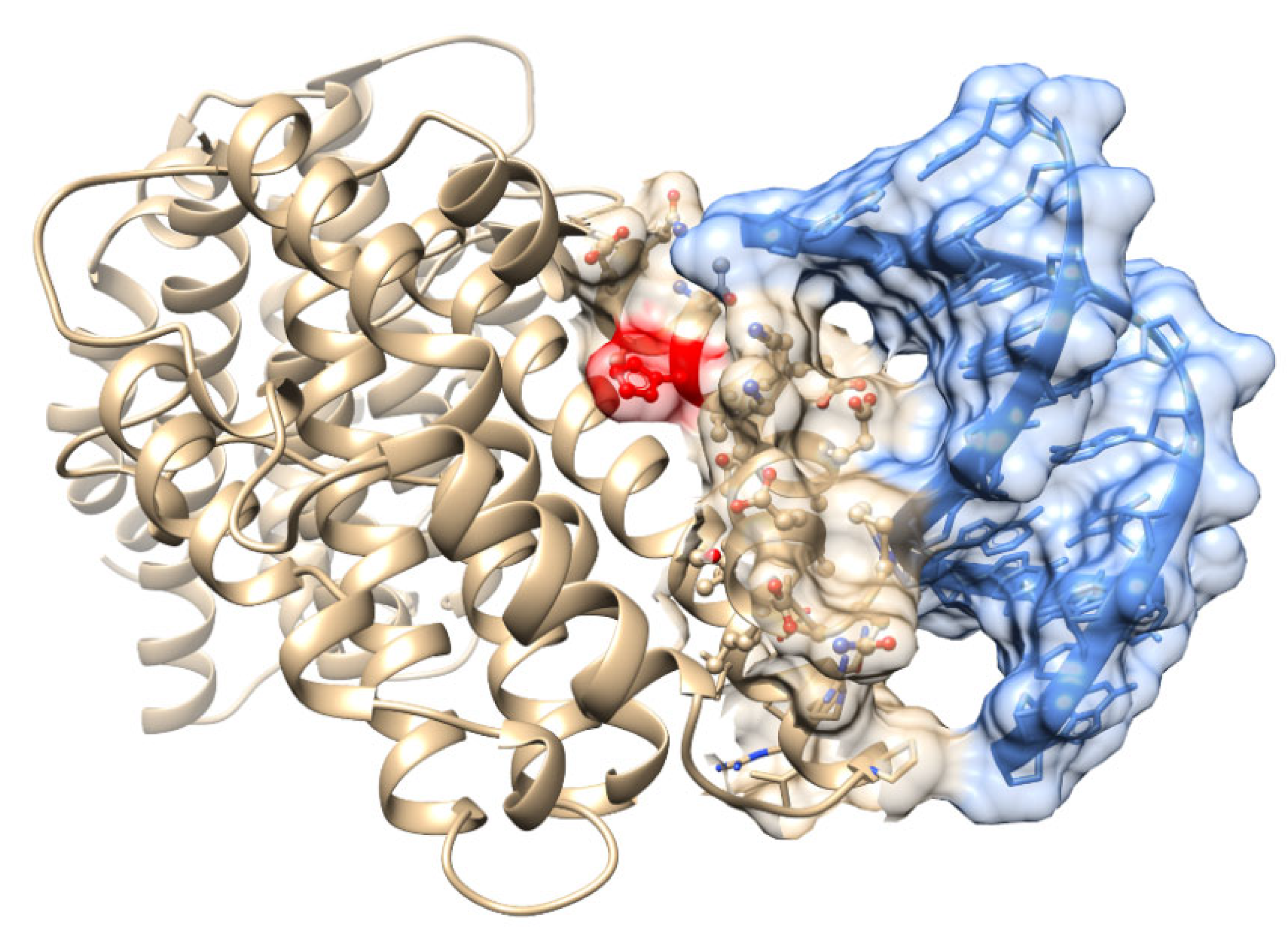
2.8. Predictive Modeling of Apo HO-1 dsDNA Binding Complex Structures
2.9. ModelX- dsDNA-Protein Docking
2.10. Identification of Compatible dsDNA Binding Targets of Apo HO-1
2.11. Visualization of HO-1 Protein and dsDNA Binding Fragment PDB Data
2.12. Transfection of Mutated HO-1 Plasmids into Hmox1 Deficient Cells
2.13. Statistical Analysis
3. Results
3.1. CHIP-Seq Reveals That HO-1 Binds to DNA
3.2. PPI Network Analysis Reveals Multiple Interactions
3.3. Functional Enrichment of Protein–Protein Interaction Network Reveals Many Biological Processes Are Impacted by HO-1
3.4. Predictive Modeling of Apo HO-1 dsDNA Binding Complex Structures Reveals Several HO-1 DNA Binding Domains
3.5. Identification of Compatible dsDNA Binding Targets of Apo HO-1 among CHIP-Seq Genes
3.5.1. HO-1 DNA-Binding Domain 1 Targets among CHIP-Seq Genes
3.5.2. HO-1 DNA-Binding Domain 2 Targets among CHIP-Seq Genes
3.5.3. HO-1 DNA-Binding Domain 3 Targets among CHIP-Seq gENE
3.6. Identification of Compatible dsDNA Binding Targets of Apo HO-1 among Motif BLAST Hit Genes
3.6.1. HO-1 DNA-Binding Domain 1 Targets among Motif BLAST Hit Genes
3.6.2. HO-1 DNA-Binding Domain 2 Targets among Motif BLAST Hit Genes
3.6.3. HO-1 DNA-Binding Domain 3 Targets among Motif BLAST Hit Genes
3.7. Functional Enrichment of HO-1 Binding Gene Targets Reveals a Role in Regulation of Transcription Factor Regulation, Cell Proliferation, and Tissue Development
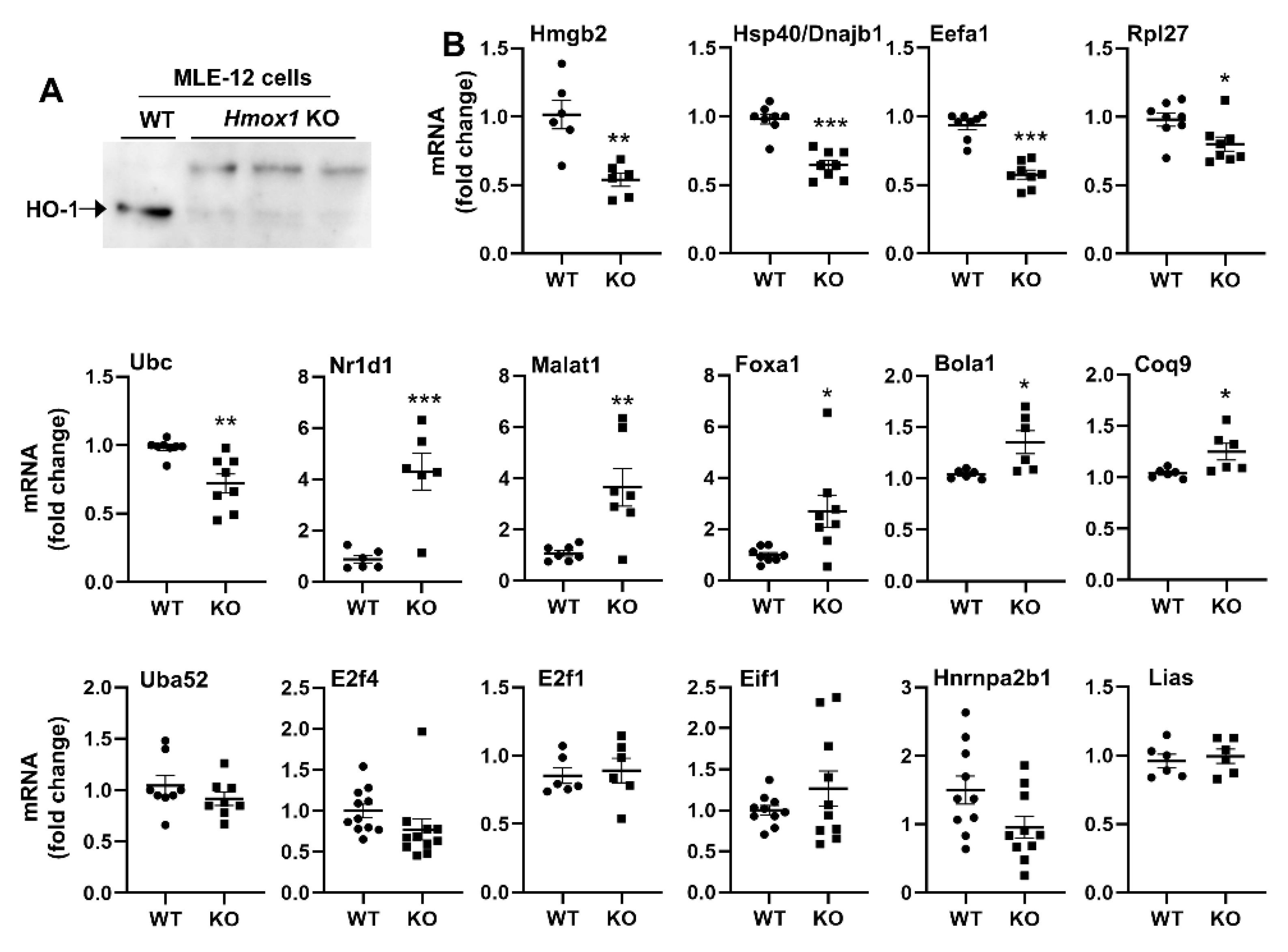
3.8. Hmox1 Deficient MLE-12 Cells Show Dysregulation of Multiple Target Genes
3.9. Mutation of Amino Acids in HO-1 DBD1 Alters Target Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carr, J.; Garcia, D.; Scaffa, A.; Peterson, A.; Ghio, A.; Dennery, P. Heme Oxygenase-1 Supports Mitochondrial Energy Production and Electron Transport Chain Activity in Cultured Lung Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 6941. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Signaling Function of Heme Oxygenase Proteins. Antioxid. Redox Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Öllinger, R.; Pratschke, J. Role of heme oxygenase-1 in transplantation. Transpl. Int. 2010, 23, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.-H.; Kim, H.P.; Kim, B.-R.; Cha, S.H.; Kim, G.A.; Ha, H.; Na, Y.E.; Cha, Y.-N. Evidence for Heme Oxygenase-1 Association with Caveolin-1 and -2 in Mouse Mesangial Cells. IUBMB Life 2003, 55, 525–532. [Google Scholar] [CrossRef]
- Jin, Y.; Kim, H.P.; Chi, M.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.K. Deletion of Caveolin-1 Protects against Oxidative Lung Injury via Up-Regulation of Heme Oxygenase-1. Am. J. Respir. Cell Mol. Biol. 2008, 39, 171–179. [Google Scholar] [CrossRef]
- Dennery, P.A. Heme Oxygenase in Neonatal Lung Injury and Repair. Antioxid. Redox Signal. 2014, 21, 1881–1892. [Google Scholar] [CrossRef]
- Paez, A.V.; Pallavicini, C.; Schuster, F.; Valacco, M.P.; Giudice, J.; Ortiz, E.G.; Anselmino, N.; Labanca, E.; Binaghi, M.; Salierno, M.; et al. Heme oxygenase-1 in the forefront of a multi-molecular network that governs cell–cell contacts and filopodia-induced zippering in prostate cancer. Cell Death Dis. 2016, 7, e2570. [Google Scholar] [CrossRef]
- Dunn, L.L.; Midwinter, R.G.; Ni, J.; Hamid, H.A.; Parish, C.R.; Stocker, R. New Insights into Intracellular Locations and Functions of Heme Oxygenase-1. Antioxid. Redox Signal. 2014, 20, 1723–1742. [Google Scholar] [CrossRef]
- Converso, D.P.; Taillé, C.; Carreras, M.C.; Jaitovich, A.; Poderoso, J.J.; Boczkowski, J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006, 20, 1236–1238. [Google Scholar] [CrossRef]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalà, A.; Volti, G.L. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.-H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme Oxygenase-1 Protein Localizes to the Nucleus and Activates Transcription Factors Important in Oxidative Stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed]
- Boname, J.M.; Bloor, S.; Wandel, M.P.; Nathan, J.; Antrobus, R.; Dingwell, K.S.; Thurston, T.L.; Smith, D.L.; Smith, J.C.; Randow, F.; et al. Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J. Cell Biol. 2020, 205, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.-F.; Chiang, M.-T.; Li, F.-A.; Yeh, C.-T.; Lee, W.-H.; Chau, L.-Y. Acetylation is essential for nuclear heme oxygenase-1-enhanced tumor growth and invasiveness. Oncogene 2017, 36, 6805–6814. [Google Scholar] [CrossRef] [PubMed]
- Reményi, A.; Schöler, H.R.; Wilmanns, M. Combinatorial control of gene expression. Nat. Struct. Mol. Biol. 2004, 11, 812–815. [Google Scholar] [CrossRef]
- Lad, L.; Friedman, J.; Li, H.; Bhaskar, B.; de Montellano, A.P.R.O.; Poulos, T.L. Crystal Structure of Human Heme Oxygenase-1 in a Complex with Biliverdin. Biochemistry 2004, 43, 3793–3801. [Google Scholar] [CrossRef]
- Schuller, D.J.; Wilks, A.; De Montellano, P.R.O.; Poulos, T.L. Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 1999, 6, 860–867. [Google Scholar] [CrossRef]
- Armanious, D.; Schuster, J.; Tollefson, G.A.; Agudelo, A.; DeWan, A.T.; Istrail, S.; Padbury, J.; Uzun, A. Proteinarium: Multi-sample protein-protein interaction analysis and visualization tool. Genomics 2020, 112, 4288–4296. [Google Scholar] [CrossRef]
- Blanco, J.D.; Radusky, L.; Climente-Gonzalez, H.; Serrano, L. FoldX accurate structural protein-DNA binding prediction using PADA1 (Protein Assisted DNA Assembly 1). Nucleic Acids Res. 2018, 46, 3852–3863. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33 (Suppl. 2), W382–W388. [Google Scholar] [CrossRef]
- Day, D.S.; Zhang, B.; Stevens, S.M.; Ferrari, F.; Larschan, E.N.; Park, P.J.; Pu, W.T. Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biol. 2016, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Zhu, L.J. Integrative Analysis of ChIP-Chip and ChIP-Seq Dataset. Methods Mol. Biol. 2013, 1067, 105–124. [Google Scholar] [CrossRef]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Negrutskii, B.; Vlasenko, D.; El’Skaya, A. From global phosphoproteomics to individual proteins: The case of translation elongation factor eEF1A. Expert Rev. Proteom. 2012, 9, 71–83. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lin, Q.S.; Weis, S.; Yang, G.; Zhuang, T.; Abate, A.; Dennery, P.A. Catalytic inactive heme oxygenase-1 protein regulates its own expression in oxidative stress. Free Radic. Biol. Med. 2008, 44, 847–855. [Google Scholar] [CrossRef]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear Heme Oxygenase-1 (HO-1) Modulates Subcellular Distribution and Activation of Nrf2, Impacting Metabolic and Anti-oxidant Defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Satterlee, J.D.; Ma, L.H.; Dallas, J.L.; Smith, K.M.; Zhang, X.; Sato, M.; la Mar, G.N. Influence of substrate modification and C-terminal truncation on the active site structure of substrate-bound heme oxygenase from Neisseriae meningitidis. A 1H NMR study. Biochemistry 2011, 50, 8823–8833. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lad, L.; Poulos, T.L.; de Montellano, P.R.O. Regiospecificity determinants of human heme oxygenase: Differential NADPH- and ascorbate-dependent heme cleavage by the R183E mutant. J. Biol. Chem. 2005, 280, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Bolotin, E.; Jiang, T.; Sladek, F.M.; Martinez, E. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 2007, 389, 52–65. [Google Scholar] [CrossRef]
- Martinez-Guzman, O.; Willoughby, M.M.; Saini, A.; Dietz, J.V.; Bohovych, I.; Medlock, A.E.; Khalimonchuk, O.; Reddi, A.R. Mitochondrial-nuclear heme trafficking in budding yeast is regulated by GTPases that control mitochondrial dynamics and ER contact sites. J. Cell Sci. 2020, 133, jcs237917. [Google Scholar] [CrossRef]
- Xia, Z.-W.; Zhou, W.-P.; Zhong, W.-W.; Zhang, X.-H.; Ding, J.-P.; Zhang, Z.-L. Comparison of the crystal structure and function to wild-type and His25Ala mutant human heme oxygenase-1. Int. J. Mol. Med. 2009, 23, 379–387. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef]
- Salinas, M.; Wang, J.; De Sagarra, M.R.; Martín, D.; Rojo, A.I.; Martin-Perez, J.; De Montellano, P.R.O.; Cuadrado, A. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004, 578, 90–94. [Google Scholar] [CrossRef]
- Salinas, M.; Diaz, R.; Abraham, N.G.; de Galarreta, C.M.R.; Cuadrado, A. Nerve Growth Factor Protects against 6-Hydroxydopamine-induced Oxidative Stress by Increasing Expression of Heme Oxygenase-1 in a Phosphatidylinositol 3-Kinase-dependent Manner. J. Biol. Chem. 2003, 278, 13898–13904. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vlahakis, J.Z.; Vukomanovic, D.; Lee, W.; Szarek, W.A.; Nakatsu, K.; Jia, Z. A novel, “double-clamp” binding mode for human heme oxygenase-1 inhibition. PLoS ONE 2012, 7, e29514. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Reizel, Y.; Morgan, A.; Gao, L.; Schug, J.; Mukherjee, S.; Garcia, M.F.; Donahue, G.; Baur, J.A.; Zaret, K.S.; Kaestner, K.H. FoxA-dependent demethylation of DNA initiates epigenetic memory of cellular identity. Dev. Cell 2021, 56, 602–612.e4. [Google Scholar] [CrossRef] [PubMed]
- Paranjapye, A.; Mutolo, M.J.; Ebron, J.S.; Leir, S.-H.; Harris, A. The FOXA1 transcriptional network coordinates key functions of primary human airway epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 319, L126–L136. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Chen, J.; Huang, J.; Lu, J.; Pei, S.; Ding, S.; Kang, L.; Xiao, R.; Zeng, Q. Functions and regulatory mechanisms of metastasis-associated lung adenocarcinoma transcript 1. J. Cell. Physiol. 2018, 234, 134–151. [Google Scholar] [CrossRef]
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W.; et al. The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 582–587. [Google Scholar] [CrossRef]
- Wible, R.S.; Ramanathan, C.; Sutter, C.H.; Olesen, K.M.; Kensler, T.W.; Liu, A.C.; Sutter, T.R. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. eLife 2018, 7, e31656. [Google Scholar] [CrossRef]
- Jeż, M.; Martyniak, A.; Andrysiak, K.; Mucha, O.; Szade, K.; Kania, A.; Chrobok, L.; Palus-Chramiec, K.; Sanetra, A.M.; Lewandowski, M.; et al. Role of Heme-Oxygenase-1 in Biology of Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. Cells 2021, 10, 522. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Description = Known Connection to HO-1 | Relationship to HO-1 |
|---|---|---|
| Bag3 | Mus musculus BCL2-associated athanogene 3 (Bag3), mRNA. | In HO-1 gene ontology maps, Hmox1->Hsf1->Bag3 |
| Coq9 | Mus musculus coenzyme Q9 (Coq9), mRNA. | Less ox stress = less Coq9 |
| Dnajb1 | Mus musculus DnaJ heat shock protein family (Hsp40) member B1 (Dajb1), transcript variant 1, mRNA. | Mediated by Nrf2 |
| Dpp10 | Mus musculus dipeptidylpeptidase 10 (Dpp10), mRNA. (Asthma) | Related to Asthma |
| Eef1a1 | Mus musculus eukaryotic translation elongation factor 1 alpha 1 (Eef1a1), mRNA. | HO-1-interacting-protein |
| Fzd2 | Mus musculus frizzled class receptor 2 (Fzd2), mRNA. | Inhibited by cigarette smoke |
| Hist1h1e | Mus musculus histone cluster 1, H1e (Hist1h1e), mRNA. | Induced by oxidation |
| Hmgb2 | Mus musculus high mobility group box 2 (Hmgb2), mRNA. | Hmbg1 inhibited by HO-1 |
| Hnrnpa2b1 | Mus musculus heterogeneous nuclear ribonucleoprotein A2/B1 (Hnrnpa2b1), transcript variant 1, mRNA. | Mediated by Nrf2 |
| Lias | LIAS (Lipoic Acid Synthetase) | Connected to Coq9 and Bola3 Iron Sulfur metabolism |
| Mat2a | Mus musculus methionine adenosyltransferase II, alpha (Mat2a), mRNA. | Negative correlation with HO-1 |
| Opa1 | Mus musculus OPA1, mitochondrial dynamin like GTPase (Opa1), transcript variant 2, mRNA. | PKC-α/HO-1 induced |
| Rn7sk | Mus musculus RNA, 7SK, nuclear (Rn7sk), small nuclear RNA. | Inhibited by HO-1 |
| HO-1 DNA Binding Domains | DBD-1 | DBD-2 | DBD-3 |
|---|---|---|---|
| Predicted binding dsDNA fragment length (number of nucleotides) | 10 | 10 | 10 |
| Protein residue contacts | 9 | 7 | 8 |
| DNA contacts | 7 | 6 | 5 |
| HO-1 DBD-1 | ||||||||
| Non-specific contacts between DNA strand and protein backbone | ||||||||
| DNA | gA6 | aB20 | ||||||
| Protein | QB197 | RB198 | IB211 | |||||
| Non-specific contacts between DNA strand and protein side chains | ||||||||
| DNA_SC | gA6 | gA12 | tA13 | cB19 | aB20 | tB21 | gB22 | |
| Protein_SC | RA100 | QB27 | RB113 | QB197 | EB201 | IB211 | QB212 | EB215 |
| HO-1 DBD-2 | ||||||||
| Contacts between DNA strand and protein backbone | ||||||||
| DNA | tB29 | cB30 | ||||||
| Protein | PA10 | AA20 | ||||||
| Contacts between DNA strand and protein side chains | ||||||||
| DNA_SC | gA8 | aA9 | gB27 | aB28 | tB29 | cB30 | ||
| Protein_SC | PA10 | AA16 | AA20 | KA22 | TA26 | QA197 | IA200 | |
| HO-1 DBD-3 | ||||||||
| Contacts between DNA strand and protein side chains | ||||||||
| DNA_SC | cA18 | aA19 | tA20 | gA21 | tB38 | |||
| Protein_SC | KA116 | RA117 | EA120 | EA125 | RA198 | EA202 | KA204 | LA208 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scaffa, A.; Tollefson, G.A.; Yao, H.; Rizal, S.; Wallace, J.; Oulhen, N.; Carr, J.F.; Hegarty, K.; Uzun, A.; Dennery, P.A. Identification of Heme Oxygenase-1 as a Putative DNA-Binding Protein. Antioxidants 2022, 11, 2135. https://doi.org/10.3390/antiox11112135
Scaffa A, Tollefson GA, Yao H, Rizal S, Wallace J, Oulhen N, Carr JF, Hegarty K, Uzun A, Dennery PA. Identification of Heme Oxygenase-1 as a Putative DNA-Binding Protein. Antioxidants. 2022; 11(11):2135. https://doi.org/10.3390/antiox11112135
Chicago/Turabian StyleScaffa, Alejandro, George A. Tollefson, Hongwei Yao, Salu Rizal, Joselynn Wallace, Nathalie Oulhen, Jennifer F. Carr, Katy Hegarty, Alper Uzun, and Phyllis A. Dennery. 2022. "Identification of Heme Oxygenase-1 as a Putative DNA-Binding Protein" Antioxidants 11, no. 11: 2135. https://doi.org/10.3390/antiox11112135
APA StyleScaffa, A., Tollefson, G. A., Yao, H., Rizal, S., Wallace, J., Oulhen, N., Carr, J. F., Hegarty, K., Uzun, A., & Dennery, P. A. (2022). Identification of Heme Oxygenase-1 as a Putative DNA-Binding Protein. Antioxidants, 11(11), 2135. https://doi.org/10.3390/antiox11112135