
Role of Nrf2 in Pancreatic Cancer



Abstract
:1. Introduction
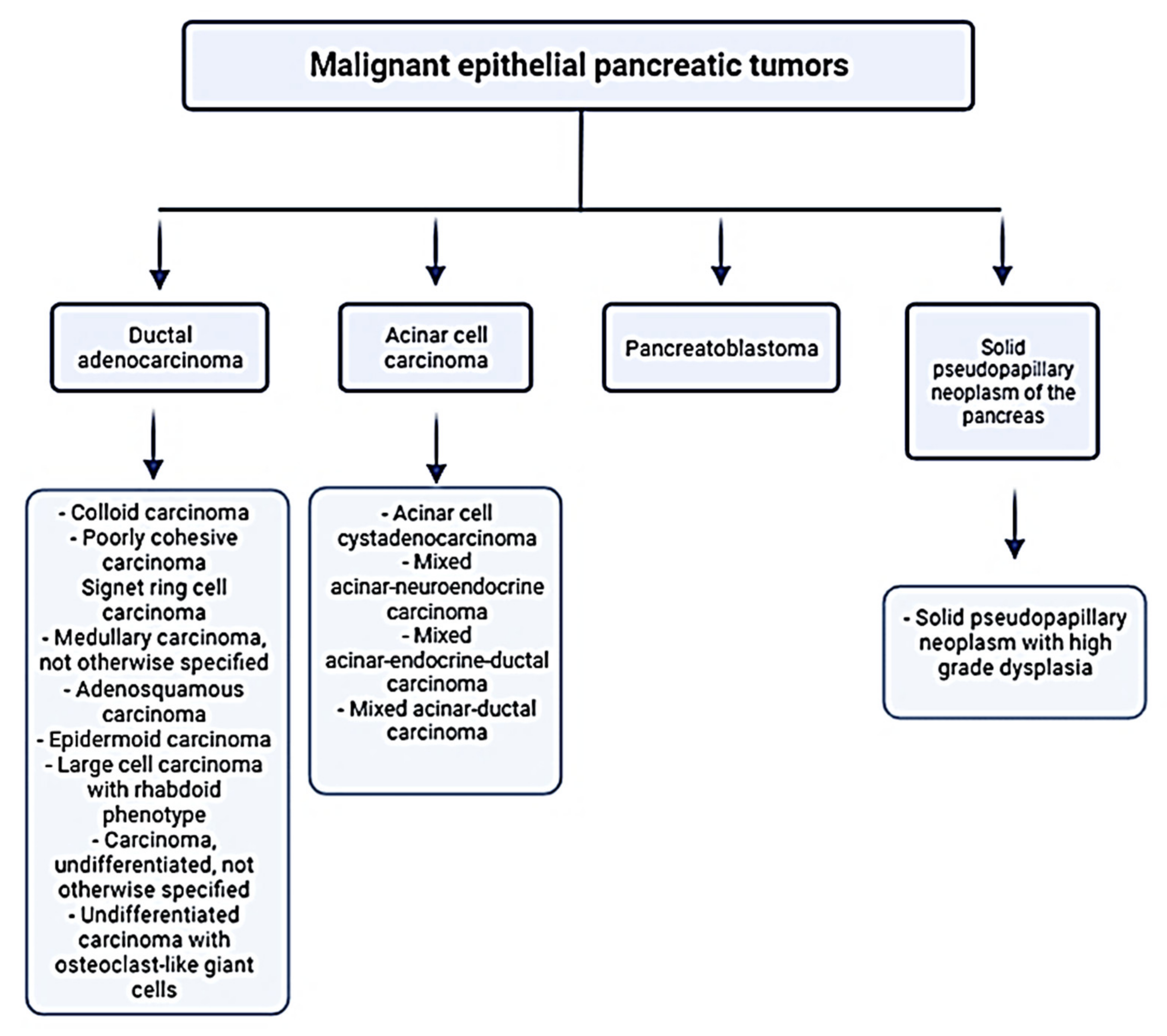
1.1. Pancreatic Cancer
1.2. Oxidative Stress and Signal Transduction in Pancreatic Cancer
2. Molecular Aspects of Nrf2 Activation
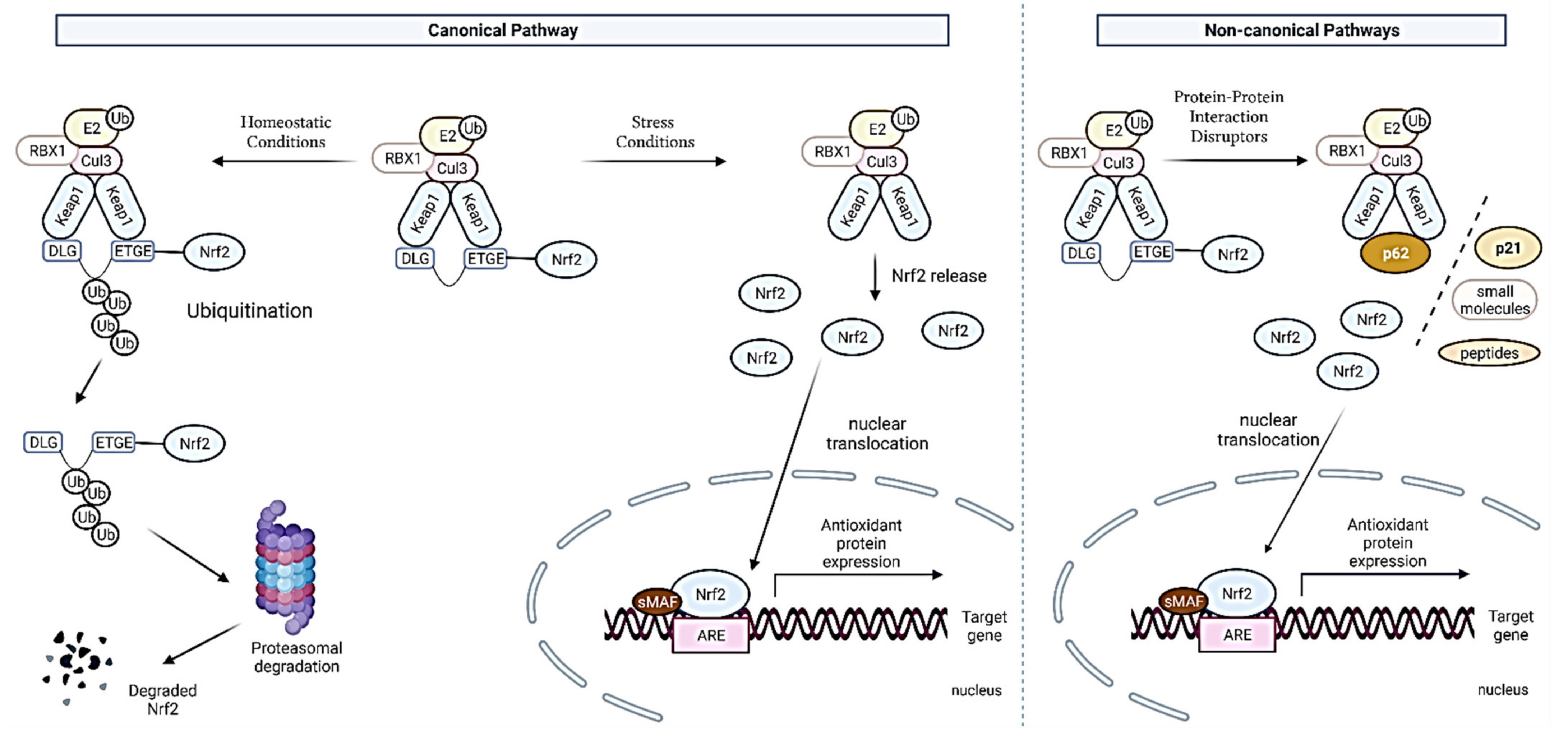
2.1. Nrf2-Keap1 Interaction Interface and Its Regulation
2.2. Canonical and Noncanonical Nrf2 Activation
3. Dual Role of Nrf2 in Pancreatic Cancers
3.1. Nrf2 as Tumor Suppressor in Pancreatic Cancer
3.2. The Carcinogenic Role of Keap1-Nrf2 Pathway in Pancreatic Cancer
4. Therapeutic Strategies Targeting Nrf2 in Pancreatic Cancers
4.1. Natural Compounds
4.1.1. Natural Compounds with Inhibitory Effects on Nrf2
Trigonelline
Brusatol
Digoxin
Ailanthone
4.1.2. Natural Compounds with Activation Effects on Nrf2
Curcumin
Sulforaphane
Esculetin
Xanthohumol
Resveratrol
Phenethyl Isothiocyanate
4.2. Synthetic Compounds
4.2.1. Nrf2 Inhibitors
Dexamethasone
Dimethyl Fumarate
4.2.2. Compounds Interfering with Oncogenic Functional Interactors of the Nrf2/Keap1 Pathway
Inhibitor of PI3K/DNA-PK–PIK-75
Inhibitor of Asparagine Synthesis Pathway-NSC84167

{kind=link}
{kind=link}
{kind=link}
| Natural Compounds | ||||
| Compound | Dosage | Model | Mechanism of Modulation | Reference |
| Trigonelline | 0.01–10 μM | Panc1, MiaPaCa-2, and Colo357 cell lines | a dose-dependent inhibition of ARE-driven luciferase expression; a decreased accumulation of Nrf2 protein in the nucleus; reduction of proteasome activity | [86] |
| Brusatol | 0.5 μM | PATU-8988, BxPC-3 and Panc1 cell lines | inhibition of the Nrf2 pathway and increased ROS accumulation; abrogation of Gemcitabine-induced Nrf2 activation; decrease in mRNA and protein levels of Nrf2 target genes | [89] |
| 2 mg/kg i.p. once/day | Panc-1 xenografts | augmented antitumor activity of Gemcitabine | [90] | |
| Digoxin | 20 and 40 and 80 nM | SW1990 and Panc1 cell lines with induced gemcitabine-resistance | decrease in total and nuclear protein levels of Nrf2; an increase in the sensitivity to gemcitabine; an increase in the number of cells undergoing apoptosis and inhibition of cell colony formation compared with gemcitabine single treatment | [92] |
| Ailanthone | 0.1–2 μg/mL | Panc1 cell line | post-translational downregulation of Nrf2 and YAP proteins, by targeting deubiquitinases | [95] |
| Curcumin | Panc1 and CFPAC-1 cell lines | enhanced antitumor effect of Sestrin2 through the Nrf-2-Keap1/HO-1/NQO-1 signaling pathway | [96] | |
| Meriva®, a patented preparation of curcumin complexed with phospholipids | clinical trials; fifty-two consecutive patients | activation of Nrf2 downregulates NF-κB controlled genes involved in inflammation, proliferation, survival, invasion, angiogenesis, and metastasis | [97] | |
| Sulforaphane | 1–100 μM | Panc1 and MiaPaCa-2 cell lines | activation of adenosine 5′-monophosphate-activated protein kinase (AMPK) by excessively generated ROS and subsequent increase in the Nrf2 nuclear translocation, which suppresses pancreatic cancer cell proliferation | [99] |
| 50 mg/kg, i.p. | transgenic mouse model | inhibition of tumor growth, consistent with the antiproliferative effects of SFN through ROS activated AMPK signaling pathway and NRF2 nuclear translocation | [99] | |
| Esculetin | 100 μM | Panc1, MiaPaCa-2, and AsPC-1 cell lines | directly binding to Keap1 and inhibition of its binding to Nrf2; reduction of ROS level, inhibition of cell growth, cell cycle arrest at G1 phase, and induction of apoptosis and loss of mitochondrial membrane potential | [100] |
| Xanthohumol | 5 and 10 μM | Panc1 cell line | enhanced binding of Nrf2 to ARE sequence and increased protein level of Nrf2 correlated with decreased NF-κB expression; protein levels and inhibited proliferation | [71] |
| 5 and 10 μM | MiaPaCa-2 cell line | enhanced binding of Nrf2 to ARE sequence and increased protein level of Nrf2; in the combination with PEITC increased Caspase-3 and LC3 protein levels and inhibition of proliferation | [70] | |
| Resveratrol | 50 and 100 μM | Panc1 and MiaPaCa-2 cell lines | reduction in the level of NAF-1 and enhancement of the Nrf2 expression by inducing the accumulation of ROS, which contribute to cell death | [102] |
| 5 and 10 μM | Panc1 and MiaPaCa-2 cell lines | promotion of apoptosis via activation of Nrf2 and consequently downregulation of NF-κB | [70,71] | |
| Phenethyl isothiocyanate | 5 μmol/L | Panc-28, MiaPaCa-2, AsPC-1 cell lines | depletion of cellular GSH | [103] |
| 5 and 10 μM | MiaPaCa-2 cell line | activation of Nrf2 and its target genes by increased levels of p-JNK and decreased levels of p-GSK3β; increased Caspase-3 and LC3 protein levels | [70] | |
| Synthetic compounds | ||||
| Compound | Dosage | Model | Mechanism of modulation | Reference |
| Dexamethasone | 1 µM | Panc1 CSLC and PSN-1 CSLC (CSLC-cancer stem-like cells) | reduction of Nrf2 expression with significant decrease in GSH; increase in the growth-inhibitory effects of Gemcitabine and 5-fluorouracil | [104] |
| Dimethyl Fumarate | 100 μM | MiaPaCa-2 cell line | a decrease in total Nrf2 and HO-1 corresponding with decreased DJ-1 protein levels | [107] |
| PIK-75 | 0.1–1 μM | MiaPaCa-2 and AsPC-1 cell lines; xenograft model | reduction of the Nrf2 protein levels and its transcriptional activity by proteasome-mediated degradation | [82] |
| NSC84167 | 1–10 μM | Panc1 and AsPC-1 cell lines; patient-derived pancreatic cancer cells and PDX tumor tissue | selectively targeting Nrf2-activated pancreatic cancer by inhibiting asparagine synthesis pathway; induction of apoptosis in Nrf2-activated pancreatic cancer cells independent of ROS | [60] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Franceschi, M.; Rodriguez-Castro, K.I.; Crafa, P.; Cambiè, G.; Miraglia, C.; Barchi, A.; Nouvenne, A.; Leandro, G.; Meschi, T.; et al. Epidemiology and Risk Factors of Pancreatic Cancer. Acta Biomed. 2018, 89, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Guinter, M.A.; Merchant, A.T.; Wirth, M.D.; Zhang, J.; Stolzenberg-Solomon, R.Z.; Steck, S.E. Dietary Patterns and Risk of Pancreatic Cancer: A Systematic Review. Nutr. Rev. 2017, 75, 883–908. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Gharibi, A.; Adamian, Y.; Kelber, J.A. Cellular and Molecular Aspects of Pancreatic Cancer. Acta Histochem. 2016, 118, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R. WHO Classification. PathologyOutlines.com Website. Available online: https://www.pathologyoutlines.com/topic/pancreaswho.html (accessed on 8 December 2021).
- Li, L.; Leung, P.S. Chapter 13—Pancreatic Cancer, Pancreatitis, and Oxidative Stress. In Gastrointestinal Tissue; Gracia-Sancho, J., Salvadó, J., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 173–186. ISBN 978-0-12-805377-5. [Google Scholar]
- Swentek, L.; Chung, D.; Ichii, H. Antioxidant Therapy in Pancreatitis. Antioxidants 2021, 10, 657. [Google Scholar] [CrossRef] [PubMed]
- Kolodecik, T.; Shugrue, C.; Ashat, M.; Thrower, E.C. Risk Factors for Pancreatic Cancer: Underlying Mechanisms and Potential Targets. Front. Physiol. 2014, 4, 415. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal Transduction in Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, H.P.; Albuquerque, M.T.O.M.; Rocha Oliveira, C.J.; Curcio, M.F. Chapter 3—Signal Transduction Pathways in Endothelial Cells: Implications for Angiogenesis. In Endothelium and Cardiovascular Diseases; Da Luz, P.L., Libby, P., Chagas, A.C.P., Laurindo, F.R.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 23–36. ISBN 978-0-12-812348-5. [Google Scholar]
- Zhang, Z.; Ji, S.; Zhang, B.; Liu, J.; Qin, Y.; Xu, J.; Yu, X. Role of Angiogenesis in Pancreatic Cancer Biology and Therapy. Biomed. Pharmacother. 2018, 108, 1135–1140. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; Ruggieri, S.; Ribatti, D. Angiogenesis in Pancreatic Cancer: Pre-Clinical and Clinical Studies. Cancers 2019, 11, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yi, J. Cancer Cell Killing via ROS: To Increase or Decrease, That Is the Question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Gupta, V.K.; Singh, R.; Sharma, B. Phytochemicals Mediated Signalling Pathways and Their Implications in Cancer Chemotherapy: Challenges and Opportunities in Phytochemicals Based Drug Development: A Review. Biochem. Compd. 2017, 5, 1–15. [Google Scholar] [CrossRef]
- Grek, C.L.; Tew, K.D. Redox Metabolism and Malignancy. Curr. Opin. Pharmacol. 2010, 10, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirshner, J.R.; He, S.; Balasubramanyam, V.; Kepros, J.; Yang, C.-Y.; Zhang, M.; Du, Z.; Barsoum, J.; Bertin, J. Elesclomol Induces Cancer Cell Apoptosis through Oxidative Stress. Mol. Cancer Ther. 2008, 7, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox Regulation in Cancer: A Double-Edged Sword with Therapeutic Potential. Oxidat. Med. Cell. Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Jung, B.-J.; Yoo, H.-S.; Shin, S.; Park, Y.-J.; Jeon, S.-M. Dysregulation of NRF2 in Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Biomol. Ther. 2018, 26, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating MRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, L.; Mesgarzadeh, J.; Xu, I.; Powers, E.T.; Wiseman, R.L.; Bollong, M.J. Defining the Functional Targets of Cap‘n’Collar Transcription Factors NRF1, NRF2, and NRF. Antioxidants 2020, 9, 1025. [Google Scholar] [CrossRef]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in Antioxidant Response Element-Mediated Gene Expression and Beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A. Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers 2020, 12, E2681. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular Mechanism of Human Nrf2 Activation and Degradation: Role of Sequential Phosphorylation by Protein Kinase CK. Free Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-Related Factor 2 (Nrf2), a NF-E2-like Basic Leucine Zipper Transcriptional Activator That Binds to the Tandem NF-E2/AP1 Repeat of the Beta-Globin Locus Control Region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [Green Version]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural Basis of Keap1 Interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Song, M.-Y.; Lee, D.-Y.; Chun, K.-S.; Kim, E.-H. The Role of NRF2/KEAP1 Signaling Pathway in Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular Basis for the Disruption of Keap1–Nrf2 Interaction via Hinge & Latch Mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in Cancers: A Double-edged Sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Jyrkkänen, H.-K.; Levonen, A.-L. Activation of Stress Signaling Pathways by Electrophilic Oxidized and Nitrated Lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer Chemoprevention Mechanisms Mediated through the Keap1-Nrf2 Pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 Is Controlled by Two Distinct β-TrCP Recognition Motifs in Its Neh6 Domain, One of Which Can Be Modulated by GSK-3 Activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yu, S.; Liu, T.; Kim, J.-H.; Blank, V.; Li, H.; Kong, A.-N.T. Heterodimerization with Small Maf Proteins Enhances Nuclear Retention of Nrf2 via Masking the NESzip Motif. Biochim. Biophys. Acta 2008, 1783, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and Non-Canonical Mechanisms of Nrf2 Activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Baer-Dubowska, W. Modulation of Nrf2 and NF-ΚB Signaling Pathways by Naturally Occurring Compounds in Relation to Cancer Prevention and Therapy. Are Combinations Better Than Single Compounds? Int. J. Mol. Sci. 2021, 22, 8223. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of P62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Zhang, D.D. Non-Canonical Activation of NRF2: New Insights and Its Relevance to Disease. Curr. Pathobiol. Rep. 2017, 5, 171–176. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting P62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Cao, W. Toll-Like Receptor Signaling Induces Nrf2 Pathway Activation through P62-Triggered Keap1 Degradation. Mol. Cell Biol. 2015, 35, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Rea, S.; Sultana, M.; Shaw, B.; Searle, M.S.; Layfield, R. ALS-FTLD Associated Mutations of SQSTM1 Impact on Keap1-Nrf2 Signalling. Mol. Cell Neurosci. 2016, 76, 52–58. [Google Scholar] [CrossRef]
- Riz, I.; Hawley, T.S.; Marsal, J.W.; Hawley, R.G. Noncanonical SQSTM1/P62-Nrf2 Pathway Activation Mediates Proteasome Inhibitor Resistance in Multiple Myeloma Cells via Redox, Metabolic and Translational Reprogramming. Oncotarget 2016, 7, 66360–66385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and P62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic Analysis of Ubiquitin Ligase KEAP1 Reveals Associated Proteins That Inhibit NRF2 Ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress-Induced DPP3-KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Karapetian, R.N.; Evstafieva, A.G.; Abaeva, I.S.; Chichkova, N.V.; Filonov, G.S.; Rubtsov, Y.P.; Sukhacheva, E.A.; Melnikov, S.V.; Schneider, U.; Wanker, E.E.; et al. Nuclear Oncoprotein Prothymosin α Is a Partner of Keap1: Implications for Expression of Oxidative Stress-Protecting Genes. Mol. Cell. Biol. 2005, 25, 1089–1099. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, B.; Nakamura, Y.; Yokoyama, S. Structural Analysis of the Complex of Keap1 with a Prothymosin α Peptide. Acta Cryst. Sect. F 2008, 64, 233–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Y.; Wei, Z.; Tao, L.; Wang, S.; Zhang, F.; Shen, C.; Wu, H.; Liu, Z.; Zhu, P.; Wang, A.; et al. Prophylaxis of Diallyl Disulfide on Skin Carcinogenic Model via P21-Dependent Nrf2 Stabilization. Sci. Rep. 2016, 6, 35676. [Google Scholar] [CrossRef]
- Sánchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of NRF2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- Telkoparan-Akillilar, P.; Panieri, E.; Cevik, D.; Suzen, S.; Saso, L. Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer. Molecules 2021, 26, 1417. [Google Scholar] [CrossRef]
- Qin, J.-J.; Cheng, X.-D.; Zhang, J.; Zhang, W.-D. Dual Roles and Therapeutic Potential of Keap1-Nrf2 Pathway in Pancreatic Cancer: A Systematic Review. Cell Commun. Signal. 2019, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Augustine, J.J.; Kang, Y.; Roife, D.; Li, X.; Deng, J.; Tan, L.; Rusling, L.A.; Weinstein, J.N.; Lorenzi, P.L.; et al. Compound NSC84167 Selectively Targets NRF2-Activated Pancreatic Cancer by Inhibiting Asparagine Synthesis Pathway. Cell Death Dis. 2021, 12, 693. [Google Scholar] [CrossRef]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 Is Overexpressed in Pancreatic Cancer: Implications for Cell Proliferation and Therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 Promotes Mutant K-Ras/P53-Driven Pancreatic Carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Genrich, G.; Kruppa, M.; Lenk, L.; Helm, O.; Broich, A.; Freitag-Wolf, S.; Röcken, C.; Sipos, B.; Schäfer, H.; Sebens, S. The Anti-Oxidative Transcription Factor Nuclear Factor E2 Related Factor-2 (Nrf2) Counteracts TGF-β1 Mediated Growth Inhibition of Pancreatic Ductal Epithelial Cells -Nrf2 as Determinant of pro-Tumorigenic Functions of TGF-β1. BMC Cancer 2016, 16, 155. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription Factor NFE2L2/NRF2 Is a Regulator of Macroautophagy Genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Saddawi-Konefka, R.; O’Sullivan, T.; Gross, E.T.; Washington, A.; Bui, J.D. Tumor-Expressed IL-17D Recruits NK Cells to Reject Tumors. Oncoimmunology 2014, 3, e954853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory Mechanisms Controlling Gene Expression Mediated by the Antioxidant Response Element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Kim, E.-H.; Jung, H.S.; Yang, D.; Park, E.-Y.; Jun, H.-S. EX4 Stabilizes and Activates Nrf2 via PKCδ, Contributing to the Prevention of Oxidative Stress-Induced Pancreatic Beta Cell Damage. Toxicol. Appl. Pharmacol. 2017, 315, 60–69. [Google Scholar] [CrossRef]
- Cykowiak, M.; Krajka-Kuźniak, V.; Baer-Dubowska, W. Combinations of Phytochemicals More Efficiently than Single Components Activate Nrf2 and Induce the Expression of Antioxidant Enzymes in Pancreatic Cancer Cells. Nutr. Cancer 2021, 1–16. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Cykowiak, M.; Szaefer, H.; Kleszcz, R.; Baer-Dubowska, W. Combination of Xanthohumol and Phenethyl Isothiocyanate Inhibits NF-ΚB and Activates Nrf2 in Pancreatic Cancer Cells. Toxicol. In Vitro 2020, 65, 104799. [Google Scholar] [CrossRef]
- Probst, B.L.; McCauley, L.; Trevino, I.; Wigley, W.C.; Ferguson, D.A. Cancer Cell Growth Is Differentially Affected by Constitutive Activation of NRF2 by KEAP1 Deletion and Pharmacological Activation of NRF2 by the Synthetic Triterpenoid, RTA 405. PLoS ONE 2015, 10, e0135257. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Matsumoto, R.; Tanaka, Y.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. Int. J. Mol. Sci. 2021, 22, 1870. [Google Scholar] [CrossRef]
- Kha, M.-L.; Hesse, L.; Deisinger, F.; Sipos, B.; Röcken, C.; Arlt, A.; Sebens, S.; Helm, O.; Schäfer, H. The Antioxidant Transcription Factor Nrf2 Modulates the Stress Response and Phenotype of Malignant as Well as Premalignant Pancreatic Ductal Epithelial Cells by Inducing Expression of the ATF3 Splicing Variant ΔZip2. Oncogene 2019, 38, 1461–1476. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble Factors from Stellate Cells Induce Pancreatic Cancer Cell Proliferation via Nrf2-Activated Metabolic Reprogramming and ROS Detoxification. Oncotarget 2016, 7, 36719–36732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 Signaling Mediates the Effects of Pancreatic Stellate Cells on Epithelial-Mesenchymal Transition via Stat3/Nrf2 Pathway in Pancreatic Cancer Cells. Biochim. Biophys. Acta Gen. Sub. 2017, 1861, 296–306. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 Activation Drive Macrophages Polarization and Cancer Cell Epithelial-Mesenchymal Transition during Interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. P62 Links the Autophagy Pathway and the Ubiqutin–Proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Yang, F.; Fu, R.; Li, X.; French, R.; Mose, E.; Pu, X.; Trinh, B.; Kumar, A.; Liu, J.; et al. Cancer Cells Escape Autophagy Inhibition via NRF2-Induced Macropinocytosis. Cancer Cell 2021, 39, 678–693.e11. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, E1755. [Google Scholar] [CrossRef] [Green Version]
- Duong, H.-Q.; Yi, Y.W.; Kang, H.J.; Hong, Y.B.; Tang, W.; Wang, A.; Seong, Y.-S.; Bae, I. Inhibition of NRF2 by PIK-75 Augments Sensitivity of Pancreatic Cancer Cells to Gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.B.; Kang, H.J.; Kwon, S.Y.; Kim, H.J.; Kwon, K.Y.; Cho, C.H.; Lee, J.-M.; Kallakury, B.V.S.; Bae, I. Nuclear Factor (Erythroid-Derived 2)-like 2 Regulates Drug Resistance in Pancreatic Cancer Cells. Pancreas 2010, 39, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, H.-Q.; You, K.S.; Oh, S.; Kwak, S.-J.; Seong, Y.-S. Silencing of NRF2 Reduces the Expression of ALDH1A1 and ALDH3A1 and Sensitizes to 5-FU in Pancreatic Cancer Cells. Antioxidants 2017, 6, E52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Buha, A.; Telkoparan-Akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential Applications of NRF2 Modulators in Cancer Therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef] [Green Version]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.-L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 Transcription Factor by the Alkaloid Trigonelline Renders Pancreatic Cancer Cells More Susceptible to Apoptosis through Decreased Proteasomal Gene Expression and Proteasome Activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 Inhibitor for Future Cancer Therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol Enhances the Efficacy of Chemotherapy by Inhibiting the Nrf2-Mediated Defense Mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Ye, W.; Huang, C.; Lou, B.; Zhang, J.; Yu, D.; Huang, X.; Chen, B.; Zhou, M. Brusatol Inhibits Growth and Induces Apoptosis in Pancreatic Cancer Cells via JNK/P38 MAPK/NF-Κb/Stat3/Bcl-2 Signaling Pathway. Biochem. Biophys. Res. Commun. 2017, 487, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, W.; Huang, C.; Yu, D.; Chen, H.; Deng, T.; Zhang, F.; Lou, B.; Zhang, J.; Shi, K.; et al. Brusatol Enhances the Chemotherapy Efficacy of Gemcitabine in Pancreatic Cancer via the Nrf2 Signalling Pathway. Oxid. Med. Cell Longev. 2018, 2018, 2360427. [Google Scholar] [CrossRef]
- Zhao, Y.-T.; Yan, J.-Y.; Han, X.-C.; Niu, F.-L.; Zhang, J.-H.; Hu, W.-N. Anti-Proliferative Effect of Digoxin on Breast Cancer Cells via Inducing Apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5837–5842. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Yang, M.; Wang, K.; Liu, Y.; Zhang, M.; Yang, Y.; Jin, C.; Wang, R.; Hu, R. Digoxin Sensitizes Gemcitabine-Resistant Pancreatic Cancer Cells to Gemcitabine via Inhibiting Nrf2 Signaling Pathway. Redox Biol. 2019, 22, 101131. [Google Scholar] [CrossRef]
- Wang, R.; Lu, Y.; Li, H.; Sun, L.; Yang, N.; Zhao, M.; Zhang, M.; Shi, Q. Antitumor Activity of the Ailanthus Altissima Bark Phytochemical Ailanthone against Breast Cancer MCF-7 Cells. Oncol. Lett. 2018, 15, 6022–6028. [Google Scholar] [CrossRef] [Green Version]
- Cucci, M.A.; Grattarola, M.; Dianzani, C.; Damia, G.; Ricci, F.; Roetto, A.; Trotta, F.; Barrera, G.; Pizzimenti, S. Ailanthone Increases Oxidative Stress in CDDP-Resistant Ovarian and Bladder Cancer Cells by Inhibiting of Nrf2 and YAP Expression through a Post-Translational Mechanism. Free Radic. Biol. Med. 2020, 150, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Grattarola, M.; Cucci, M.A.; Roetto, A.; Dianzani, C.; Barrera, G.; Pizzimenti, S. Post-Translational down-Regulation of Nrf2 and YAP Proteins, by Targeting Deubiquitinases, Reduces Growth and Chemoresistance in Pancreatic Cancer Cells. Free Radic. Biol. Med. 2021, 174, 202–210. [Google Scholar] [CrossRef]
- Fu, H.; Ni, X.; Ni, F.; Li, D.; Sun, H.; Kong, H.; Shan, Y.; Dai, S. Study of the Mechanism by Which Curcumin Cooperates with Sestrin2 to Inhibit the Growth of Pancreatic Cancer. Gastroenterol. Res. Pract. 2021, 2021, 7362233. [Google Scholar] [CrossRef] [PubMed]
- Pastorelli, D.; Fabricio, A.S.C.; Giovanis, P.; D’Ippolito, S.; Fiduccia, P.; Soldà, C.; Buda, A.; Sperti, C.; Bardini, R.; Da Dalt, G.; et al. Phytosome Complex of Curcumin as Complementary Therapy of Advanced Pancreatic Cancer Improves Safety and Efficacy of Gemcitabine: Results of a Prospective Phase II Trial. Pharmacol. Res. 2018, 132, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, P.; Kim, J.S. Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers. Molecules 2018, 23, E2983. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jiang, Z.; Zhou, C.; Chen, K.; Li, X.; Wang, Z.; Wu, Z.; Ma, J.; Ma, Q.; Duan, W. Activation of Nrf2 by Sulforaphane Inhibits High Glucose-Induced Progression of Pancreatic Cancer via AMPK Dependent Signaling. Cell Physiol. Biochem. 2018, 50, 1201–1215. [Google Scholar] [CrossRef]
- Arora, R.; Sawney, S.; Saini, V.; Steffi, C.; Tiwari, M.; Saluja, D. Esculetin Induces Antiproliferative and Apoptotic Response in Pancreatic Cancer Cells by Directly Binding to KEAP. Mol. Cancer 2016, 15, 64. [Google Scholar] [CrossRef] [Green Version]
- Girisa, S.; Saikia, Q.; Bordoloi, D.; Banik, K.; Monisha, J.; Daimary, U.D.; Verma, E.; Ahn, K.S.; Kunnumakkara, A.B. Xanthohumol from Hop: Hope for Cancer Prevention and Treatment. IUBMB Life 2021, 73, 1016–1044. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yan, B.; Chen, K.; Jiang, Z.; Zhou, C.; Cao, J.; Qian, W.; Li, J.; Sun, L.; Ma, J.; et al. Resveratrol-Induced Downregulation of NAF-1 Enhances the Sensitivity of Pancreatic Cancer Cells to Gemcitabine via the ROS/Nrf2 Signaling Pathways. Oxid. Med. Cell Longev. 2018, 2018, 9482018. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.-N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of Overcoming Intrinsic Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma through the Redox Modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Okada, M.; Kitanaka, C. Dexamethasone Sensitizes Cancer Stem Cells to Gemcitabine and 5-Fluorouracil by Increasing Reactive Oxygen Species Production through NRF2 Reduction. Life 2021, 11, 885. [Google Scholar] [CrossRef] [PubMed]
- Kourakis, S.; Timpani, C.A.; de Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility? Pharmaceuticals 2020, 13, E306. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-Inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, E630. [Google Scholar] [CrossRef] [PubMed]
- Bennett Saidu, N.E.; Bretagne, M.; Mansuet, A.L.; Just, P.-A.; Leroy, K.; Cerles, O.; Chouzenoux, S.; Nicco, C.; Damotte, D.; Alifano, M.; et al. Dimethyl Fumarate Is Highly Cytotoxic in KRAS Mutated Cancer Cells but Spares Non-Tumorigenic Cells. Oncotarget 2018, 9, 9088–9099. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 Regulates Serine Biosynthesis in Non-Small Cell Lung Cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cykowiak, M.; Krajka-Kuźniak, V. Role of Nrf2 in Pancreatic Cancer. Antioxidants 2022, 11, 98. https://doi.org/10.3390/antiox11010098
Cykowiak M, Krajka-Kuźniak V. Role of Nrf2 in Pancreatic Cancer. Antioxidants. 2022; 11(1):98. https://doi.org/10.3390/antiox11010098
Chicago/Turabian StyleCykowiak, Marta, and Violetta Krajka-Kuźniak. 2022. "Role of Nrf2 in Pancreatic Cancer" Antioxidants 11, no. 1: 98. https://doi.org/10.3390/antiox11010098
APA StyleCykowiak, M., & Krajka-Kuźniak, V. (2022). Role of Nrf2 in Pancreatic Cancer. Antioxidants, 11(1), 98. https://doi.org/10.3390/antiox11010098