
Overexpression of NOX2 Exacerbates AngII-Mediated Cardiac Dysfunction and Metabolic Remodelling

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Echocardiography and Blood Pressure Measurements
2.3. Isolated Heart Perfusions
2.4. Real-Time Quantitative PCR
2.5. Respirometry in Frozen Samples
2.6. Blood Glucose Levels
2.7. Statistics
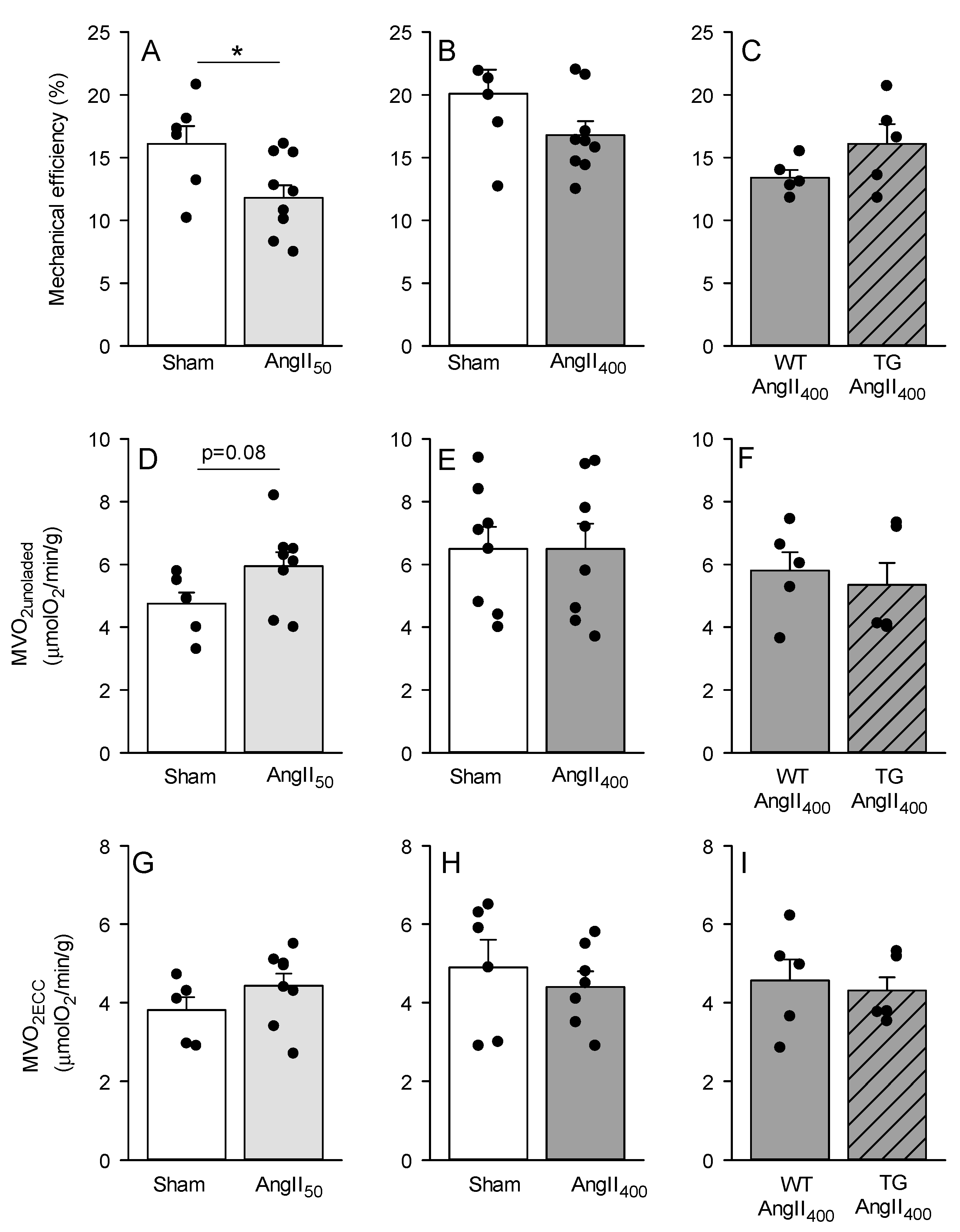
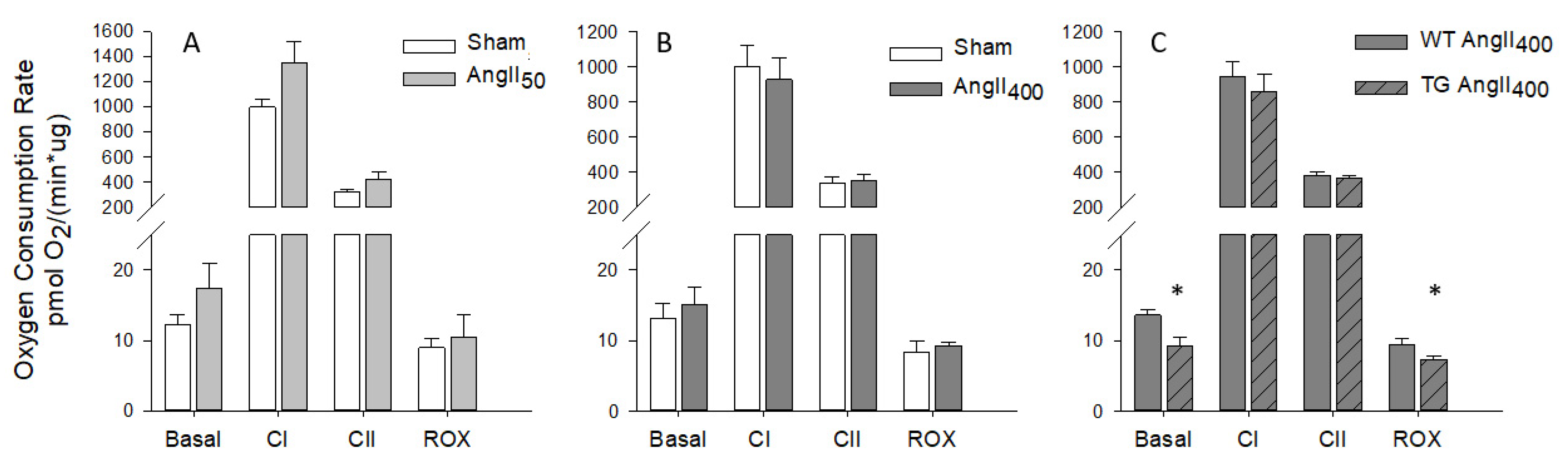
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domenighetti, A.A.; Wang, Q.; Egger, M.; Richards, S.M.; Pedrazzini, T.; Delbridge, L.M. Angiotensin II–mediated phenotypic cardiomyocyte remodeling leads to age-dependent cardiac dysfunction and failure. Hypertension 2005, 46, 426–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzolai, L.; Nussberger, J.r.; Aubert, J.-F.; Brunner, D.B.; Gabbiani, G.; Brunner, H.R.; Pedrazzini, T. Blood pressure–independent cardiac hypertrophy induced by locally activated renin-angiotensin system. Hypertension 1998, 31, 1324–1330. [Google Scholar] [CrossRef] [Green Version]
- Zablocki, D.; Sadoshima, J. Angiotensin II and oxidative stress in the failing heart. Antioxid. Redox Signal. 2013, 19, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Hauck, L.; Grothe, D.; Billia, F. p21CIP1/WAF1-dependent inhibition of cardiac hypertrophy in response to Angiotensin II involves Akt/Myc and pRb signaling. Peptides 2016, 83, 38–48. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, H.; Guang, G.-c.; Deng, Z.-r. Overexpressed connective tissue growth factor in cardiomyocytes attenuates left ventricular remodeling induced by angiotensin II perfusion. Clin. Exp. Hypertens. 2017, 39, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.-h.; Penninger, J.M.; Kassiri, Z. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Kawada, N.; Imai, E.; Karber, A.; Welch, W.J.; Wilcox, C.S. A mouse model of angiotensin II slow pressor response: Role of oxidative stress. J. Am. Soc. Nephrol. 2002, 13, 2860–2868. [Google Scholar] [CrossRef] [Green Version]
- Simon, G.; Abraham, G.; Cserep, G. Pressor and subpressor angiotensin II administration two experimental models of hypertension. Am. J. Hypertens. 1995, 8, 645–650. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- How, O.-J.; Aasum, E.; Severson, D.L.; Chan, W.A.; Essop, M.F.; Larsen, T.S. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 2006, 55, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.R.; Waggoner, A.D.; Schechtman, K.B.; Meyer, T.; Gropler, R.J.; Barzilai, B.; Dávila-Román, V.G. Alterations in left ventricular structure and function in young healthy obese women: Assessment by echocardiography and tissue Doppler imaging. J. Am. Coll. Cardiol. 2004, 43, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Aasum, E.; Hafstad, A.D.; Larsen, T.S. Changes in substrate metabolism in isolated mouse hearts following ischemia-reperfusion. In Biochemistry of Diabetes and Atherosclerosis; Springer: Berlin/Heidelberg, Germany, 2003; pp. 97–103. [Google Scholar]
- Hafstad, A.D.; Lund, J.; Hadler-Olsen, E.; Höper, A.C.; Larsen, T.S.; Aasum, E. High-and moderate-intensity training normalizes ventricular function and mechanoenergetics in mice with diet-induced obesity. Diabetes 2013, 62, 2287–2294. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-angiotensin system and cardiovascular functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 2012, 5, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; de Mattos, A.B.M.; Shao, D.; Li, T.; Nabben, M.; Kim, M.; Wang, W.; Tian, R.; Kolwicz Jr, S.C. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J. Mol. Cell. Cardiol. 2016, 100, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Brand, S.; Amann, K.; Schupp, N. Angiotensin II-induced hypertension dose-dependently leads to oxidative stress and DNA damage in mouse kidneys and hearts. J. Hypertens. 2013, 31, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Kinugawa, S.; Suga, T.; Yokota, T.; Hirabayashi, K.; Kuroda, S.; Okita, K.; Tsutsui, H. Angiotensin II-induced reduction in exercise capacity is associated with increased oxidative stress in skeletal muscle. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, 1202–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002, 105, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Byrne Jonathan, A.; Grieve, D.J.; Bendall, J.K.; Li, J.-M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II–induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W. Contractile function during angiotensin-II activation: Increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafstad, A.D.; Hansen, S.S.; Lund, J.; Santos, C.X.; Boardman, N.T.; Shah, A.M.; Aasum, E. NADPH Oxidase 2 Mediates Myocardial Oxygen Wasting in Obesity. Antioxidants 2020, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, T.M.; Boardman, N.T.; Hafstad, A.D.; Aasum, E. Isolated perfused working hearts provide valuable additional information during phenotypic assessment of the diabetic mouse heart. PLoS ONE 2018, 13, e0204843. [Google Scholar] [CrossRef] [PubMed]
- How, O.-J.; Aasum, E.; Kunnathu, S.; Severson, D.L.; Myhre, E.S.; Larsen, T.S. Influence of substrate supply on cardiac efficiency, as measured by pressure-volume analysis in ex vivo mouse hearts. Am. J. Physiol.-Heart Circ. Physiol. 2005, 288, 2979–2985. [Google Scholar] [CrossRef] [Green Version]
- Boardman, N.; Hafstad, A.D.; Larsen, T.S.; Severson, D.L.; Aasum, E. Increased O2 cost of basal metabolism and excitation-contraction coupling in hearts from type 2 diabetic mice. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafstad, A.D.; Boardman, N.T.; Lund, J.; Hagve, M.; Khalid, A.M.; Wisløff, U.; Larsen, T.S.; Aasum, E. High intensity interval training alters substrate utilization and reduces oxygen consumption in the heart. J. Appl. Physiol. 2011, 111, 1235–1241. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Benador, I.Y.; Petcherski, A.; Veliova, M.; Benavides, G.A.; Lagarrigue, S.; Caudal, A.; Vergnes, L.; Murphy, A.N.; Karamanlidis, G. A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 2020, 39, e104073. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Khalid, A.M.; How, O.-J.; Larsen, T.S.; Aasum, E. Glucose and insulin improve cardiac efficiency and postischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, 1288–1294. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, M.; Yamaki, A.; Furuya, M.; Inomata, N.; Minamitake, Y.; Ohsuye, K.; Kangawa, K. Ghrelin improves body weight loss and skeletal muscle catabolism associated with angiotensin II-induced cachexia in mice. Regul. Pept. 2012, 178, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, T.; He, Y.; Schiffrin, E.L.; Touyz, R.M. Differential regulation of thioredoxin and NAD (P) H oxidase by angiotensin II in male and female mice. J. Hypertens. 2007, 25, 1263–1271. [Google Scholar] [CrossRef]
- Glenn, D.J.; Cardema, M.C.; Ni, W.; Zhang, Y.; Yeghiazarians, Y.; Grapov, D.; Fiehn, O.; Gardner, D.G. Cardiac steatosis potentiates angiotensin II effects in the heart. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, 339–350. [Google Scholar] [CrossRef]
- Regan, J.A.; Mauro, A.G.; Carbone, S.; Marchetti, C.; Gill, R.; Mezzaroma, E.; Valle Raleigh, J.; Salloum, F.N.; Van Tassell, B.W.; Abbate, A. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H771–H778. [Google Scholar] [CrossRef] [Green Version]
- Joseph, L.C.; Avula, U.M.R.; Wan, E.Y.; Reyes, M.V.; Lakkadi, K.R.; Subramanyam, P.; Nakanishi, K.; Homma, S.; Muchir, A.; Pajvani, U.B. Dietary saturated fat promotes arrhythmia by activating NOX2 (NADPH Oxidase 2). Circ. Arrhythmia Electrophysiol. 2019, 12, e007573. [Google Scholar] [CrossRef] [PubMed]
- Donoso, P.; Finkelstein, J.P.; Montecinos, L.; Said, M.; Sánchez, G.; Vittone, L.; Bull, R. Stimulation of NOX2 in isolated hearts reversibly sensitizes RyR2 channels to activation by cytoplasmic calcium. J. Mol. Cell. Cardiol. 2014, 68, 38–46. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, S.; Hu, N.; Gu, M.; Chu, C.; Li, Y.; Lu, X.; Huang, C. Rhein reduces fat weight in db/db mouse and prevents diet-induced obesity in C57Bl/6 mouse through the inhibition of PPARγ signaling. PPAR Res. 2012, 2012, 374936. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Mori, J.; Alrob, O.A.; Wagg, C.S.; Harris, R.A.; Lopaschuk, G.D.; Oudit, G.Y. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: A critical role of PDK4. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, 1103–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellieux, C.; Aasum, E.; Larsen, T.S.; Montessuit, C.; Papageorgiou, I.; Pedrazzini, T.; Lerch, R. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J. Mol. Cell. Cardiol. 2006, 41, 459–466. [Google Scholar] [CrossRef]
- Aikawa, R.; Nawano, M.; Gu, Y.; Katagiri, H.; Asano, T.; Zhu, W.; Nagai, R.; Komuro, I. Insulin prevents cardiomyocytes from oxidative stress–induced apoptosis through activation of PI3 kinase/Akt. Circulation 2000, 102, 2873–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, C.; Dong, B.; Xing, F.; Huang, H.; Yao, F.; Ma, Y.; He, J.; Dong, Y. AMPK attenuates ventricular remodeling and dysfunction following aortic banding in mice via the Sirt3/Oxidative stress pathway. Eur. J. Pharmacol. 2017, 814, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sun, G.-b.; Sun, X.; Wang, H.-w.; Meng, X.-b.; Qin, M.; Sun, J.; Luo, Y.; Sun, X.-b. Cardioprotective effect of salvianolic acid B against arsenic trioxide-induced injury in cardiac H9c2 cells via the PI3K/Akt signal pathway. Toxicol. Lett. 2013, 216, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Pellieux, C.; Montessuit, C.; Papageorgiou, I.; Lerch, R. Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-α. Cardiovasc. Res. 2009, 82, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Levinsson, A.; Dubé, M.P.; Tardif, J.C.; de Denus, S. Sex, drugs, and heart failure: A sex-sensitive review of the evidence base behind current heart failure clinical guidelines. ESC Heart Fail. 2018, 5, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Pamidimukkala, J.; Hay, M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am. J. Physiol.-Heart Circ. Physiol. 2005, 288, 2177–2184. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Sham | AngII50 | Sham | AngII400 | WT AngII400 | TG AngII400 | |
|---|---|---|---|---|---|---|
| n | 9 | 11 | 10 | 10 | 6 | 6 |
| MAP (mmHg) | n.m. | n.m. | 99 ± 6 | 105 ± 6 | 86 ± 3 | 90 ± 4 # |
| SBP | n.m. | n.m. | 136 ± 12 | 131 ± 7 | 107 ± 3 # | 110 ± 4 # |
| Body weight (g) | 26 ± 0.3 | 27 ± 0.2 | 25 ± 0.3 | 25 ± 0.4 | 27.2 ± 0.8 | 27.0 ± 0.2 |
| Liver weight (g) | 1.33 ± 0.7 | 1.48 ± 0.05 | 0.99 ± 0.03 | 1.00 ± 0.04 | 0.95 ± 0.07 | 0.96 ± 0.04 |
| Blood glucose (mM) | 5.2 ± 0.3 | 4.6 ± 0.3 | 5.8 ± 0.5 | 5.5 ± 0.3 | 6.2 ± 0.6 | 6.2 ± 0.5 |
| HW/BW (mg) | 5.1 ± 0.2 | 4.9 ± 0.1 | 5.1 ± 0.1 | 5.5 ± 0.1 * | 6.0 ± 0.2 | 5.7 ± 0.3 |
| nppaheart | 1.0 ± 0.1 | 1.2 ± 0.2 | 1.0 ± 0.2 | 2.5 ± 0.3 * | 1.0 ± 0.2 | 0.7 ± 0.2 |
| nppbheart | 1.0 ± 0.1 | 1.3 ± 0.2 | 1.0 ± 0.1 | 1.4 ± 0.1 * | 1.0 ± 0.1 | 0.9 ± 0.1 |
| Sham | AngII400 | |||
|---|---|---|---|---|
| Baseline | Week 2 | Baseline | Week 2 | |
| n | 7 | 7 | 7 | 7 |
| Heart rate (BPM) | 460 ± 21 | 451 ± 13 | 470 ± 9 | 458 ± 19 |
| LVPW;d (mm) | 0.71 ± 0.04 | 0.74 ± 0.02 | 0.77 ± 0.02 | 0.81 ± 0.02 * |
| LVID;d (mm) | 3.7 ± 0.1 | 3.9 ± 0.1 | 3.7 ± 0.1 | 4.0 ± 0.1 # |
| LV mass (mg) | 74 ± 7 | 88 ± 3 * | 82 ± 2 | 102 ± 3 #,* |
| LV Mass/BW (mg/g) | 3.1 ± 0.2 | 3.9 ± 0.1 | 3.6 ± 0.1 | 4.4 ± 0.2 |
| LVEDV (μL) | 57 ± 3 | 65 ± 4 | 59 ± 3 | 69 ± 2 # |
| LVESV (μL) | 19 ± 1 | 22 ± 3 | 20 ± 2 | 24 ± 2 |
| SV (μL) | 38 ± 2 | 43 ± 2 # | 39 ± 2 | 45 ± 2 # |
| FS (%) | 37 ± 1 | 36 ± 1 | 36 ± 1 | 36 ± 2 |
| EF (%) | 67 ± 1 | 67 ± 2 | 66 ± 2 | 66 ± 3 |
| LV Volume/LV Mass (µL/mg) | 0.78 ± 0.05 | 0.74 ± 0.04 | 0.69 ± 0.04 | 0.68 ± 0.01 |
| WT AngII400 | TG AngII400 | |||
|---|---|---|---|---|
| Baseline | Week 2 | Baseline | Week 2 | |
| n | 4 | 6 | 6 | 5 |
| Heart rate (BPM) | 491 ± 12 | 526 ± 14 | 484 ± 7 | 515 ± 13 # |
| LVPW;d (mm) | 0.82 ± 0.05 | 0.92 ± 0.05 # | 0.77 ± 0.02 | 0.89 ± 0.02 # |
| LVID;d (mm) | 4.1 ± 0.1 | 4.0 ± 0.1 | 4.2 ± 0.1 | 4.3 ± 0.2 |
| LV mass (mg) | 101 ± 5 | 118 ± 4 # | 95 ± 4 | 113 ± 6 # |
| LV Mass/BW (mg/g) | 3.6 ± 0.1 | 4.3 ± 0.1 # | 3.5 ± 0.1 | 4.2 ± 0.1 # |
| LVEDV (μL) | 73 ± 3 | 71 ± 6 | 77 ± 2 | 84 ± 8 |
| LVESV (μL) | 27 ± 1 | 30 ± 4 | 32 ± 2 | 43 ± 5 #,* |
| SV (μL) | 45 ± 2 | 40 ± 2 | 45 ± 2 | 41 ± 3 |
| FS (%) | 33 ± 1 | 30 ± 2 | 31 ± 1 | 25 ± 1 #,* |
| EF (%) | 62 ± 2 | 58 ± 2 | 59 ± 2 | 50 ± 2 #,* |
| LV Volume/LV Mass (µL/mg) | 0.72 ± 0.04 | 0.60 ± 0.05 | 0.83 ± 0.02 * | 0.74 ± 0.04 #,* |
| E/A | 1.3 ± 0.1 | 1.5 ± 0.2 | 1.4 ± 0.0 | 1.4 ± 0.2 |
| E/E’ | 28 ± 2 | 30 ± 2 | 29 ± 1 | 34 ± 2 # |
| Deceleration time (ms) | 24 ± 1 | 20 ± 2 | 23 ± 2 | 19 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, S.S.; Pedersen, T.M.; Marin, J.; Boardman, N.T.; Shah, A.M.; Aasum, E.; Hafstad, A.D. Overexpression of NOX2 Exacerbates AngII-Mediated Cardiac Dysfunction and Metabolic Remodelling. Antioxidants 2022, 11, 143. https://doi.org/10.3390/antiox11010143
Hansen SS, Pedersen TM, Marin J, Boardman NT, Shah AM, Aasum E, Hafstad AD. Overexpression of NOX2 Exacerbates AngII-Mediated Cardiac Dysfunction and Metabolic Remodelling. Antioxidants. 2022; 11(1):143. https://doi.org/10.3390/antiox11010143
Chicago/Turabian StyleHansen, Synne S., Tina M. Pedersen, Julie Marin, Neoma T. Boardman, Ajay M. Shah, Ellen Aasum, and Anne D. Hafstad. 2022. "Overexpression of NOX2 Exacerbates AngII-Mediated Cardiac Dysfunction and Metabolic Remodelling" Antioxidants 11, no. 1: 143. https://doi.org/10.3390/antiox11010143
APA StyleHansen, S. S., Pedersen, T. M., Marin, J., Boardman, N. T., Shah, A. M., Aasum, E., & Hafstad, A. D. (2022). Overexpression of NOX2 Exacerbates AngII-Mediated Cardiac Dysfunction and Metabolic Remodelling. Antioxidants, 11(1), 143. https://doi.org/10.3390/antiox11010143