
Redox Regulation in Aging Lungs and Therapeutic Implications of Antioxidants in COPD




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Redox Regulation in the Mammalian Respiratory System
2.1. The Sources and Roles of Oxidative Stress in the Respiratory System
2.2. The Antioxidant Defense System
3. Impaired Redox Regulation in Aging and Patients with COPD
3.1. Aging Is a Major Risk Factor for Chronic Lung Diseases, Including COPD
3.2. Redox Dysregulation in Aging Lungs
3.3. Redox Dysregulation in COPD Patient Lungs
4. Antioxidant Drugs
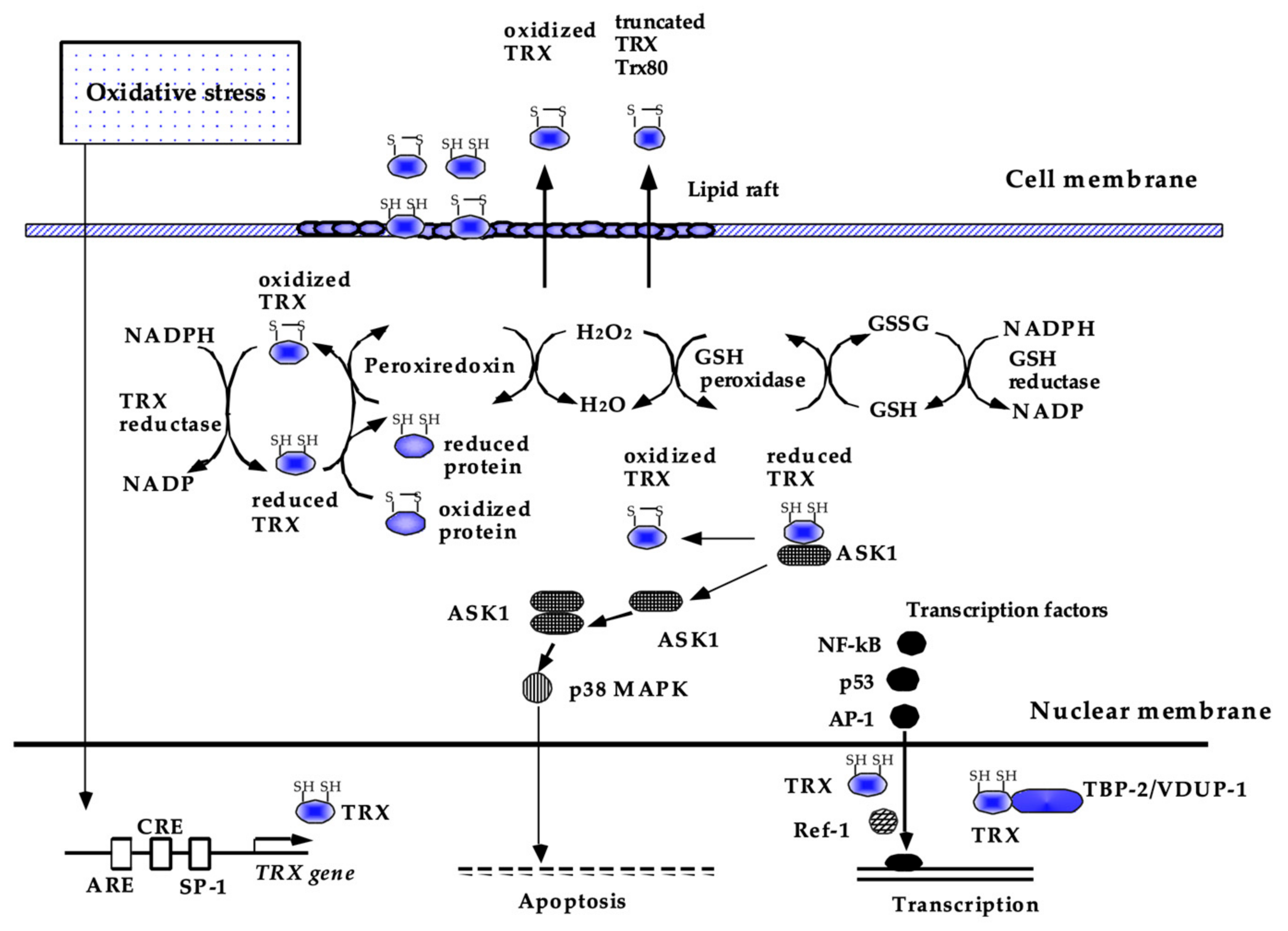
4.1. Thioredoxin (TRX)
4.2. N-Acetylcysteine (NAC)
4.3. The Therapeutic Potential of Current COPD Drugs for Oxidative Stress
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agarwal, A.; Aponte-Mellado, A.; Premkumar, B.J.; Shaman, A.; Gupta, S. The effects of oxidative stress on female reproduction: A review. Reprod. Biol. Endocrinol. 2012, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Airway Redox Homeostasis and Inflammation Gone Awry: From Molecular Pathogenesis to Emerging Therapeutics in Respiratory Pathology. Int. J. Mol. Sci. 2020, 21, 9317. [Google Scholar] [CrossRef]
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Prevention, Diagnosis and Management of COPD. 2020. Available online: https://goldcopd.org/gold-reports/ (accessed on 1 September 2021).
- Pryor, W.A.; Prier, D.G.; Church, D.F. Electron-spin resonance study of mainstream and sidestream cigarette smoke: Nature of the free radicals in gas-phase smoke and in cigarette tar. Environ. Health Perspect. 1983, 47, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef]
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489. [Google Scholar] [CrossRef]
- Wickens, A.P. Ageing and the free radical theory. Respir. Physiol. 2001, 128, 379–391. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Hoshino, T.; Imaoka, H.; Ichiki, H.; Okamoto, M.; Kawayama, T.; Yodoi, J.; Kato, S.; Aizawa, H. Thioredoxin prevents the development and progression of elastase-induced emphysema. Biochem. Biophys. Res. Commun. 2007, 354, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Mishima, M. Redox-based therapeutics for lung diseases. Antioxid. Redox Signal. 2008, 10, 701–704. [Google Scholar] [CrossRef]
- Matera, M.G.; Calzetta, L.; Cazzola, M. Oxidation pathway and exacerbations in COPD: The role of NAC. Expert Rev. Respir. Med. 2016, 10, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Watson, W.H.; Ritzenthaler, J.D.; Roman, J. Lung extracellular matrix and redox regulation. Redox Biol. 2016, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef]
- Miki, H.; Funato, Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012, 151, 255–261. [Google Scholar] [CrossRef]
- Tew, K.D.; Townsend, D.M. Glutathione-s-transferases as determinants of cell survival and death. Antioxid. Redox Signal. 2012, 17, 1728–1737. [Google Scholar] [CrossRef]
- Hoshino, Y.; Shioji, K.; Nakamura, H.; Masutani, H.; Yodoi, J. From oxygen sensing to heart failure: Role of thioredoxin. Antioxid. Redox Signal. 2007, 9, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, J.; Wang, J.; Li, S.; Fukunaga, A.; Yodoi, J.; Tian, H. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct. Target. Ther. 2020, 5, 248. [Google Scholar] [CrossRef]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radic. Biol. Med. 2000, 29, 403–409. [Google Scholar] [CrossRef]
- Barnes, P.J. Novel signal transduction modulators for the treatment of airway diseases. Pharmacol. Ther. 2006, 109, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Leikauf, G.D.; Simpson, L.G.; Santrock, J.; Zhao, Q.; Abbinante-Nissen, J.; Zhou, S.; Driscoll, K.E. Airway epithelial cell responses to ozone injury. Environ. Health Perspect. 1995, 103 (Suppl. 2), 91–95. [Google Scholar] [CrossRef]
- Marumo, S.; Hoshino, Y.; Kiyokawa, H.; Tanabe, N.; Sato, A.; Ogawa, E.; Muro, S.; Hirai, T.; Mishima, M. p38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm. Med. 2014, 14, 79. [Google Scholar] [CrossRef]
- Holmgren, A.; Buchanan, B.B.; Wolosiuk, R.A. Photosynthetic regulatory protein from rabbit liver is identical with thioredoxin. FEBS Lett. 1977, 82, 351–354. [Google Scholar] [CrossRef]
- Yatmaz, S.; Seow, H.J.; Gualano, R.C.; Wong, Z.X.; Stambas, J.; Selemidis, S.; Crack, P.J.; Bozinovski, S.; Anderson, G.P.; Vlahos, R. Glutathione peroxidase-1 reduces influenza A virus-induced lung inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 48, 17–26. [Google Scholar] [CrossRef]
- Chae, H.Z.; Robison, K.; Poole, L.B.; Church, G.; Storz, G.; Rhee, S.G. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 7017–7021. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.T.; Markkanen, P.M.; Peltoniemi, M.; Kang, S.W.; Kinnula, V.L. Variable overoxidation of peroxiredoxins in human lung cells in severe oxidative stress. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L997–L1001. [Google Scholar] [CrossRef]
- Das, K.C.; Das, C.K. Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: Redox independent functions. Biochem. Biophys. Res. Commun. 2000, 277, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Stadtman, T.C. A new selenoprotein from human lung adenocarcinoma cells: Purification, properties, and thioredoxin reductase activity. Proc. Natl. Acad. Sci. USA 1996, 93, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.O. Antioxidants and respiratory disease: The uric acid paradox. Thorax 2014, 69, 978–979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hemila, H.; Louhiala, P. Vitamin C may affect lung infections. J. R. Soc. Med. 2007, 100, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Burdeos, G.C.; Itaya, M.; Nakagawa, K.; Miyazawa, T. Vitamin E: Regulatory Redox Interactions. IUBMB Life 2019, 71, 430–441. [Google Scholar] [CrossRef]
- Abed, D.A.; Goldstein, M.; Albanyan, H.; Jin, H.; Hu, L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 2015, 5, 285–299. [Google Scholar] [CrossRef]
- Sun, X.; Chen, L.; He, Z. PI3K/Akt-Nrf2 and Anti-Inflammation Effect of Macrolides in Chronic Obstructive Pulmonary Disease. Curr. Drug Metab. 2019, 20, 301–304. [Google Scholar] [CrossRef]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Konigshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Birch, J.; Anderson, R.K.; Correia-Melo, C.; Jurk, D.; Hewitt, G.; Marques, F.M.; Green, N.J.; Moisey, E.; Birrell, M.A.; Belvisi, M.G.; et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1124–L1137. [Google Scholar] [CrossRef]
- Brandenberger, C.; Muhlfeld, C. Mechanisms of lung aging. Cell Tissue Res. 2017, 367, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Quirk, J.D.; Sukstanskii, A.L.; Woods, J.C.; Lutey, B.A.; Conradi, M.S.; Gierada, D.S.; Yusen, R.D.; Castro, M.; Yablonskiy, D.A. Experimental evidence of age-related adaptive changes in human acinar airways. J. Appl. Physiol. 2016, 120, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, M.; Matsushita, T.; Hosokawa, M.; Takeda, T. Age-related changes in lung structure and function in the senescence-accelerated mouse (SAM): SAM-P/1 as a new murine model of senile hyperinflation of lung. Am. J. Respir. Crit. Care Med. 1994, 149, 776–782. [Google Scholar] [CrossRef]
- Sato, A.; Hirai, T.; Imura, A.; Kita, N.; Iwano, A.; Muro, S.; Nabeshima, Y.; Suki, B.; Mishima, M. Morphological mechanism of the development of pulmonary emphysema in klotho mice. Proc. Natl. Acad. Sci. USA 2007, 104, 2361–2365. [Google Scholar] [CrossRef]
- Ravikumar, P.; Ye, J.; Zhang, J.; Pinch, S.N.; Hu, M.C.; Kuro-o, M.; Hsia, C.C.; Moe, O.W. alpha-Klotho protects against oxidative damage in pulmonary epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L566–L575. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Yuan, C.; Zhang, J.; Li, L.; Yu, L.; Wiegman, C.H.; Barnes, P.J.; Adcock, I.M.; Huang, M.; Yao, X. Klotho expression is reduced in COPD airway epithelial cells: Effects on inflammation and oxidant injury. Clin. Sci. 2015, 129, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Pako, J.; Barta, I.; Balogh, Z.; Kerti, M.; Drozdovszky, O.; Bikov, A.; Antus, B.; Horvath, I.; Varga, J. Assessment of the Anti-Aging Klotho Protein in Patients with COPD Undergoing Pulmonary Rehabilitation. COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 176–180. [Google Scholar] [CrossRef][Green Version]
- Lange, P.; Celli, B.; Agusti, A.; Boje Jensen, G.; Divo, M.; Faner, R.; Guerra, S.; Marott, J.L.; Martinez, F.D.; Martinez-Camblor, P.; et al. Lung-Function Trajectories Leading to Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2015, 373, 111–122. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Reichert, S.; Stier, A. Does oxidative stress shorten telomeres in vivo? A review. Biol. Lett. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.E.; Chen, J.J.; Podlevsky, J.D.; Alder, J.K.; Hansel, N.N.; Mathias, R.A.; Qi, X.; Rafaels, N.M.; Wise, R.A.; Silverman, E.K.; et al. Telomerase mutations in smokers with severe emphysema. J. Clin. Investig. 2015, 125, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Cordoba-Lanus, E.; Cazorla-Rivero, S.; Espinoza-Jimenez, A.; de-Torres, J.P.; Pajares, M.J.; Aguirre-Jaime, A.; Celli, B.; Casanova, C. Telomere shortening and accelerated aging in COPD: Findings from the BODE cohort. Respir. Res. 2017, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef]
- Meyer, K.C.; Rosenthal, N.S.; Soergel, P.; Peterson, K. Neutrophils and low-grade inflammation in the seemingly normal aging human lung. Mech. Ageing Dev. 1998, 104, 169–181. [Google Scholar] [CrossRef]
- Moliva, J.I.; Rajaram, M.V.; Sidiki, S.; Sasindran, S.J.; Guirado, E.; Pan, X.J.; Wang, S.H.; Ross, P., Jr.; Lafuse, W.P.; Schlesinger, L.S.; et al. Molecular composition of the alveolar lining fluid in the aging lung. Age 2014, 36, 9633. [Google Scholar] [CrossRef]
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 level and the development of disability in older persons. J. Am. Geriatr. Soc. 1999, 47, 639–646. [Google Scholar] [CrossRef]
- Giovannini, S.; Onder, G.; Liperoti, R.; Russo, A.; Carter, C.; Capoluongo, E.; Pahor, M.; Bernabei, R.; Landi, F. Interleukin-6, C-reactive protein, and tumor necrosis factor-alpha as predictors of mortality in frail, community-living elderly individuals. J. Am. Geriatr. Soc. 2011, 59, 1679–1685. [Google Scholar] [CrossRef]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Baraldo, S.; Bazzan, E.; Zanin, M.E.; Turato, G.; Garbisa, S.; Maestrelli, P.; Papi, A.; Miniati, M.; Fabbri, L.M.; Zuin, R.; et al. Matrix metalloproteinase-2 protein in lung periphery is related to COPD progression. Chest 2007, 132, 1733–1740. [Google Scholar] [CrossRef]
- Ranes, J.; Stoller, J.K. A review of alpha-1 antitrypsin deficiency. Semin. Respir. Crit. Care Med. 2005, 26, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Marwick, J.A.; Chung, K.F. Glucocorticoid insensitivity as a future target of therapy for chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2010, 5, 297–309. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2011, 163, 29–43. [Google Scholar] [CrossRef]
- Vernooy, J.H.; Lindeman, J.H.; Jacobs, J.A.; Hanemaaijer, R.; Wouters, E.F. Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with COPD. Chest 2004, 126, 1802–1810. [Google Scholar] [CrossRef]
- Omachi, T.A.; Eisner, M.D.; Rames, A.; Markovtsova, L.; Blanc, P.D. Matrix metalloproteinase-9 predicts pulmonary status declines in alpha1-antitrypsin deficiency. Respir. Res. 2011, 12, 35. [Google Scholar] [CrossRef]
- Harman, D. The Free Radical Theory of Aging: Effect of Age on Serum Copper Levels. J. Gerontol. 1965, 20, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N. The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef]
- Doonan, R.; McElwee, J.J.; Matthijssens, F.; Walker, G.A.; Houthoofd, K.; Back, P.; Matscheski, A.; Vanfleteren, J.R.; Gems, D. Against the oxidative damage theory of aging: Superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008, 22, 3236–3241. [Google Scholar] [CrossRef]
- Mesquita, A.; Weinberger, M.; Silva, A.; Sampaio-Marques, B.; Almeida, B.; Leao, C.; Costa, V.; Rodrigues, F.; Burhans, W.C.; Ludovico, P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 15123–15128. [Google Scholar] [CrossRef]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Feghali-Bostwick, C.; Lasky, J.A.; Sanchez, C.G. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 595–602. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Y.; Roth, M.; Zhang, L.; Shi, R.; Yang, X.; Li, Y.; Zhang, J. Sirtuin 3 Inhibits Airway Epithelial Mitochondrial Oxidative Stress in Cigarette Smoke-Induced COPD. Oxid. Med. Cell. Longev. 2020, 2020, 7582980. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent Notch signaling. Cell Stem Cell 2014, 15, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Swamy, S.M.; Rajasekaran, N.S.; Thannickal, V.J. Nuclear Factor-Erythroid-2-Related Factor 2 in Aging and Lung Fibrosis. Am. J. Pathol. 2016, 186, 1712–1723. [Google Scholar] [CrossRef]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Rajendrasozhan, S.; Yao, H.; Chung, S.; Sundar, I.K.; Huyck, H.L.; Pryhuber, G.S.; Kinnula, V.L.; Rahman, I. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 2011, 187, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Paredi, P.; Kharitonov, S.A.; Barnes, P.J. Analysis of expired air for oxidation products. Am. J. Respir. Crit. Care Med. 2002, 166, S31–S37. [Google Scholar] [CrossRef] [PubMed]
- Paredi, P.; Kharitonov, S.A.; Leak, D.; Ward, S.; Cramer, D.; Barnes, P.J. Exhaled ethane, a marker of lipid peroxidation, is elevated in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2000, 162, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154, 813–816. [Google Scholar] [CrossRef]
- Corradi, M.; Pignatti, P.; Manini, P.; Andreoli, R.; Goldoni, M.; Poppa, M.; Moscato, G.; Balbi, B.; Mutti, A. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur. Respir. J. 2004, 24, 1011–1017. [Google Scholar] [CrossRef]
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; Macnee, W.; Rahman, I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 633–642. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Ilumets, H.; Myllarniemi, M.; Sovijarvi, A.; Rytila, P. 8-Isoprostane as a marker of oxidative stress in nonsymptomatic cigarette smokers and COPD. Eur. Respir. J. 2007, 29, 51–55. [Google Scholar] [CrossRef]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003, 58, 294–298. [Google Scholar] [CrossRef]
- Seemungal, T.A.; Donaldson, G.C.; Paul, E.A.; Bestall, J.C.; Jeffries, D.J.; Wedzicha, J.A. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.A.; Johansen, M.B.; Olsen, M.; Xu, X.; Parker, J.M.; Molfino, N.A.; Lash, T.L.; Sorensen, H.T.; Christiansen, C.F. The impact of exacerbation frequency on mortality following acute exacerbations of COPD: A registry-based cohort study. BMJ Open 2014, 4, e006720. [Google Scholar] [CrossRef] [PubMed]
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300. [Google Scholar] [CrossRef]
- Tanabe, N.; Muro, S.; Hirai, T.; Oguma, T.; Terada, K.; Marumo, S.; Kinose, D.; Ogawa, E.; Hoshino, Y.; Mishima, M. Impact of exacerbations on emphysema progression in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1653–1659. [Google Scholar] [CrossRef]
- Woodhead, M.; Blasi, F.; Ewig, S.; Huchon, G.; Ieven, M.; Ortqvist, A.; Schaberg, T.; Torres, A.; van der Heijden, G.; Verheij, T.J.; et al. Guidelines for the management of adult lower respiratory tract infections. Eur. Respir. J. 2005, 26, 1138–1180. [Google Scholar] [CrossRef]
- Tanabe, N.; Hoshino, Y.; Marumo, S.; Kiyokawa, H.; Sato, S.; Kinose, D.; Uno, K.; Muro, S.; Hirai, T.; Yodoi, J.; et al. Thioredoxin-1 protects against neutrophilic inflammation and emphysema progression in a mouse model of chronic obstructive pulmonary disease exacerbation. PLoS ONE 2013, 8, e79016. [Google Scholar] [CrossRef]
- Lomas, D.A. Does Protease-Antiprotease Imbalance Explain Chronic Obstructive Pulmonary Disease? Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 2), S130–S137. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Pavlisko, E.; Voynow, J.A. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstruct. Pulmon. Dis. 2011, 6, 413–421. [Google Scholar] [CrossRef]
- Rahman, I.; Morrison, D.; Donaldson, K.; MacNee, W. Systemic oxidative stress in asthma, COPD, and smokers. Am. J. Respir. Crit. Care Med. 1996, 154, 1055–1060. [Google Scholar] [CrossRef]
- Liu, J.; Huang, J.; Liu, H.; Chen, C.; Xu, J.; Zhong, L. Elevated serum 4HNE plus decreased serum thioredoxin: Unique feature and implications for acute exacerbation of chronic obstructive pulmonary disease. PLoS ONE 2021, 16, e0245810. [Google Scholar] [CrossRef]
- Bernardo, I.; Bozinovski, S.; Vlahos, R. Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol. Ther. 2015, 155, 60–79. [Google Scholar] [CrossRef]
- Barreiro, E.; Peinado, V.I.; Galdiz, J.B.; Ferrer, E.; Marin-Corral, J.; Sanchez, F.; Gea, J.; Barbera, J.A. Cigarette smoke-induced oxidative stress: A role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am. J. Respir. Crit. Care Med. 2010, 182, 477–488. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Muro, S.; Oguma, T.; Sato, S.; Tanabe, N.; Takahashi, T.; Kudo, M.; Kinose, D.; Kondoh, H.; Kubo, T.; et al. Impact of COPD exacerbations on osteoporosis assessed by chest CT scan. COPD J. Chronic Obstr. Pulm. Dis. 2012, 9, 235–242. [Google Scholar] [CrossRef]
- Tanimura, K.; Sato, S.; Sato, A.; Tanabe, N.; Hasegawa, K.; Uemasu, K.; Hamakawa, Y.; Oguma, T.; Muro, S.; Hirai, T. Accelerated Loss of Antigravity Muscles Is Associated with Mortality in Patients with COPD. Respiration 2020, 99, 298–306. [Google Scholar] [CrossRef] [PubMed]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Matsumoto, K.; Gon, Y.; Furuichi, S.; Maruoka, S.; Takeshita, I.; Hirota, K.; Yodoi, J.; Horie, T. Thioredoxin negatively regulates p38 MAP kinase activation and IL-6 production by tumor necrosis factor-alpha. Biochem. Biophys. Res. Commun. 1999, 258, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hoshino, Y.; Hara, T.; Muro, S.; Nakamura, H.; Mishima, M.; Yodoi, J. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J. Pharmacol. Exp. Ther. 2008, 325, 380–388. [Google Scholar] [CrossRef]
- Laurent, T.C.; Moore, E.C.; Reichard, P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia Coli B. J. Biol. Chem. 1964, 239, 3436–3444. [Google Scholar] [CrossRef]
- Nakamura, H.; Matsuda, M.; Furuke, K.; Kitaoka, Y.; Iwata, S.; Toda, K.; Inamoto, T.; Yamaoka, Y.; Ozawa, K.; Yodoi, J. Adult T cell leukemia-derived factor/human thioredoxin protects endothelial F-2 cell injury caused by activated neutrophils or hydrogen peroxide. Immunol. Lett. 1994, 42, 75–80. [Google Scholar] [CrossRef]
- Nakamura, H.; Nakamura, K.; Yodoi, J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997, 15, 351–369. [Google Scholar] [CrossRef]
- Nakamura, H.; Herzenberg, L.A.; Bai, J.; Araya, S.; Kondo, N.; Nishinaka, Y.; Herzenberg, L.A.; Yodoi, J. Circulating thioredoxin suppresses lipopolysaccharide-induced neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2001, 98, 15143–15148. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Iwata, S.; Nakamura, K.; Hori, T.; Mori, K.; Yodoi, J. Thiol-mediated redox regulation of apoptosis. Possible roles of cellular thiols other than glutathione in T cell apoptosis. J. Immunol. 1995, 154, 3194–3203. [Google Scholar]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Matsui, M.; Iwata, S.; Nishiyama, A.; Mori, K.; Yodoi, J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA 1997, 94, 3633–3638. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Ogiwara, H.; Hayashi, T.; Mitsui, A.; Kawabe, T.; Yodoi, J. Human thioredoxin/adult T cell leukemia-derived factor activates the enhancer binding protein of human immunodeficiency virus type 1 by thiol redox control mechanism. Int. Immunol. 1992, 4, 811–819. [Google Scholar] [CrossRef]
- Ueno, M.; Masutani, H.; Arai, R.J.; Yamauchi, A.; Hirota, K.; Sakai, T.; Inamoto, T.; Yamaoka, Y.; Yodoi, J.; Nikaido, T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 1999, 274, 35809–35815. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef]
- Kondo, N.; Ishii, Y.; Kwon, Y.W.; Tanito, M.; Horita, H.; Nishinaka, Y.; Nakamura, H.; Yodoi, J. Redox-sensing release of human thioredoxin from T lymphocytes with negative feedback loops. J. Immunol. 2004, 172, 442–448. [Google Scholar] [CrossRef]
- Kasuno, K.; Nakamura, H.; Ono, T.; Muso, E.; Yodoi, J. Protective roles of thioredoxin, a redox-regulating protein, in renal ischemia/reperfusion injury. Kidney Int. 2003, 64, 1273–1282. [Google Scholar] [CrossRef]
- Nakamura, H.; Tamura, S.; Watanabe, I.; Iwasaki, T.; Yodoi, J. Enhanced resistancy of thioredoxin-transgenic mice against influenza virus-induced pneumonia. Immunol. Lett. 2002, 82, 165–170. [Google Scholar] [CrossRef]
- Okuyama, H.; Nakamura, H.; Shimahara, Y.; Uyama, N.; Kwon, Y.W.; Kawada, N.; Yamaoka, Y.; Yodoi, J. Overexpression of thioredoxin prevents thioacetamide-induced hepatic fibrosis in mice. J. Hepatol. 2005, 42, 117–123. [Google Scholar] [CrossRef]
- Ueda, S.; Nakamura, T.; Yamada, A.; Teratani, A.; Matsui, N.; Furukawa, S.; Hoshino, Y.; Narita, M.; Yodoi, J.; Nakamura, H. Recombinant human thioredoxin suppresses lipopolysaccharide-induced bronchoalveolar neutrophil infiltration in rat. Life Sci. 2006, 79, 1170–1177. [Google Scholar] [CrossRef]
- Hattori, I.; Takagi, Y.; Nakamura, H.; Nozaki, K.; Bai, J.; Kondo, N.; Sugino, T.; Nishimura, M.; Hashimoto, N.; Yodoi, J. Intravenous administration of thioredoxin decreases brain damage following transient focal cerebral ischemia in mice. Antioxid. Redox Signal. 2004, 6, 81–87. [Google Scholar] [CrossRef]
- Ohashi, S.; Nishio, A.; Nakamura, H.; Kido, M.; Ueno, S.; Uza, N.; Inoue, S.; Kitamura, H.; Kiriya, K.; Asada, M.; et al. Protective roles of redox-active protein thioredoxin-1 for severe acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G772–G781. [Google Scholar] [CrossRef]
- Farina, A.R.; Tacconelli, A.; Cappabianca, L.; Masciulli, M.P.; Holmgren, A.; Beckett, G.J.; Gulino, A.; Mackay, A.R. Thioredoxin alters the matrix metalloproteinase/tissue inhibitors of metalloproteinase balance and stimulates human SK-N-SH neuroblastoma cell invasion. Eur. J. Biochem. 2001, 268, 405–413. [Google Scholar] [CrossRef]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “inflame-aging”. Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef]
- Miwa, K.; Kishimoto, C.; Nakamura, H.; Makita, T.; Ishii, K.; Okuda, N.; Yodoi, J.; Sasayama, S. Serum thioredoxin and alpha-tocopherol concentrations in patients with major risk factors. Circ. J. 2005, 69, 291–294. [Google Scholar] [CrossRef]
- Lehtonen, S.T.; Ohlmeier, S.; Kaarteenaho-Wiik, R.; Harju, T.; Paakko, P.; Soini, Y.; Kinnula, V.L. Does the oxidative stress in chronic obstructive pulmonary disease cause thioredoxin/peroxiredoxin oxidation? Antioxid. Redox Signal. 2008, 10, 813–819. [Google Scholar] [CrossRef]
- Hurst, J.R.; Vestbo, J.; Anzueto, A.; Locantore, N.; Mullerova, H.; Tal-Singer, R.; Miller, B.; Lomas, D.A.; Agusti, A.; Macnee, W.; et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N. Engl. J. Med. 2010, 363, 1128–1138. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Balansky, R.B.; D’Agostini, F.; Zanacchi, P.; De Flora, S. Protection by N-acetylcysteine of the histopathological and cytogenetical damage produced by exposure of rats to cigarette smoke. Cancer Lett. 1992, 64, 123–131. [Google Scholar] [CrossRef]
- Huber, G.L.; Davies, P.; Zwilling, G.R.; Pochay, V.E.; Hinds, W.C.; Nicholas, H.A.; Mahajan, V.K.; Hayashi, M.; First, M.W. A morphologic and physiologic bioassay for quantifying alterations in the lung following experimental chronic inhalation of tobacco smoke. Bull. Eur. Physiopathol. Respir. 1981, 17, 269–327. [Google Scholar]
- Jeffery, P.K.; Rogers, D.F.; Ayers, M.M. Effect of oral acetylcysteine on tobacco smoke-induced secretory cell hyperplasia. Eur. J. Respir. Dis. Suppl. 1985, 139, 117–122. [Google Scholar] [PubMed]
- Rogers, D.F.; Godfrey, R.W.; Majumdar, S.; Jeffery, P.K. Oral N-acetylcysteine speeds reversal of cigarette smoke-induced mucous cell hyperplasia in the rat. Exp. Lung Res. 1988, 14, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Messier, E.M.; Day, B.J.; Bahmed, K.; Kleeberger, S.R.; Tuder, R.M.; Bowler, R.P.; Chu, H.W.; Mason, R.J.; Kosmider, B. N-acetylcysteine protects murine alveolar type II cells from cigarette smoke injury in a nuclear erythroid 2-related factor-2-independent manner. Am. J. Respir. Cell Mol. Biol. 2013, 48, 559–567. [Google Scholar] [CrossRef]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Decramer, M.; Rutten-van Molken, M.; Dekhuijzen, P.N.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.; Olivieri, D.; Del Donno, M.; De Backer, W.; et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): A randomised placebo-controlled trial. Lancet 2005, 365, 1552–1560. [Google Scholar] [CrossRef]
- Zheng, J.P.; Wen, F.Q.; Bai, C.X.; Wan, H.Y.; Kang, J.; Chen, P.; Yao, W.Z.; Ma, L.J.; Xia, Q.K.; Gao, Y.; et al. High-dose N-acetylcysteine in the prevention of COPD exacerbations: Rationale and design of the PANTHEON Study. COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Zheng, J.; Criner, G.J.; Fabbri, L.M.; Calverley, P.M.A. Impact of smoking status and concomitant medications on the effect of high-dose N-acetylcysteine on chronic obstructive pulmonary disease exacerbations: A post-hoc analysis of the PANTHEON study. Respir. Med. 2019, 147, 37–43. [Google Scholar] [CrossRef]
- Cazzola, M.; Calzetta, L.; Page, C.; Jardim, J.; Chuchalin, A.G.; Rogliani, P.; Matera, M.G. Influence of N-acetylcysteine on chronic bronchitis or COPD exacerbations: A meta-analysis. Eur. Respir. Rev. 2015, 24, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef]
- Walters, J.A.; Tan, D.J.; White, C.J.; Gibson, P.G.; Wood-Baker, R.; Walters, E.H. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2014, CD001288. [Google Scholar] [CrossRef]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000, 20, 6891–6903. [Google Scholar] [CrossRef] [PubMed]
- Renkema, T.E.; Schouten, J.P.; Koeter, G.H.; Postma, D.S. Effects of long-term treatment with corticosteroids in COPD. Chest 1996, 109, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Ichinose, M.; Yamagata, S.; Koarai, A.; Shirato, K.; Hattori, T. Correlation between change in pulmonary function and suppression of reactive nitrogen species production following steroid treatment in COPD. Thorax 2003, 58, 299–305. [Google Scholar] [CrossRef][Green Version]
- Profita, M.; Albano, G.D.; Montalbano, A.M.; Di Sano, C.; Anzalone, G.; Gagliardo, R.; Riccobono, L.; Bonanno, A.; Siena, L.; Pieper, M.P.; et al. Acetylcholine leads to signal transducer and activator of transcription 1 (STAT-1) mediated oxidative/nitrosative stress in human bronchial epithelial cell line. Biochim. Biophys. Acta 2013, 1832, 1949–1958. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiyokawa, H.; Hoshino, Y.; Sakaguchi, K.; Muro, S.; Yodoi, J. Redox Regulation in Aging Lungs and Therapeutic Implications of Antioxidants in COPD. Antioxidants 2021, 10, 1429. https://doi.org/10.3390/antiox10091429
Kiyokawa H, Hoshino Y, Sakaguchi K, Muro S, Yodoi J. Redox Regulation in Aging Lungs and Therapeutic Implications of Antioxidants in COPD. Antioxidants. 2021; 10(9):1429. https://doi.org/10.3390/antiox10091429
Chicago/Turabian StyleKiyokawa, Hirofumi, Yuma Hoshino, Kazuhiro Sakaguchi, Shigeo Muro, and Junji Yodoi. 2021. "Redox Regulation in Aging Lungs and Therapeutic Implications of Antioxidants in COPD" Antioxidants 10, no. 9: 1429. https://doi.org/10.3390/antiox10091429
APA StyleKiyokawa, H., Hoshino, Y., Sakaguchi, K., Muro, S., & Yodoi, J. (2021). Redox Regulation in Aging Lungs and Therapeutic Implications of Antioxidants in COPD. Antioxidants, 10(9), 1429. https://doi.org/10.3390/antiox10091429