
Reactive Oxygen Species and Antioxidative Defense in Chronic Obstructive Pulmonary Disease



Abstract
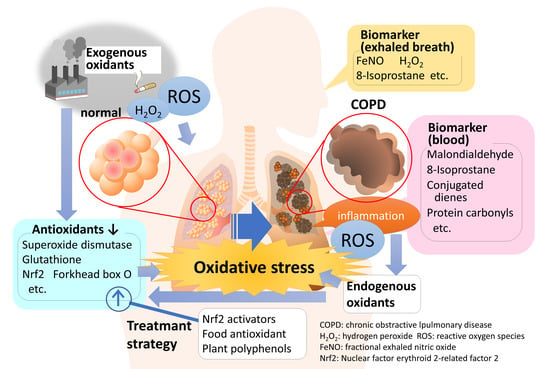
1. Introduction
2. Cigarette Smoke and Oxidative Stress in COPD
3. Other Sources of Oxidative Stress in COPD
4. Oxidative Stress in the Development and Progression of COPD
5. Oxidative Stress and Vascular Endothelial Dysfunction
6. Citrulline–Arginine and Arginine–Ornithine Pathways
7. Oxidative Stress and Oxidative Stress Biomarkers
8. Evaluation of Oxidative Stress Status in the Blood of Patients with COPD
9. Oxidative Stress Biomarkers in Exhaled Breath of Patients with COPD
10. Oxidative Stress and Genetic Factors
11. Treatment Strategy for COPD Focusing on Oxidative Stress
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chapman, K.R.; Mannino, D.M.; Soriano, J.B.; Vermeire, P.A.; Buist, A.S.; Thun, M.J.; Connell, C.; Jemal, A.; Lee, T.A.; Miravitlles, M.; et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 188–207. [Google Scholar] [CrossRef]
- Soriano, J.B.; Abajobir, A.A.; Abate, K.H.; Abera, S.F.; Agrawal, A.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; Aichour, M.T.; Alam, K.; et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir. Med. 2017, 5, 691–706. [Google Scholar] [CrossRef]
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2021 Report). Available online: https://goldcopd.org/2021-gold-reports/ (accessed on 15 July 2021).
- Szalontai, K.; Gemes, N.; Furak, J.; Varga, T.; Neuperger, P.; Balog, J.A.; Puskas, L.G.; Szebeni, G.J. Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer. J. Clin. Med. 2021, 10, 2889. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, A.; Miyahara, N.; Oda, N.; Morichika, D.; Ichihara, E.; Oze, I.; Tanimoto, Y.; Ichikawa, H.; Fujii, U.; Tanimoto, M.; et al. Protective Effects of Bisoprolol against Acute Exacerbation in Moderate-to-Severe Chronic Obstructive Pulmonary Disease. Acta Med. Okayama 2017, 71, 453–457. [Google Scholar] [CrossRef]
- Oda, N.; Miyahara, N.; Ichikawa, H.; Tanimoto, Y.; Kajimoto, K.; Sakugawa, M.; Kawai, H.; Taniguchi, A.; Morichika, D.; Tanimoto, M.; et al. Long-term effects of beta-blocker use on lung function in Japanese patients with chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1119–1124. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef]
- Fischer, B.M.; Voynow, J.A.; Ghio, A.J. COPD: Balancing oxidants and antioxidants. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef]
- Hou, H.H.; Cheng, S.L.; Chung, K.P.; Kuo, M.Y.; Yeh, C.C.; Chang, B.E.; Lu, H.H.; Wang, H.C.; Yu, C.J. Elastase induces lung epithelial cell autophagy through placental growth factor: A new insight of emphysema pathogenesis. Autophagy 2014, 10, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Rennard, S.I.; Vestbo, J. COPD: The dangerous underestimate of 15%. Lancet 2006, 367, 1216–1219. [Google Scholar] [CrossRef]
- Rutgers, S.R.; Postma, D.S.; ten Hacken, N.H.; Kauffman, H.F.; van Der Mark, T.W.; Koëter, G.H.; Timens, W. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000, 55, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, E.; Miyahara, N.; Kanehiro, A.; Waseda, K.; Taniguchi, A.; Ikeda, G.; Koga, H.; Nishimori, H.; Tanimoto, Y.; Kataoka, M.; et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir. Res. 2013, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Fujii, U.; Miyahara, N.; Taniguchi, A.; Waseda, K.; Morichika, D.; Kurimoto, E.; Koga, H.; Kataoka, M.; Gelfand, E.W.; Cua, D.J.; et al. IL-23 Is Essential for the Development of Elastase-Induced Pulmonary Inflammation and Emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 55, 697–707. [Google Scholar] [CrossRef]
- Waseda, K.; Miyahara, N.; Taniguchi, A.; Kurimoto, E.; Ikeda, G.; Koga, H.; Fujii, U.; Yamamoto, Y.; Gelfand, E.W.; Yamamoto, H.; et al. Emphysema requires the receptor for advanced glycation end-products triggering on structural cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 482–491. [Google Scholar] [CrossRef]
- Morichika, D.; Miyahara, N.; Fujii, U.; Taniguchi, A.; Oda, N.; Senoo, S.; Kataoka, M.; Tanimoto, M.; Kakuta, H.; Kiura, K.; et al. A retinoid X receptor partial agonist attenuates pulmonary emphysema and airway inflammation. Respir. Res. 2019, 20, 2. [Google Scholar] [CrossRef]
- Morichika, D.; Taniguchi, A.; Oda, N.; Fujii, U.; Senoo, S.; Itano, J.; Kanehiro, A.; Kitaguchi, Y.; Yasuo, M.; Hanaoka, M.; et al. Loss of IL-33 enhances elastase-induced and cigarette smoke extract-induced emphysema in mice. Respir. Res. 2021, 22, 150. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef]
- Cho, H.Y.; Kleeberger, S.R. Nrf2 protects against airway disorders. Toxicol. Appl. Pharmacol. 2010, 244, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Stämpfli, M.R.; Anderson, G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef]
- Pryor, W.A. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ. Health Perspect. 1997, 105 (Suppl. 4), 875–882. [Google Scholar] [CrossRef]
- Churg, A.; Cosio, M.; Wright, J.L. Mechanisms of cigarette smoke-induced COPD: Insights from animal models. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L612–L631. [Google Scholar] [CrossRef]
- MacNee, W. Oxidants and COPD. Curr. Drug Targets Inflamm. Allergy 2005, 4, 627–641. [Google Scholar] [CrossRef]
- MacNee, W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 50–60. [Google Scholar] [CrossRef]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef]
- Pryor, W.A. Cigarette smoke and the involvement of free radical reactions in chemical carcinogenesis. Br. J. Cancer Suppl. 1987, 8, 19–23. [Google Scholar]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef]
- Song, Q.; Chen, P.; Liu, X.M. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir. Res. 2021, 22, 39. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef]
- Gensch, E.; Gallup, M.; Sucher, A.; Li, D.; Gebremichael, A.; Lemjabbar, H.; Mengistab, A.; Dasari, V.; Hotchkiss, J.; Harkema, J.; et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J. Biol. Chem. 2004, 279, 39085–39093. [Google Scholar] [CrossRef]
- Di, Y.P.; Zhao, J.; Harper, R. Cigarette smoke induces MUC5AC protein expression through the activation of Sp1. J. Biol. Chem. 2012, 287, 27948–27958. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Schwalm, K.; Smith, K.R.; Schuyler, M.; Tesfaigzi, Y. Cigarette smoke suppresses Bik to cause epithelial cell hyperplasia and mucous cell metaplasia. Am. J. Respir. Crit. Care Med. 2011, 183, 1531–1538. [Google Scholar] [CrossRef]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Siddharthan, T.; Gupte, A.; Barnes, P.J. Chronic Obstructive Pulmonary Disease Endotypes in Low- and Middle-Income Country Settings: Precision Medicine for All. Am. J. Respir. Crit. Care Med. 2020, 202, 171–172. [Google Scholar] [CrossRef]
- Hagstad, S.; Backman, H.; Bjerg, A.; Ekerljung, L.; Ye, X.; Hedman, L.; Lindberg, A.; Torén, K.; Lötvall, J.; Rönmark, E.; et al. Prevalence and risk factors of COPD among never-smokers in two areas of Sweden—Occupational exposure to gas, dust or fumes is an important risk factor. Respir. Med. 2015, 109, 1439–1445. [Google Scholar] [CrossRef]
- Thomsen, M.; Nordestgaard, B.G.; Vestbo, J.; Lange, P. Characteristics and outcomes of chronic obstructive pulmonary disease in never smokers in Denmark: A prospective population study. Lancet Respir. Med. 2013, 1, 543–550. [Google Scholar] [CrossRef]
- Shin, S.; Bai, L.; Burnett, R.T.; Kwong, J.C.; Hystad, P.; van Donkelaar, A.; Lavigne, E.; Weichenthal, S.; Copes, R.; Martin, R.V.; et al. Air Pollution as a Risk Factor for Incident Chronic Obstructive Pulmonary Disease and Asthma. A 15-Year Population-based Cohort Study. Am. J. Respir. Crit. Care Med. 2021, 203, 1138–1148. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, Z.; Lau, A.K.H.; Lin, C.Q.; Chuang, Y.C.; Chan, J.; Jiang, W.K.; Tam, T.; Yeoh, E.K.; Chan, T.C.; et al. Effect of long-term exposure to fine particulate matter on lung function decline and risk of chronic obstructive pulmonary disease in Taiwan: A longitudinal, cohort study. Lancet Planet. Health 2018, 2, e114–e125. [Google Scholar] [CrossRef]
- Andersen, Z.J.; Hvidberg, M.; Jensen, S.S.; Ketzel, M.; Loft, S.; Sørensen, M.; Tjønneland, A.; Overvad, K.; Raaschou-Nielsen, O. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution: A cohort study. Am. J. Respir. Crit. Care Med. 2011, 183, 455–461. [Google Scholar] [CrossRef]
- Brassington, K.; Selemidis, S.; Bozinovski, S.; Vlahos, R. New frontiers in the treatment of comorbid cardiovascular disease in chronic obstructive pulmonary disease. Clin. Sci. 2019, 133, 885–904. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, M.C.; Brown, R.; Ryan, S.; Mall, M.A.; Weldon, S.; Taggart, C.C. Proteases, Mucus, and Mucosal Immunity in Chronic Lung Disease. Int. J. Mol. Sci. 2021, 22, 5018. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576. [Google Scholar] [CrossRef]
- Hwang, J.W.; Rajendrasozhan, S.; Yao, H.; Chung, S.; Sundar, I.K.; Huyck, H.L.; Pryhuber, G.S.; Kinnula, V.L.; Rahman, I. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 2011, 187, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Role of Nrf2 in the pathogenesis of respiratory diseases. Respir. Investig. 2020, 58, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, Y.D.; Moon, U.Y.; Kim, J.H.; Jeon, J.H.; Lee, J.G.; Bae, Y.S.; Yoon, J.H. The role of Nox4 in oxidative stress-induced MUC5AC overexpression in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, E.; Chen, J.; Brodsky, S.V.; Smirnova, I.; Li, H.; Goligorsky, M.S. Endothelial cell dysfunction: The syndrome in making. Kidney Int. 2005, 67, 1654–1658. [Google Scholar] [CrossRef]
- Zatz, R.; Baylis, C. Chronic nitric oxide inhibition model six years on. Hypertension 1998, 32, 958–964. [Google Scholar] [CrossRef]
- Tsukahara, H.; Hiraoka, M.; Kobata, R.; Hata, I.; Ohshima, Y.; Jiang, M.Z.; Noiri, E.; Mayumi, M. Increased oxidative stress in rats with chronic nitric oxide depletion: Measurement of urinary 8-hydroxy-2’-deoxyguanosine excretion. Redox Rep. 2000, 5, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef]
- Schulze, F.; Wesemann, R.; Schwedhelm, E.; Sydow, K.; Albsmeier, J.; Cooke, J.P.; Böger, R.H. Determination of asymmetric dimethylarginine (ADMA) using a novel ELISA assay. Clin. Chem. Lab. Med. 2004, 42, 1377–1383. [Google Scholar] [CrossRef]
- Tsukahara, H.; Ohta, N.; Tokuriki, S.; Nishijima, K.; Kotsuji, F.; Kawakami, H.; Ohta, N.; Sekine, K.; Nagasaka, H.; Mayumi, M. Determination of asymmetric dimethylarginine, an endogenous nitric oxide synthase inhibitor, in umbilical blood. Metabolism 2008, 57, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Celli, B.R.; Owen, C.A. COPD as an endothelial disorder: Endothelial injury linking lesions in the lungs and other organs? (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018758528. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Fois, A.G.; Mangoni, A.A.; Paliogiannis, P.; Sotgiu, E.; Zinellu, E.; Marras, V.; Pirina, P.; Carru, C. Systemic concentrations of asymmetric dimethylarginine (ADMA) in chronic obstructive pulmonary disease (COPD): State of the art. Amino Acids 2018, 50, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.; Altintas, N.; Cem Mutlu, L.; Bilir, B.; Oran, M.; Tülübaş, F.; Topçu, B.; Tayfur, İ.; Küçükyalçin, V.; Kaplan, G.; et al. Asymmetric dimethylarginine contributes to airway nitric oxide deficiency in patients with COPD. Clin. Respir. J. 2017, 11, 318–327. [Google Scholar] [CrossRef]
- Vögeli, A.; Ottiger, M.; Meier, M.A.; Steuer, C.; Bernasconi, L.; Huber, A.; Christ-Crain, M.; Henzen, C.; Hoess, C.; Thomann, R.; et al. Asymmetric Dimethylarginine Predicts Long-Term Outcome in Patients with Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Lung 2017, 195, 717–727. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef]
- Kubo, M.; Ogino, K. Analytical Procedures for Nitrative/Nitrosative Stress. In Studies on Pediatric Disorders; Tsukahara, H., Kaneko, K., Eds.; Springer: New York, NY, USA, 2014; pp. 149–158. [Google Scholar]
- Meurs, H.; Zaagsma, J.; Maarsingh, H.; van Duin, M. Recent Patents in Allergy/Immunology: Use of arginase inhibitors in the treatment of asthma and allergic rhinitis. Allergy 2019, 74, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Grenfell, A.; Nieto-Velázquez, N.; Torres-Ramos, Y.; Montoya-Estrada, A.; Ramírez-Venegas, A.; Ochoa-Cautiño, L.; Flores-Trujillo, F.; Hicks, J.J. Increased platelet and erythrocyte arginase activity in chronic obstructive pulmonary disease associated with tobacco or wood smoke exposure. J. Investig. Med. 2011, 59, 587–592. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, M.P.; Meurs, H.; Gosens, R. Targeting arginase and nitric oxide metabolism in chronic airway diseases and their co-morbidities. Curr. Opin. Pharmacol. 2018, 40, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Pera, T.; Zuidhof, A.B.; Smit, M.; Menzen, M.H.; Klein, T.; Flik, G.; Zaagsma, J.; Meurs, H.; Maarsingh, H. Arginase inhibition prevents inflammation and remodeling in a guinea pig model of chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 2014, 349, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.A.; Maarsingh, H.; Holguin, F.; Grasemann, H. Arginine Therapy for Lung Diseases. Front. Pharmacol. 2021, 12, 627503. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Toyokuni, S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol. Int. 1999, 49, 91–102. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Ash, D.; Nagarkoti, S.; Belin de Chantemèle, E.J.; Fulton, D.J.R.; Fukai, T. Interplay Between Reactive Oxygen/Reactive Nitrogen Species and Metabolism in Vascular Biology and Disease. Antioxid. Redox Signal. 2021, 34, 1319–1354. [Google Scholar] [CrossRef]
- Tsukahara, H. Oxidative Stress Biomarkers: Current Status and Future Perspective. In Studies on Pediatric Disorders; Tsukahara, H., Kaneko, K., Eds.; Springer: New York, NY, USA, 2014; pp. 87–113. [Google Scholar]
- Noiri, E.; Tsukahara, H. Parameters for measurement of oxidative stress in diabetes mellitus: Applicability of enzyme-linked immunosorbent assay for clinical evaluation. J. Investig. Med. 2005, 53, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Tsukahara, H.; Ueno, M.; Maeda, M.; Kawakami, H.; Sekine, K.; Mayumi, M. Evaluation of a urinary multi-parameter biomarker set for oxidative stress in children, adolescents and young adults. Free Radic. Res. 2006, 40, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K. Rapid Diagnostic Tests for Oxidative Stress Status. In Studies on Pediatric Disorders; Tsukahara, H., Kaneko, K., Eds.; Springer: New York, NY, USA, 2014; pp. 137–148. [Google Scholar]
- Dodig, S.; Richter, D.; Zrinski-Topić, R. Inflammatory markers in childhood asthma. Clin. Chem. Lab. Med. 2011, 49, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Popov, T.A. Human exhaled breath analysis. Ann. Allergy Asthma Immunol. 2011, 106, 451–456. [Google Scholar] [CrossRef]
- Gelb, A.F.; Barnes, P.J.; George, S.C.; Ricciardolo, F.L.; DiMaria, G.; Zamel, N. Review of exhaled nitric oxide in chronic obstructive pulmonary disease. J. Breath Res. 2012, 6, 047101. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Milevoj Kopčinović, L.; Domijan, A.M.; Posavac, K.; Čepelak, I.; Žanić Grubišić, T.; Rumora, L. Systemic redox imbalance in stable chronic obstructive pulmonary disease. Biomarkers 2016, 21, 692–698. [Google Scholar] [CrossRef]
- Sunnetcioglu, A.; Alp, H.H.; Sertogullarından, B.; Balaharoglu, R.; Gunbatar, H. Evaluation of Oxidative Damage and Antioxidant Mechanisms in COPD, Lung Cancer, and Obstructive Sleep Apnea Syndrome. Respir. Care 2016, 61, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Arja, C.; Surapaneni, K.M.; Raya, P.; Adimoolam, C.; Balisetty, B.; Kanala, K.R. Oxidative stress and antioxidant enzyme activity in South Indian male smokers with chronic obstructive pulmonary disease. Respirology 2013, 18, 1069–1075. [Google Scholar] [CrossRef]
- Ahmad, A.; Shameem, M.; Husain, Q. Altered oxidant-antioxidant levels in the disease prognosis of chronic obstructive pulmonary disease. Int. J. Tuberc. Lung Dis. 2013, 17, 1104–1109. [Google Scholar] [CrossRef]
- Torres-Ramos, Y.D.; Guzman-Grenfell, A.M.; Montoya-Estrada, A.; Ramirez-Venegas, A.; Martinez, R.S.; Flores-Trujillo, F.; Ochoa-Cautino, L.; Hicks, J.J. RBC membrane damage and decreased band 3 phospho-tyrosine phosphatase activity are markers of COPD progression. Front. Biosci. (Elite Ed.) 2010, 2, 1385–1393. [Google Scholar] [CrossRef]
- Maury, J.; Gouzi, F.; De Rigal, P.; Heraud, N.; Pincemail, J.; Molinari, N.; Pomiès, P.; Laoudj-Chenivesse, D.; Mercier, J.; Préfaut, C.; et al. Heterogeneity of Systemic Oxidative Stress Profiles in COPD: A Potential Role of Gender. Oxid. Med. Cell. Longev. 2015, 2015, 201843. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, M.; Ciebiada, M.; Pękala-Wojciechowska, A.; Pawłowski, M.; Nielepkowicz-Goździńska, A.; Antczak, A. Evaluation of Markers of Inflammation and Oxidative Stress in COPD Patients with or without Cardiovascular Comorbidities. Heart Lung Circ. 2015, 24, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Raj, H.G.; Chhabra, S.K. Increased oxidative stress and altered levels of antioxidants in chronic obstructive pulmonary disease. Inflammation 2005, 29, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ceylan, E.; Kocyigit, A.; Gencer, M.; Aksoy, N.; Selek, S. Increased DNA damage in patients with chronic obstructive pulmonary disease who had once smoked or been exposed to biomass. Respir. Med. 2006, 100, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Stanojkovic, I.; Kotur-Stevuljevic, J.; Milenkovic, B.; Spasic, S.; Vujic, T.; Stefanovic, A.; Llic, A.; Ivanisevic, J. Pulmonary function, oxidative stress and inflammatory markers in severe COPD exacerbation. Respir. Med. 2011, 105 (Suppl. 1), S31–S37. [Google Scholar] [CrossRef]
- Stanojkovic, I.; Kotur-Stevuljevic, J.; Spasic, S.; Milenkovic, B.; Vujic, T.; Stefanovic, A.; Ivanisevic, J. Relationship between bone resorption, oxidative stress and inflammation in severe COPD exacerbation. Clin. Biochem. 2013, 46, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Shimoi, K.; Kasai, H.; Yokota, N.; Toyokuni, S.; Kinae, N. Comparison between high-performance liquid chromatography and enzyme-linked immunosorbent assay for the determination of 8-hydroxy-2′-deoxyguanosine in human urine. Cancer Epidemiol. Biomark. Prev. 2002, 11, 767–770. [Google Scholar]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef]
- Rahman, I.; Swarska, E.; Henry, M.; Stolk, J.; MacNee, W. Is there any relationship between plasma antioxidant capacity and lung function in smokers and in patients with chronic obstructive pulmonary disease? Thorax 2000, 55, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Ben Moussa, S.; Sfaxi, I.; Tabka, Z.; Ben Saad, H.; Rouatbi, S. Oxidative stress and lung function profiles of male smokers free from COPD compared to those with COPD: A case-control study. Libyan J. Med. 2014, 9, 23873. [Google Scholar] [CrossRef] [PubMed]
- Folchini, F.; Nonato, N.L.; Feofiloff, E.; D’Almeida, V.; Nascimento, O.; Jardim, J.R. Association of oxidative stress markers and C-reactive protein with multidimensional indexes in COPD. Chronic Respir. Dis. 2011, 8, 101–108. [Google Scholar] [CrossRef]
- Nicks, M.E.; O’Brien, M.M.; Bowler, R.P. Plasma antioxidants are associated with impaired lung function and COPD exacerbations in smokers. COPD 2011, 8, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Santos, M.C.; Oliveira, A.L.; Viegas-Crespo, A.M.; Vicente, L.; Barreiros, A.; Monteiro, P.; Pinheiro, T.; Bugalho De Almeida, A. Systemic markers of the redox balance in chronic obstructive pulmonary disease. Biomarkers 2004, 9, 461–469. [Google Scholar] [CrossRef]
- Lakhdar, R.; Denden, S.; Mouhamed, M.H.; Chalgoum, A.; Leban, N.; Knani, J.; Lefranc, G.; Miled, A.; Ben Chibani, J.; Khelil, A.H. Correlation of EPHX1, GSTP1, GSTM1, and GSTT1 genetic polymorphisms with antioxidative stress markers in chronic obstructive pulmonary disease. Exp. Lung Res. 2011, 37, 195–204. [Google Scholar] [CrossRef]
- Kluchová, Z.; Petrášová, D.; Joppa, P.; Dorková, Z.; Tkáčová, R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol. Res. 2007, 56, 51–56. [Google Scholar] [CrossRef]
- Hanta, I.; Kocabas, A.; Canacankatan, N.; Kuleci, S.; Seydaoglu, G. Oxidant-antioxidant balance in patients with COPD. Lung 2006, 184, 51–55. [Google Scholar] [CrossRef]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Strange, R.C.; Spiteri, M.A.; Ramachandran, S.; Fryer, A.A. Glutathione-S-transferase family of enzymes. Mutat. Res. 2001, 482, 21–26. [Google Scholar] [CrossRef]
- Vibhuti, A.; Arif, E.; Deepak, D.; Singh, B.; Qadar Pasha, M.A. Correlation of oxidative status with BMI and lung function in COPD. Clin. Biochem. 2007, 40, 958–963. [Google Scholar] [CrossRef]
- Rajkovic, M.G.; Rumora, L.; Barisic, K. The paraoxonase 1, 2 and 3 in humans. Biochem. Med. 2011, 21, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Isik, B.; Isik, R.S.; Ceylan, A.; Calik, O. Trace elements and oxidative stress in chronic obstructive pulmonary disease. Saudi Med. J. 2005, 26, 1882–1885. [Google Scholar]
- Tsukagoshi, H.; Shimizu, Y.; Iwamae, S.; Hisada, T.; Ishizuka, T.; Iizuka, K.; Dobashi, K.; Mori, M. Evidence of oxidative stress in asthma and COPD: Potential inhibitory effect of theophylline. Respir. Med. 2000, 94, 584–588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vibhuti, A.; Arif, E.; Mishra, A.; Deepak, D.; Singh, B.; Rahman, I.; Mohammad, G.; Pasha, M.A. CYP1A1, CYP1A2 and CYBA gene polymorphisms associated with oxidative stress in COPD. Clin. Chim. Acta 2010, 411, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, A.; Górecki, D.; Szpinda, M.; Mila-Kierzenkowska, C.; Woźniak, B. Oxidant-antioxidant balance in the blood of patients with chronic obstructive pulmonary disease after smoking cessation. Oxid. Med. Cell Longev. 2013, 2013, 897075. [Google Scholar] [CrossRef]
- ben Anes, A.; Fetoui, H.; Bchir, S.; ben Nasr, H.; Chahdoura, H.; Chabchoub, E.; Yacoub, S.; Garrouch, A.; Benzarti, M.; Tabka, Z.; et al. Increased oxidative stress and altered levels of nitric oxide and peroxynitrite in Tunisian patients with chronic obstructive pulmonary disease: Correlation with disease severity and airflow obstruction. Biol. Trace Elem. Res. 2014, 161, 20–31. [Google Scholar] [CrossRef]
- Zinellu, A.; Fois, A.G.; Sotgia, S.; Sotgiu, E.; Zinellu, E.; Bifulco, F.; Mangoni, A.A.; Pirina, P.; Carru, C. Arginines Plasma Concentration and Oxidative Stress in Mild to Moderate COPD. PLoS ONE 2016, 11, e0160237. [Google Scholar] [CrossRef]
- Singh, S.; Verma, S.K.; Kumar, S.; Ahmad, M.K.; Nischal, A.; Singh, S.K.; Dixit, R.K. Evaluation of Oxidative Stress and Antioxidant Status in Chronic Obstructive Pulmonary Disease. Scand J. Immunol. 2017, 85, 130–137. [Google Scholar] [CrossRef]
- Avci, E.; Avci, G.A. Important Biomarkers that Play a Role in the Chronic Obstructive Pulmonary Disease Process. J. Med. Biochem. 2018, 37, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Fois, A.G.; Sotgia, S.; Zinellu, E.; Bifulco, F.; Pintus, G.; Mangoni, A.A.; Carru, C.; Pirina, P. Plasma protein thiols: An early marker of oxidative stress in asthma and chronic obstructive pulmonary disease. Eur. J. Clin. Investig. 2016, 46, 181–188. [Google Scholar] [CrossRef]
- Ben Moussa, S.; Rouatbi, S.; Ben Saad, H. Incapacity, Handicap, and Oxidative Stress Markers of Male Smokers With and Without COPD. Respir. Care 2016, 61, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Fermoselle, C.; Mateu-Jimenez, M.; Sanchez-Font, A.; Pijuan, L.; Gea, J.; Curull, V. Oxidative stress and inflammation in the normal airways and blood of patients with lung cancer and COPD. Free Radic. Biol. Med. 2013, 65, 859–871. [Google Scholar] [CrossRef]
- Puig-Vilanova, E.; Rodriguez, D.A.; Lloreta, J.; Ausin, P.; Pascual-Guardia, S.; Broquetas, J.; Roca, J.; Gea, J.; Barreiro, E. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic. Biol. Med. 2015, 79, 91–108. [Google Scholar] [CrossRef]
- Gopal, P.; Reynaert, N.L.; Scheijen, J.L.; Engelen, L.; Schalkwijk, C.G.; Franssen, F.M.; Wouters, E.F.; Rutten, E.P. Plasma advanced glycation end-products and skin autofluorescence are increased in COPD. Eur. Respir. J. 2014, 43, 430–438. [Google Scholar] [CrossRef]
- Foschino Barbaro, M.P.; Carpagnano, G.E.; Spanevello, A.; Cagnazzo, M.G.; Barnes, P.J. Inflammation, oxidative stress and systemic effects in mild chronic obstructive pulmonary disease. Int. J. Immunopathol. Pharmacol. 2007, 20, 753–763. [Google Scholar] [CrossRef]
- Ekin, S.; Arısoy, A.; Gunbatar, H.; Sertogullarindan, B.; Sunnetcioglu, A.; Sezen, H.; Asker, S.; Yıldız, H. The relationships among the levels of oxidative and antioxidative parameters, FEV1 and prolidase activity in COPD. Redox Rep. 2017, 22, 74–77. [Google Scholar] [CrossRef]
- Yasuo, M.; Droma, Y.; Kitaguchi, Y.; Ito, M.; Imamura, H.; Kawakubo, M.; Hanaoka, M. The relationship between acrolein and oxidative stress in COPD: In systemic plasma and in local lung tissue. Int. J. Chron Obs. Pulmon Dis. 2019, 14, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Zinellu, E.; Sotgiu, E.; Fois, A.G.; Paliogiannis, P.; Scano, V.; Piras, B.; Sotgia, S.; Mangoni, A.A.; Carru, C.; et al. Systemic transsulfuration pathway thiol concentrations in chronic obstructive pulmonary disease patients. Eur. J. Clin. Investig. 2020, 50, e13267. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G. Systemic poly(ADP-ribose) polymerase-1 activation, chronic inflammation, and oxidative stress in COPD patients. Free Radic. Biol. Med. 2003, 35, 140–148. [Google Scholar] [CrossRef]
- Tavilani, H.; Nadi, E.; Karimi, J.; Goodarzi, M.T. Oxidative stress in COPD patients, smokers, and non-smokers. Respir. Care 2012, 57, 2090–2094. [Google Scholar] [CrossRef]
- Montuschi, P.; Barnes, P.J. Analysis of exhaled breath condensate for monitoring airway inflammation. Trends Pharmacol. Sci. 2002, 23, 232–237. [Google Scholar] [CrossRef]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Kostikas, K.; Papatheodorou, G.; Psathakis, K.; Panagou, P.; Loukides, S. Oxidative stress in expired breath condensate of patients with COPD. Chest 2003, 124, 1373–1380. [Google Scholar] [CrossRef]
- Gerritsen, W.B.; Asin, J.; Zanen, P.; van den Bosch, J.M.; Haas, F.J. Markers of inflammation and oxidative stress in exacerbated chronic obstructive pulmonary disease patients. Respir. Med. 2005, 99, 84–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graille, M.; Wild, P.; Sauvain, J.J.; Hemmendinger, M.; Guseva Canu, I.; Hopf, N.B. Urinary 8-isoprostane as a biomarker for oxidative stress. A systematic review and meta-analysis. Toxicol. Lett. 2020, 328, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003, 58, 294–298. [Google Scholar] [CrossRef]
- Makris, D.; Paraskakis, E.; Korakas, P.; Karagiannakis, E.; Sourvinos, G.; Siafakas, N.M.; Tzanakis, N. Exhaled breath condensate 8-isoprostane, clinical parameters, radiological indices and airway inflammation in COPD. Respiration 2008, 75, 138–144. [Google Scholar] [CrossRef]
- Pinamonti, S.; Muzzoli, M.; Chicca, M.C.; Papi, A.; Ravenna, F.; Fabbri, L.M.; Ciaccia, A. Xanthine oxidase activity in bronchoalveolar lavage fluid from patients with chronic obstructive pulmonary disease. Free Radic. Biol. Med. 1996, 21, 147–155. [Google Scholar] [CrossRef]
- Corradi, M.; Rubinstein, I.; Andreoli, R.; Manini, P.; Caglieri, A.; Poli, D.; Alinovi, R.; Mutti, A. Aldehydes in exhaled breath condensate of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Grisham, M.B.; Johnson, G.G.; Lancaster, J.R., Jr. Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzymol. 1996, 268, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Gaston, B.; Singel, D.; Doctor, A.; Stamler, J.S. S-nitrosothiol signaling in respiratory biology. Am. J. Respir. Crit. Care Med. 2006, 173, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Corradi, M.; Montuschi, P.; Donnelly, L.E.; Pesci, A.; Kharitonov, S.A.; Barnes, P.J. Increased nitrosothiols in exhaled breath condensate in inflammatory airway diseases. Am. J. Respir. Crit. Care Med. 2001, 163, 854–858. [Google Scholar] [CrossRef]
- Cho, M.H.; McDonald, M.-L.N.; Zhou, X.; Mattheisen, M.; Castaldi, P.J.; Hersh, C.P.; DeMeo, D.L.; Sylvia, J.S.; Ziniti, J.; Laird, N.M.; et al. Risk loci for chronic obstructive pulmonary disease: A genome-wide association study and meta-analysis. Lancet Respir. Med. 2014, 2, 214–225. [Google Scholar] [CrossRef]
- Postma, D.S.; Kerkhof, M.; Boezen, H.M.; Koppelman, G.H. Asthma and chronic obstructive pulmonary disease: Common genes, common environments? Am. J. Respir. Crit. Care Med. 2011, 183, 1588–1594. [Google Scholar] [CrossRef]
- Malhotra, D.; Thimmulappa, R.K.; Mercado, N.; Ito, K.; Kombairaju, P.; Kumar, S.; Ma, J.; Feller-Kopman, D.; Wise, R.; Barnes, P.; et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J. Clin. Investig. 2011, 121, 4289–4302. [Google Scholar] [CrossRef] [PubMed]
- Nakamaru, Y.; Vuppusetty, C.; Wada, H.; Milne, J.C.; Ito, M.; Rossios, C.; Elliot, M.; Hogg, J.; Kharitonov, S.; Goto, H.; et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 2009, 23, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24, 3145–3159. [Google Scholar] [CrossRef]
- Sundar, I.K.; Yao, H.; Rahman, I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid. Redox Signal. 2013, 18, 1956–1971. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kalpha reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Obeidat, M.; Miller, S.; Probert, K.; Billington, C.K.; Henry, A.P.; Hodge, E.; Nelson, C.P.; Stewart, C.E.; Swan, C.; Wain, L.V.; et al. GSTCD and INTS12 regulation and expression in the human lung. PLoS ONE 2013, 8, e74630. [Google Scholar] [CrossRef]
- Wu, L.; Ma, L.; Nicholson, L.F.; Black, P.N. Advanced glycation end products and its receptor (RAGE) are increased in patients with COPD. Respir. Med. 2011, 105, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kaur, S.; Sarkar, M.; Sarin, B.C.; Changotra, H. The AGE-RAGE Axis and RAGE Genetics in Chronic Obstructive Pulmonary Disease. Clin. Rev. Allergy Immunol. 2021, 60, 244–258. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J.; Hong, S.H.; Rahman, I.; Yang, S.R. Inhibition of RAGE Attenuates Cigarette Smoke-Induced Lung Epithelial Cell Damage via RAGE-Mediated Nrf2/DAMP Signaling. Front. Pharmacol. 2018, 9, 684. [Google Scholar] [CrossRef]
- Hawkins, G.A.; Mora, A.L. FAM13A, A Fatty Acid Oxidation Switch in Mitochondria. Friend or Foe in Chronic Obstructive Pulmonary Disease Pathogenesis? Am. J. Respir. Cell Mol. Biol 2017, 56, 689–691. [Google Scholar] [CrossRef]
- Escribano, A.; Amor, M.; Pastor, S.; Castillo, S.; Sanz, F.; Codoñer-Franch, P.; Dasí, F. Decreased glutathione and low catalase activity contribute to oxidative stress in children with α-1 antitrypsin deficiency. Thorax 2015, 70, 82–83. [Google Scholar] [CrossRef]
- Wu, W.; Peden, D.; Diaz-Sanchez, D. Role of GSTM1 in resistance to lung inflammation. Free Radic. Biol. Med. 2012, 53, 721–729. [Google Scholar] [CrossRef]
- Laukkanen, M.O. Extracellular Superoxide Dismutase: Growth Promoter or Tumor Suppressor? Oxid. Med. Cell. Longev. 2016, 2016, 3612589. [Google Scholar] [CrossRef]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Repapi, E.; Sayers, I.; Wain, L.V.; Burton, P.R.; Johnson, T.; Obeidat, M.; Zhao, J.H.; Ramasamy, A.; Zhai, G.; Vitart, V.; et al. Genome-wide association study identifies five loci associated with lung function. Nat. Genet. 2010, 42, 36–44. [Google Scholar] [CrossRef]
- Biswas, S.K.; Rahman, I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Mol. Asp. Med. 2009, 30, 60–76. [Google Scholar] [CrossRef]
- Decramer, M.; Rutten-van Mölken, M.; Dekhuijzen, P.N.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.; Olivieri, D.; Del Donno, M.; De Backer, W.; et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): A randomised placebo-controlled trial. Lancet 2005, 365, 1552–1560. [Google Scholar] [CrossRef]
- Zheng, J.P.; Wen, F.Q.; Bai, C.X.; Wan, H.Y.; Kang, J.; Chen, P.; Yao, W.Z.; Ma, L.J.; Li, X.; Raiteri, L.; et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2014, 2, 187–194. [Google Scholar] [CrossRef]
- Zheng, J.P.; Kang, J.; Huang, S.G.; Chen, P.; Yao, W.Z.; Yang, L.; Bai, C.X.; Wang, C.Z.; Wang, C.; Chen, B.Y.; et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study): A randomised placebo-controlled study. Lancet 2008, 371, 2013–2018. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef]
- Wise, R.A.; Holbrook, J.T.; Criner, G.; Sethi, S.; Rayapudi, S.; Sudini, K.R.; Sugar, E.A.; Burke, A.; Thimmulappa, R.; Singh, A.; et al. Lack of Effect of Oral Sulforaphane Administration on Nrf2 Expression in COPD: A Randomized, Double-Blind, Placebo Controlled Trial. PLoS ONE 2016, 11, e0163716. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Hanson, C.; Lyden, E.; Furtado, J.; Campos, H.; Sparrow, D.; Vokonas, P.; Litonjua, A.A. Serum tocopherol levels and vitamin E intake are associated with lung function in the normative aging study. Clin. Nutr. 2016, 35, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Hwang, J.W.; Kirkham, P.A.; Rahman, I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr. Med. Chem. 2013, 20, 1496–1530. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed]
- Scoditti, E.; Massaro, M.; Garbarino, S.; Toraldo, D.M. Role of Diet in Chronic Obstructive Pulmonary Disease Prevention and Treatment. Nutrients 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Faris, A.N.; Comstock, A.T.; Chattoraj, S.S.; Chattoraj, A.; Burgess, J.R.; Curtis, J.L.; Martinez, F.J.; Zick, S.; Hershenson, M.B.; et al. Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir. Res. 2010, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Meja, K.K.; Rajendrasozhan, S.; Adenuga, D.; Biswas, S.K.; Sundar, I.K.; Spooner, G.; Marwick, J.A.; Chakravarty, P.; Fletcher, D.; Whittaker, P.; et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am. J. Respir. Cell Mol. Biol. 2008, 39, 312–323. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
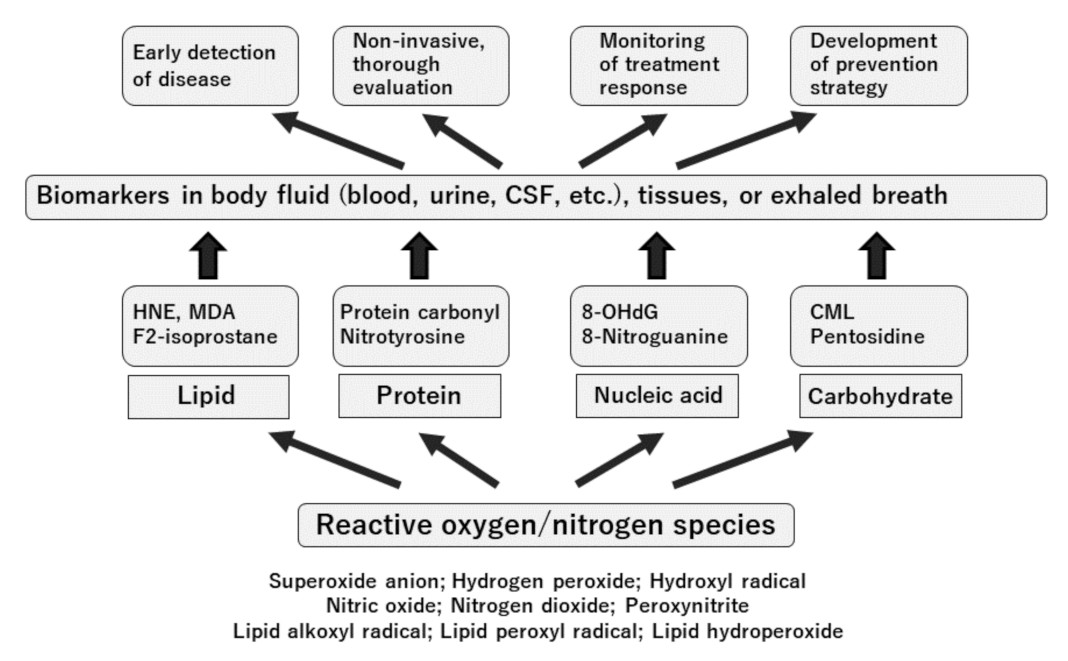{kind=link}
| Formation of Modified Molecules by Reactive Oxygen Species | COPD vs. Healthy | In COPD Stages | |
|---|---|---|---|
| Malondialdehyde | ↑[84,85,91,92,108,111,112,113,114,115,116,117] /n.d.[118,119] | ↑[85,115,116]/n.d.[114] | |
| 8-isoprostane | ↑[89] | ||
| Conjugated dienes | ↑[113] | ||
| Protein carbonyls | ↑[[91,114,120,121] /n.d.[122] | n.d.[114] | |
| Advanced oxidation protein products | ↑[92,114] | n.d.[114] | |
| Total hydroperoxides | ↑[92,93,123,124] | ||
| Oxidatively damaged DNA | ↑[91,114,125] /n.d.[84,120] | ||
| Antioxidative Molecules and Enzymes | COPD vs. Healthy | In COPD Stages | |
| Protein sulfhydryl groups | ↓[96,115,118] /n.d.[114,119] | ↓[115,126] | |
| Reduced Glutathione | Erythrocyte | ↓[85] | ↓[85] |
| Plasma | ↓[108,112,114,116,119] /n.d.[118] | n.d.[114,116] | |
| Biological antioxidative potential | ↓[92,114,127,128] | n.d.[114] | |
| Superoxide dismutase activity | Erythrocyte | ↑[113]/↓[85,116] /n.d.[114] | ↓[116] |
| Plasma | ↓[92,128] /n.d.[120,123] | ||
| Catalase activity | Erythrocyte | ↓[85,116] /n.d.[113,114] | ↓[85,116] /n.d.[114] |
| Plasma | ↓[108,112,128] /n.d.[120] | ||
| Glutathione peroxidase activity | Erythrocyte | ↓[85,113,116] | ↓[116,127] |
| Plasma | ↓[108,112] /↑[114] | ||
| Gene (Status) | Characteristics | Phenotype/Role in COPD |
|---|---|---|
| CYP1A1 | CYP1A1: Production of aromatic hydrocarbon hydroxylase (xenobiotic-metabolizing enzyme) | Gene polymorphisms associated with increased MDA (oxidative stress marker) [112] |
| CYP1A2 | CYP1A2: Xenobiotic-metabolizing enzyme, induced by cigarette smoke | |
| CYBA | CYBA: Formation of NADPH oxidase | |
| GSTM1 | GSTM1, GSTP1: Detoxification of electrophilic compounds, including products of oxidative stress [156] | Rapid decline in lung function [144] |
| GSTP1 | Rapid decline in lung function, FEV1 decline, Emphysema [144] | |
| SOD3 | SOD3: Catalyzes the dismutation of O2− into H2O2 [157] | Rapid decline in lung function [144] |
| HDAC2(downregulation) | HDAC2: Facilitating the formation of transcription repressor complexes [158] | Impairment of Nrf2 activation in the lung [145] |
| SIRT1 | SIRT1, 6: Type III HDAC that catalyze NAD+-dependent deacetylation | Promotes proteosomal degradation [148] |
| SIRT1, SIRT6 (downregulation) | Accelerating ageing of the lung and increased oxidative stress [146,147] | |
| GSTCD | GSTCD: Detoxification of products of oxidative stress and synthesis of steroid hormones, lacking key functional domains important for GST activity [150] | Related to FEV1 (Positive correlation with mRNA expression) [150,159] |
| AGER | RAGE: multiligand receptor, one of its ligands, AGE, is induced by oxidative stress | FEV1 decline [150] |
| FAM13A | FAM13A: Regulating CPT1A expression and fatty acid oxidation [154] | Regulation oxidative stress [143] |
| SERPINA1 | Encoding alpha-1-antitrypsin (AAT), inhibiting proteolytic enzymes | A major genetic risk for COPD, contributing to oxidative stress [155] |
| Antioxidants | Examples | Studies in COPD |
|---|---|---|
| Thiol compounds | N-acetylcysteine Carbocysteine Erdosteine | Reduced exacerbation Reduced exacerbation Reduced exacerbation |
| Nrf2 activators | Sulforaphane Bardoxolone methyl Dimethyl fumarate | Clinical trial negative Effective in animal models Not tested |
| Plant-derived polyphenols | Resveratrol Quercetin Curcumin | Anti-inflammatory in vitro No clinical trials Anti-inflammatory in vivo |
| Dietary antioxidants | Vitamin C (ascorbic acid) Vitamin E (α-tocopherol) | No clinical trials No clinical trials |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taniguchi, A.; Tsuge, M.; Miyahara, N.; Tsukahara, H. Reactive Oxygen Species and Antioxidative Defense in Chronic Obstructive Pulmonary Disease. Antioxidants 2021, 10, 1537. https://doi.org/10.3390/antiox10101537
Taniguchi A, Tsuge M, Miyahara N, Tsukahara H. Reactive Oxygen Species and Antioxidative Defense in Chronic Obstructive Pulmonary Disease. Antioxidants. 2021; 10(10):1537. https://doi.org/10.3390/antiox10101537
Chicago/Turabian StyleTaniguchi, Akihiko, Mitsuru Tsuge, Nobuaki Miyahara, and Hirokazu Tsukahara. 2021. "Reactive Oxygen Species and Antioxidative Defense in Chronic Obstructive Pulmonary Disease" Antioxidants 10, no. 10: 1537. https://doi.org/10.3390/antiox10101537
APA StyleTaniguchi, A., Tsuge, M., Miyahara, N., & Tsukahara, H. (2021). Reactive Oxygen Species and Antioxidative Defense in Chronic Obstructive Pulmonary Disease. Antioxidants, 10(10), 1537. https://doi.org/10.3390/antiox10101537