
Exacerbated Age-Related Hippocampal Alterations of Microglia Morphology, β-Amyloid and Lipofuscin Deposition and Presenilin Overexpression in Per1−/−-Mice


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunofluorescence
2.3. Quantifying Lipofuscin Accumulation in the Hippocampus
3. Results
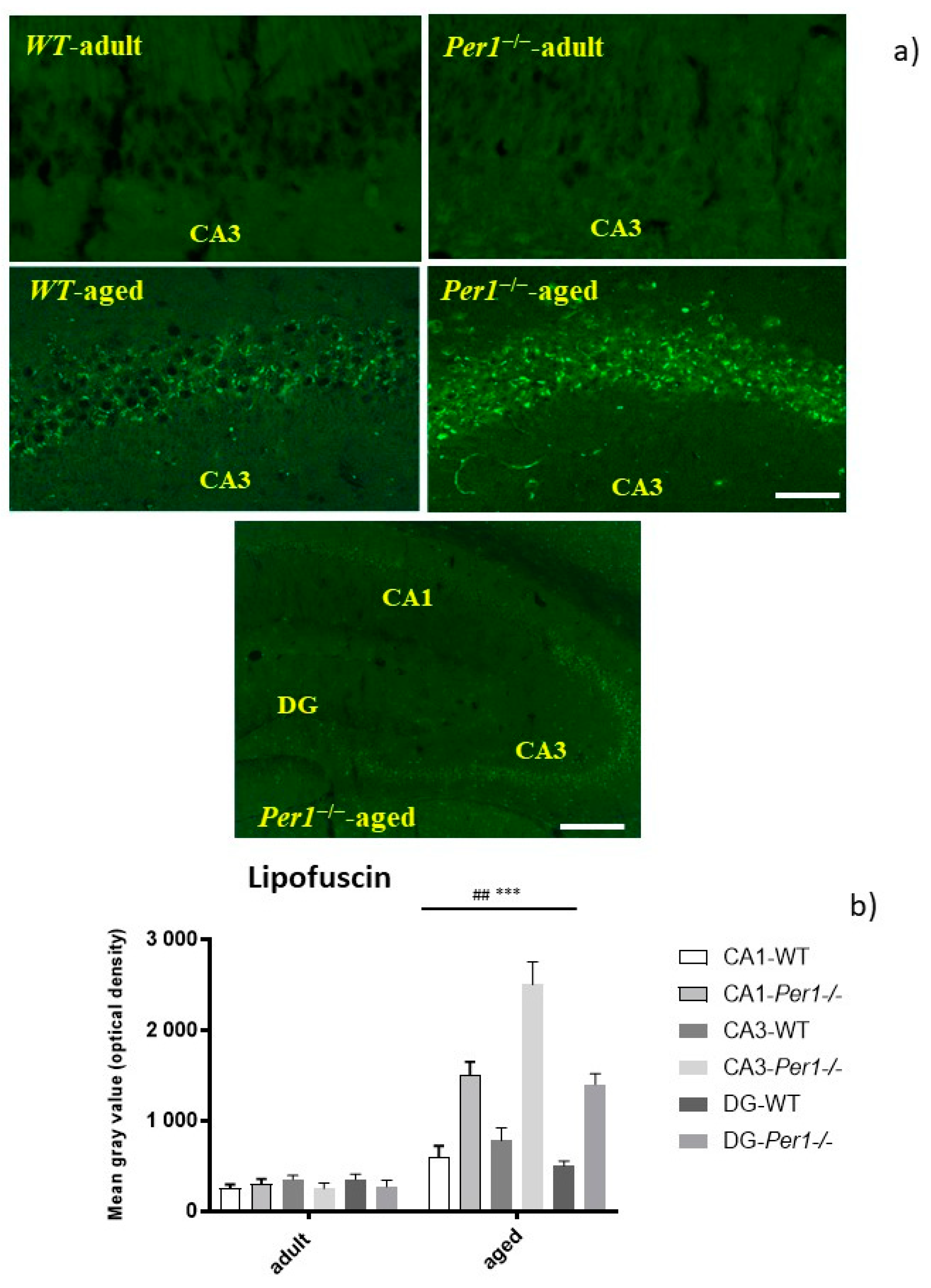
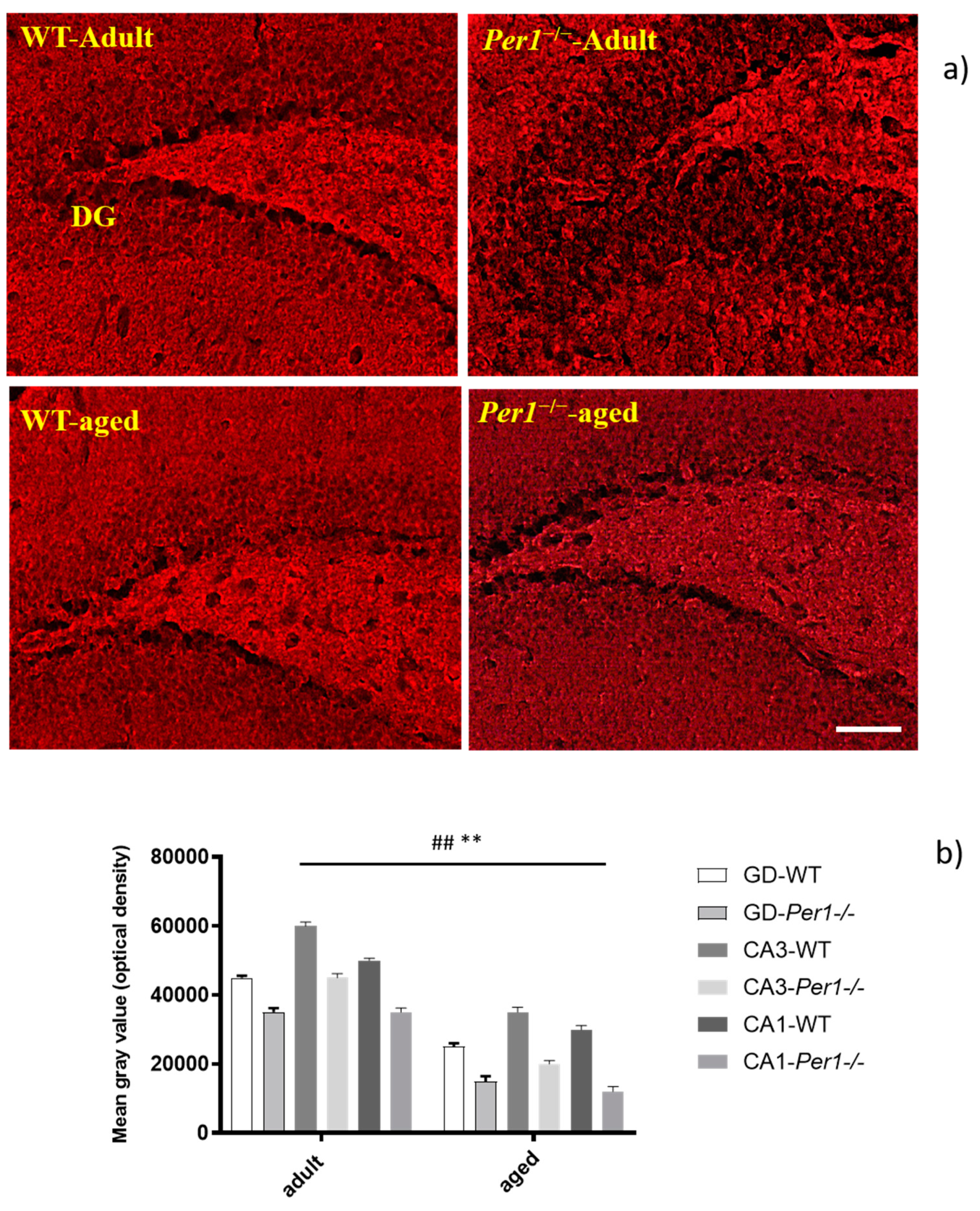
3.1. Lipofuscin Accumulation in the Hippocampus of Aged Mice
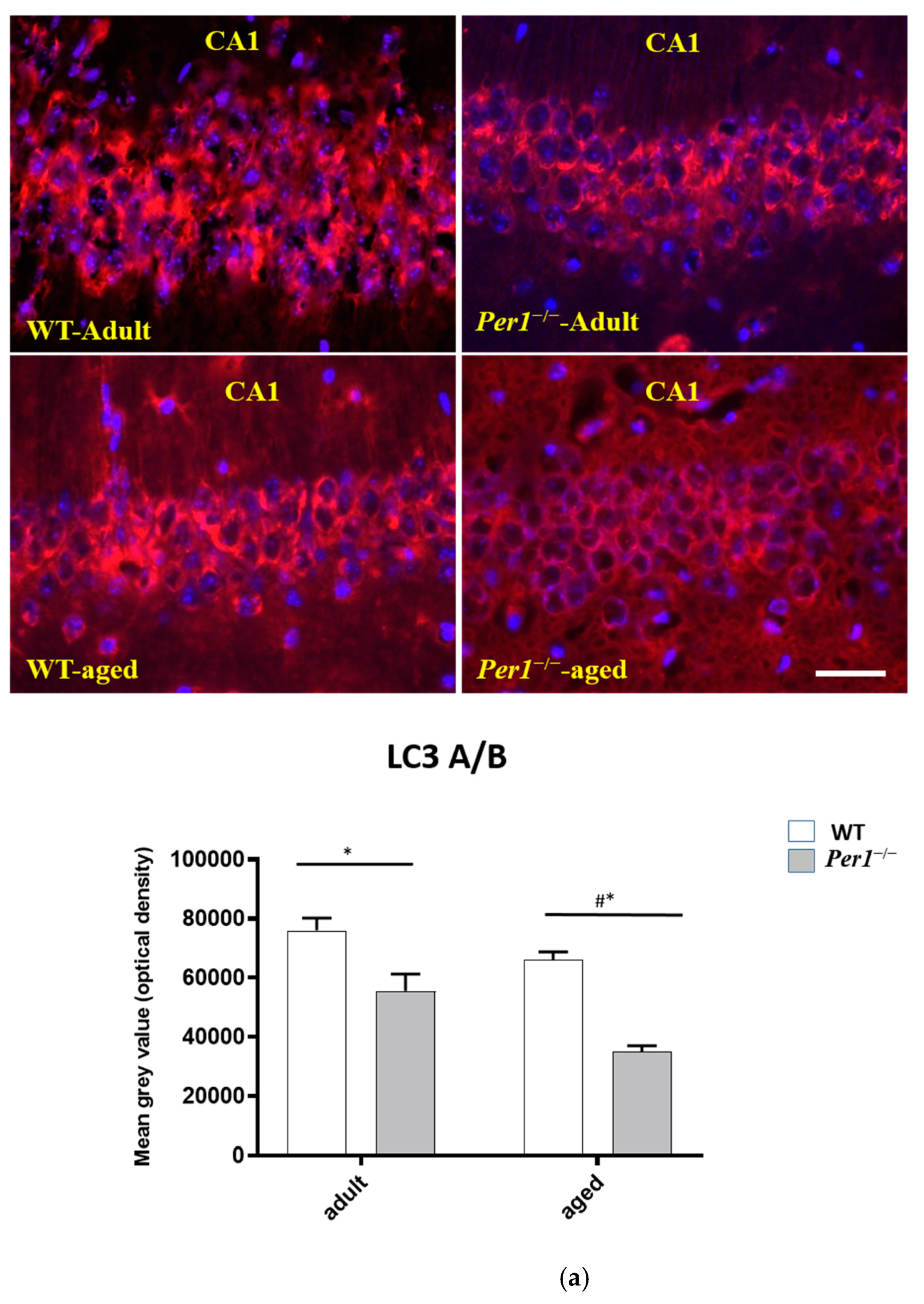
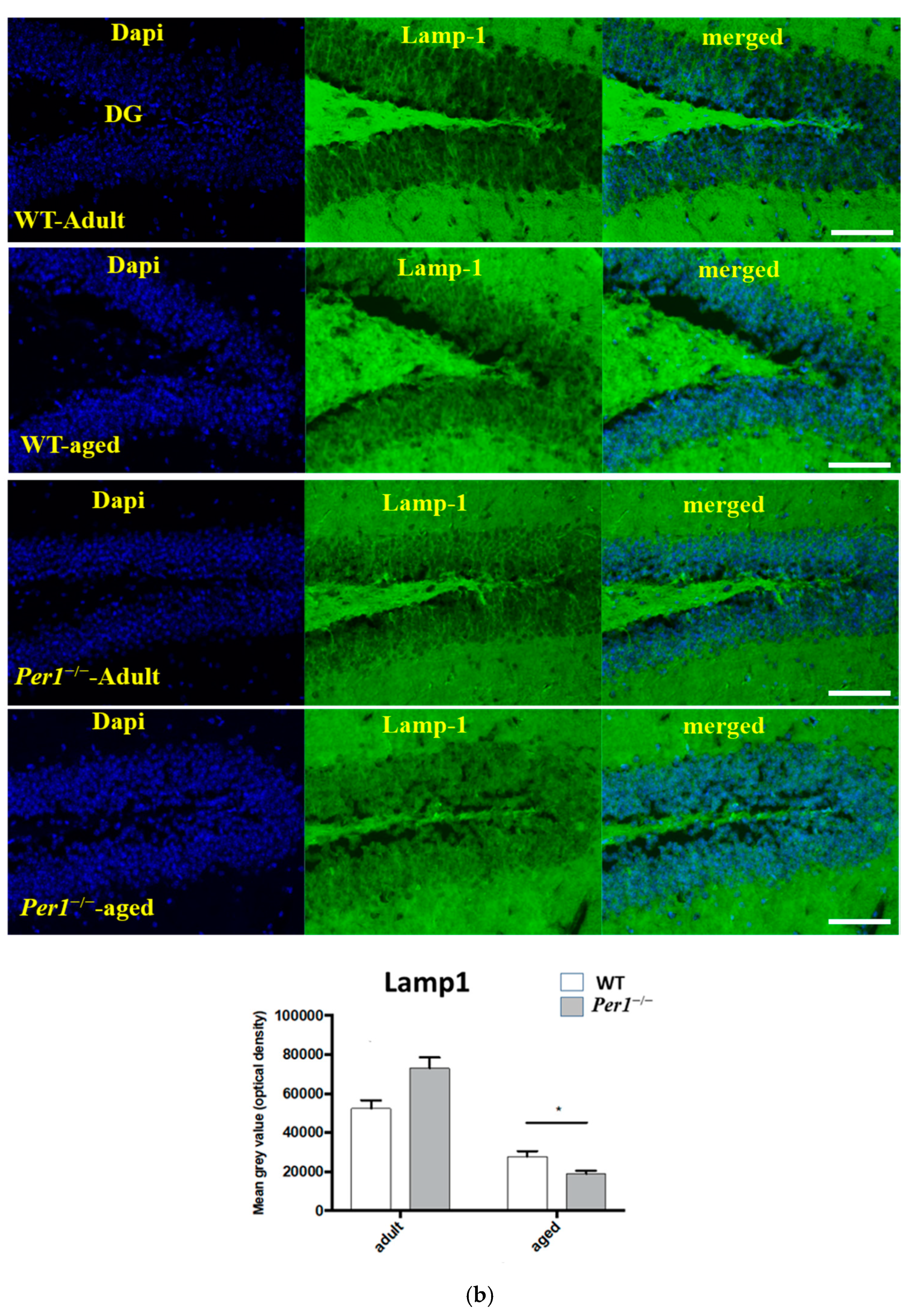
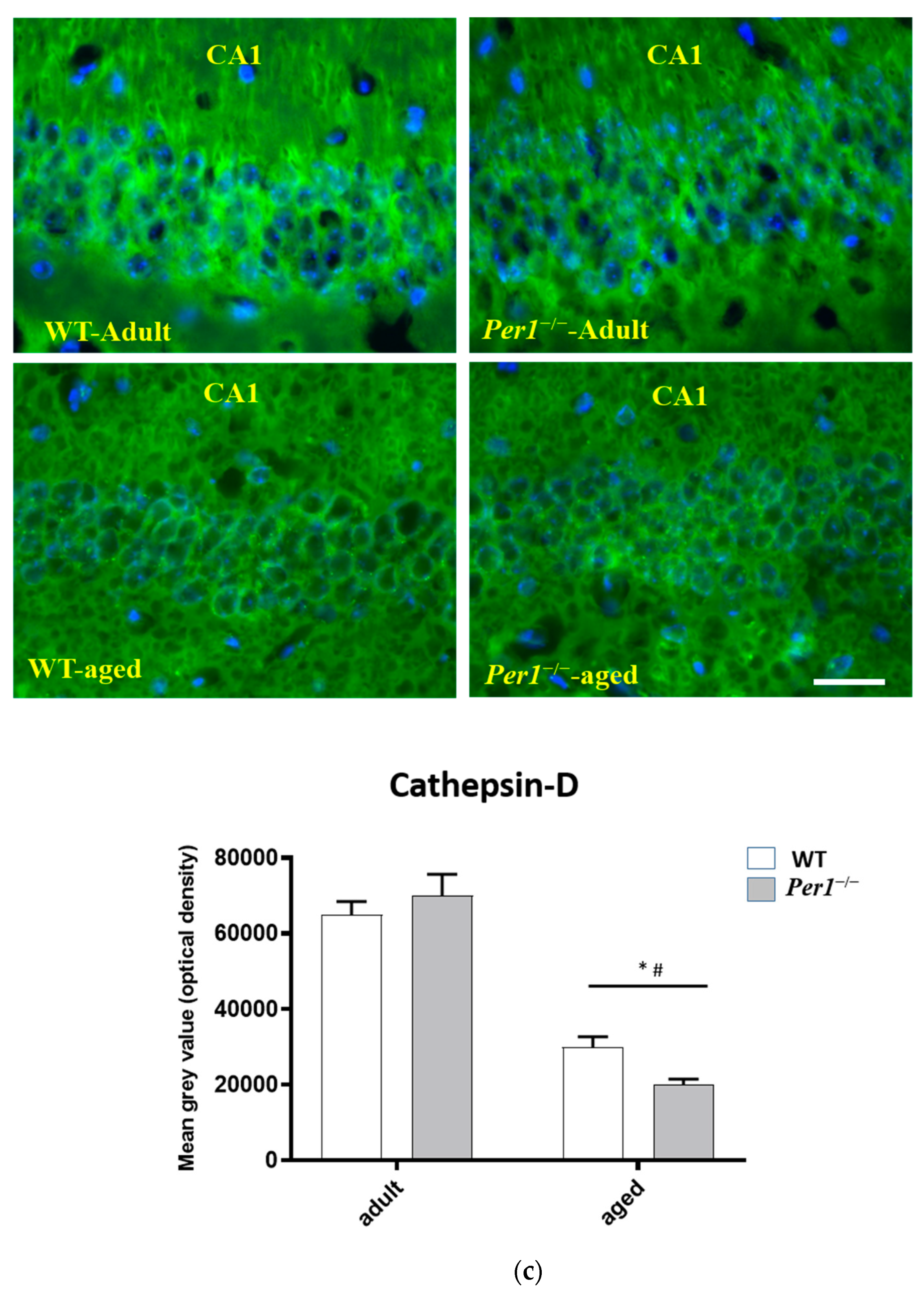
3.2. LC3, LAMP-1 and Cathepsin-B Quantification in the Hippocampus
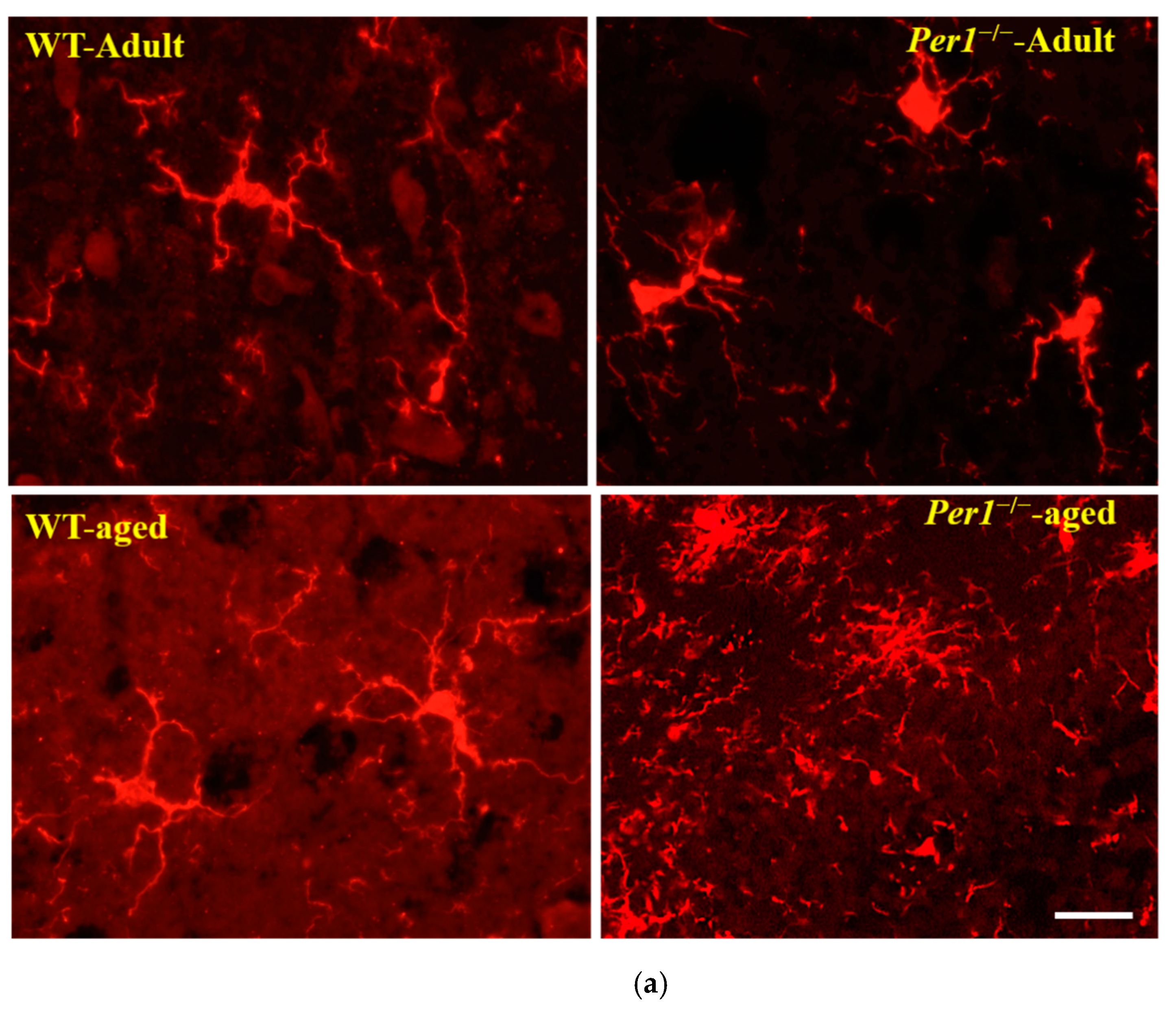
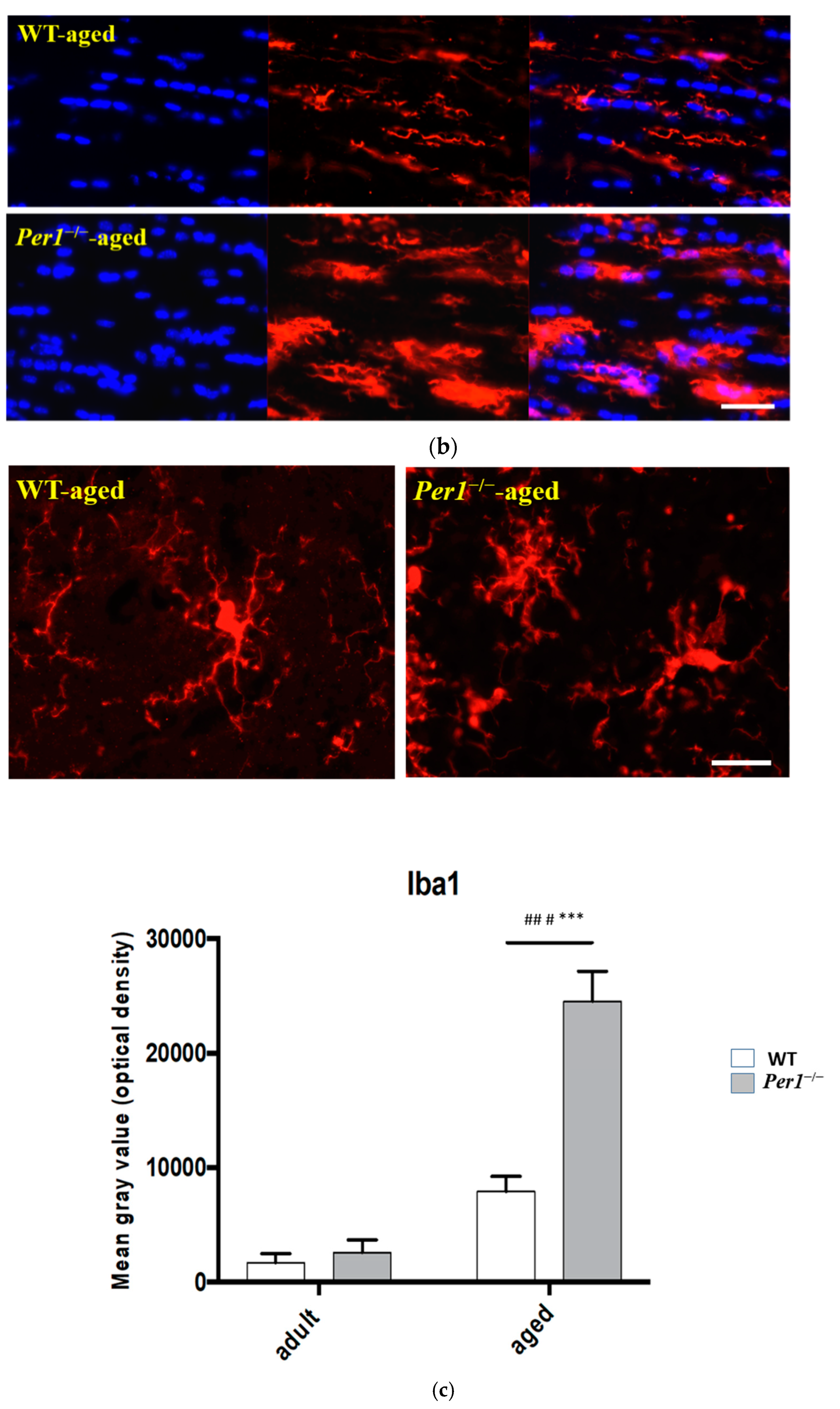
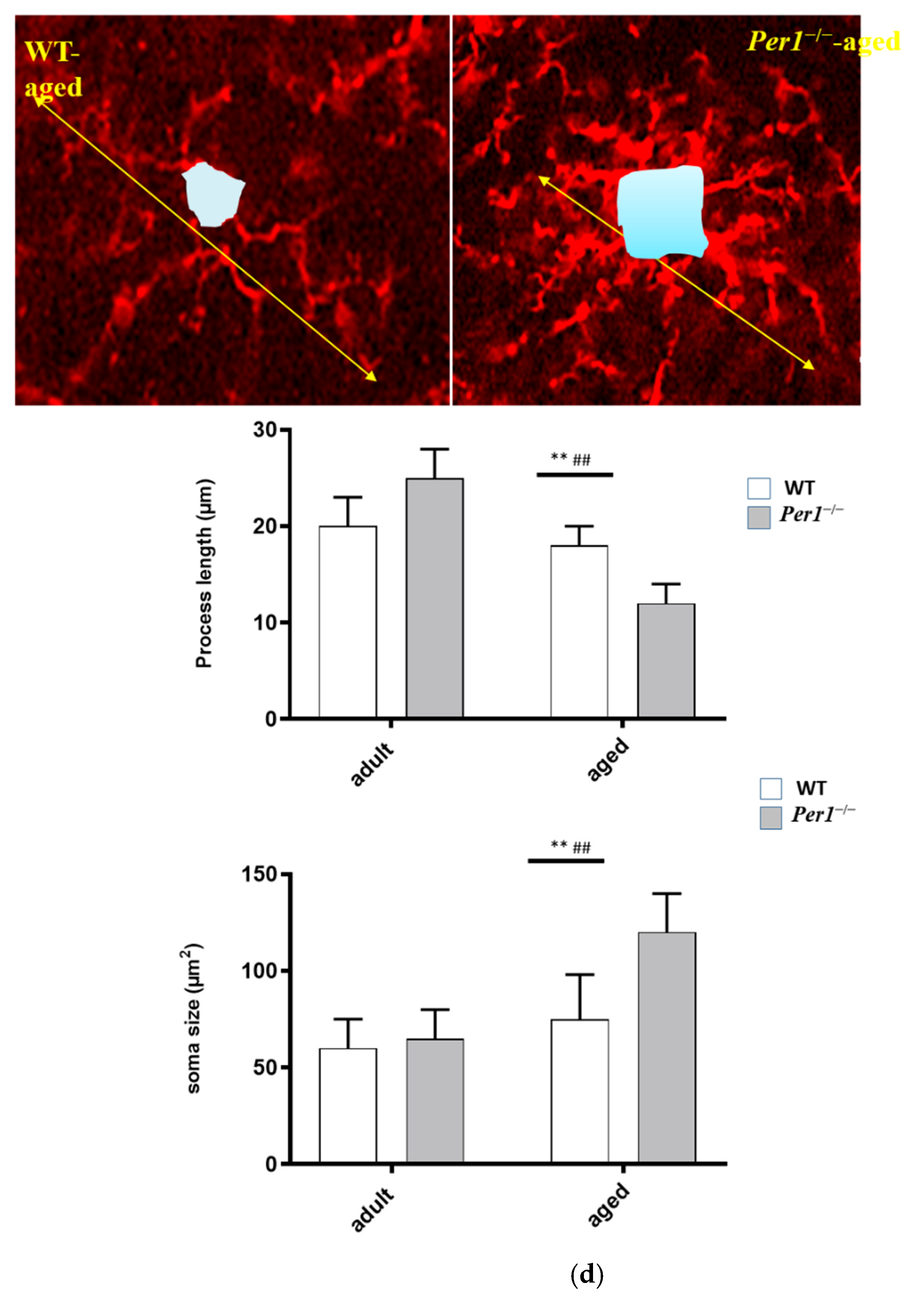
3.3. Aging-Related Morphological Changes of Microglia in the Hippocampus
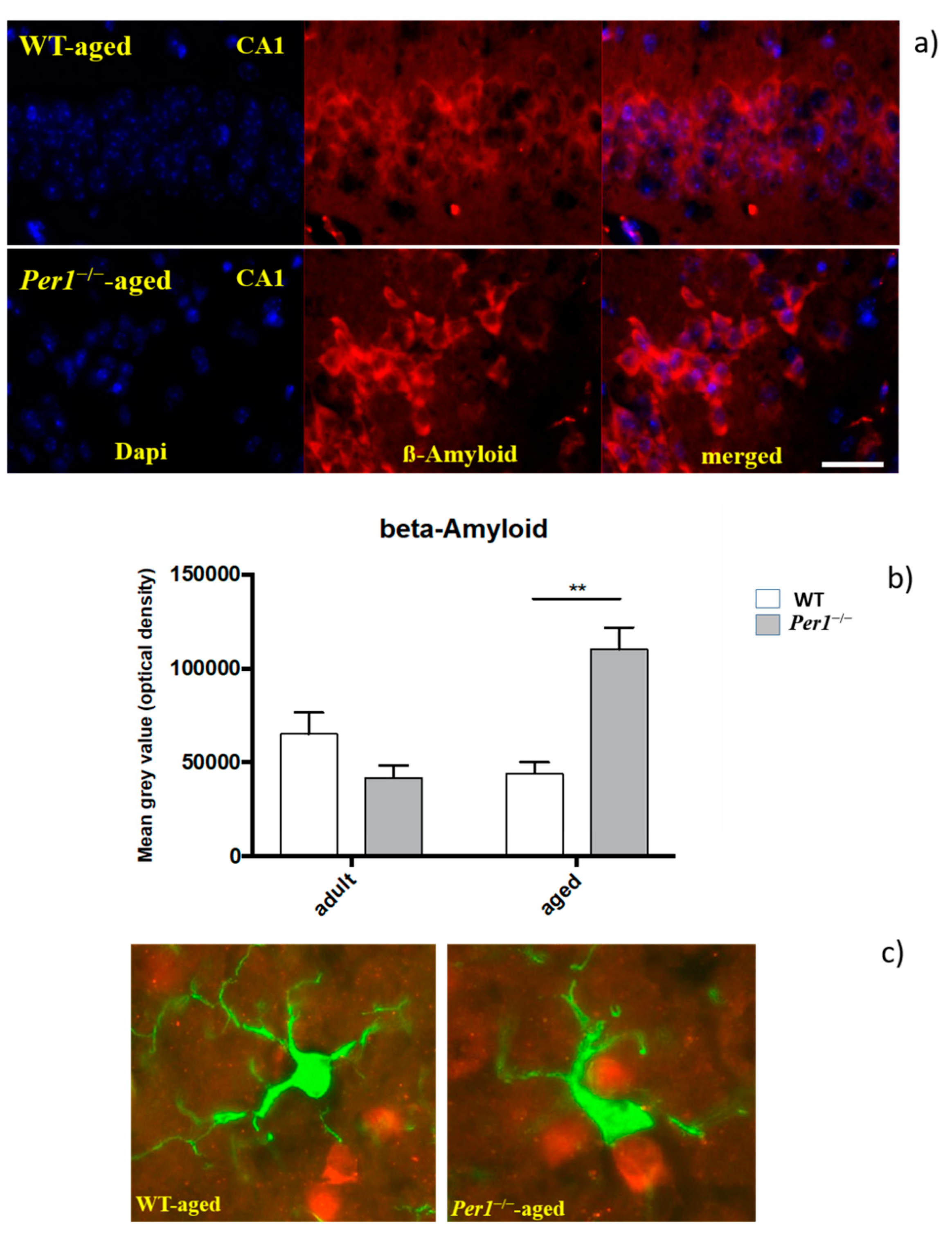
3.4. β-Amyloid (Aβ42)
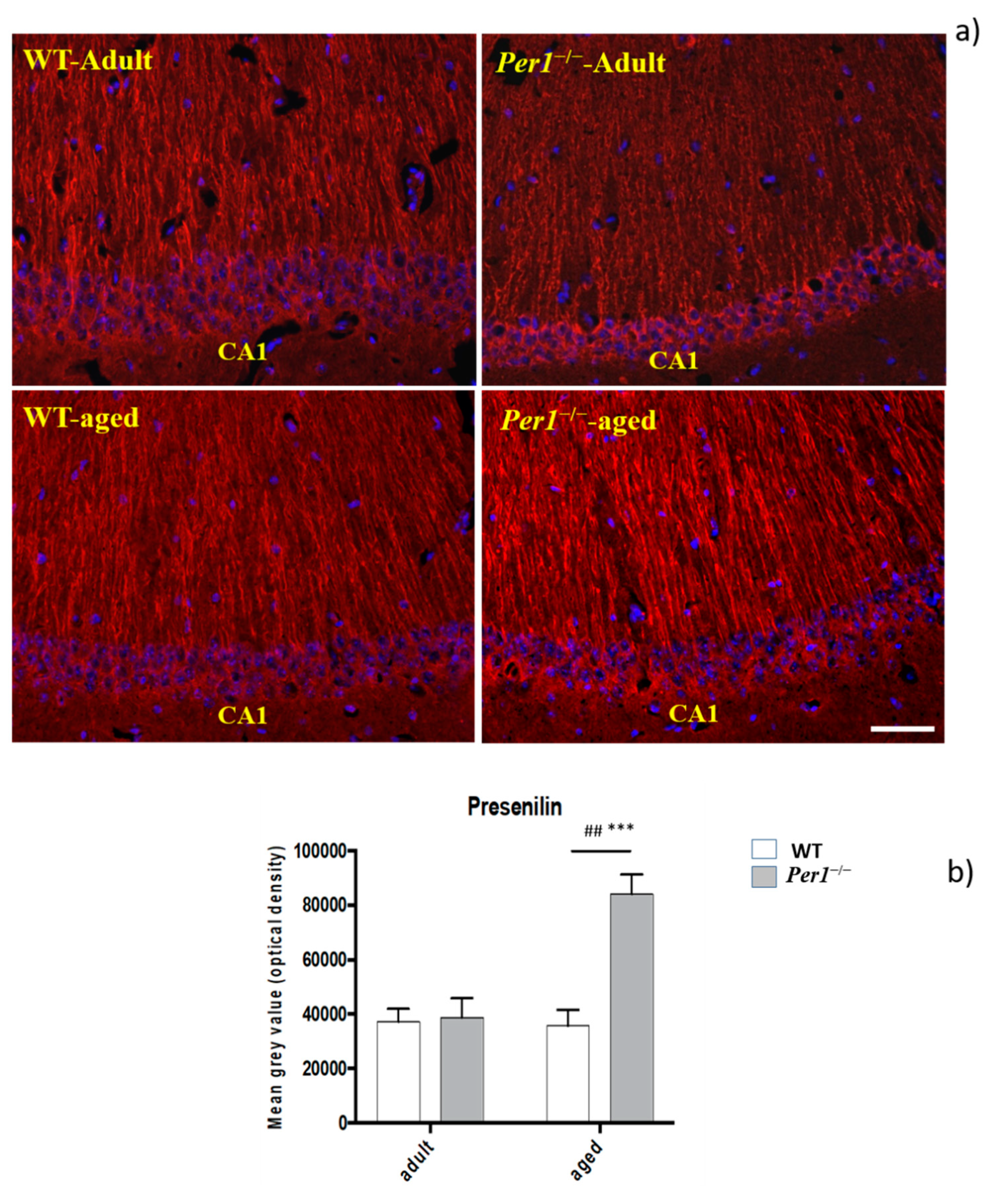
3.5. Presenilin-2
3.6. Synaptophysin Quantification in the Hippocampus
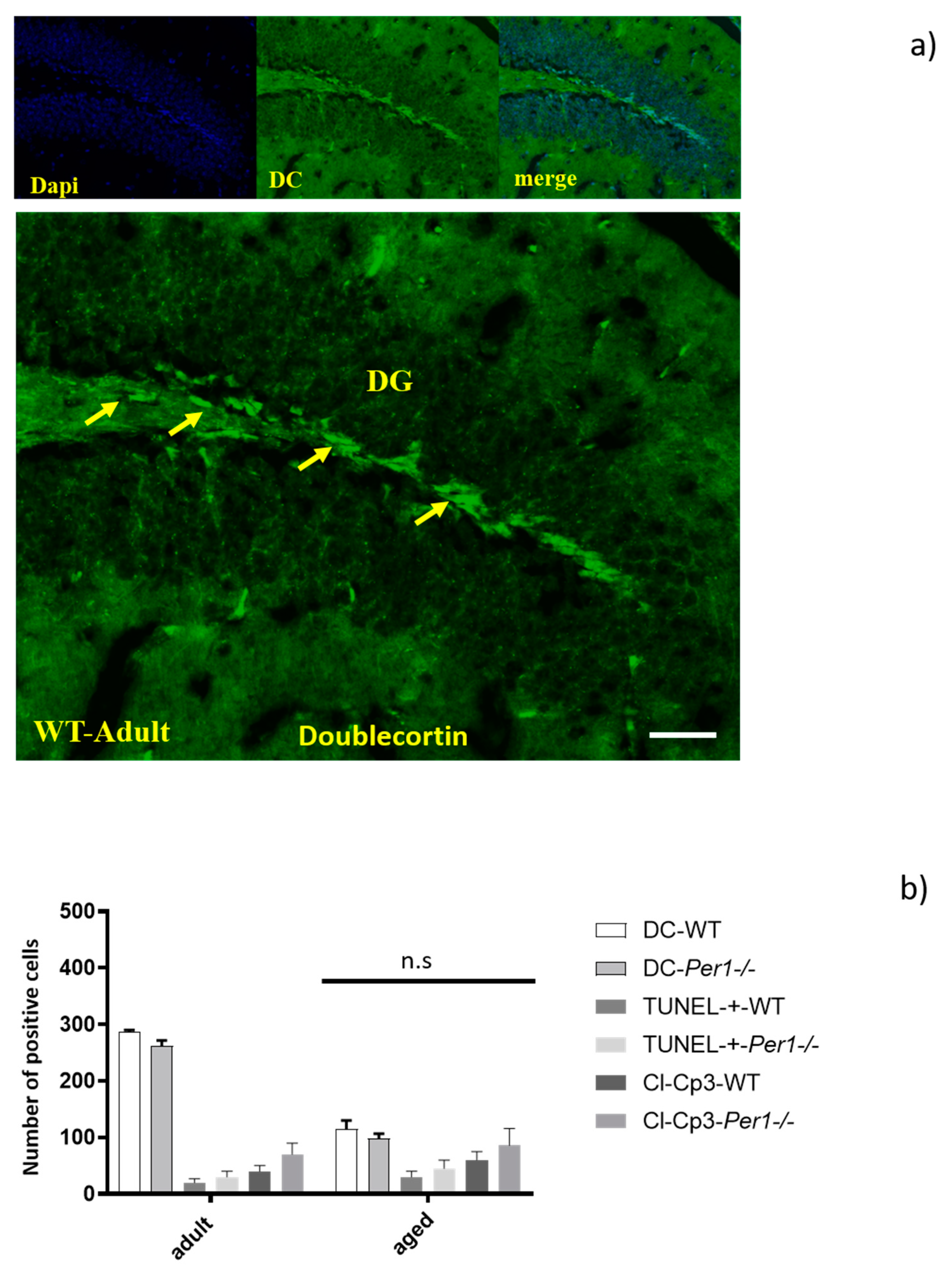
3.7. Adult Neurogenesis in the Hippocampus by Quantification of Doublecortin-Positive Cells
4. Discussion
4.1. Enhanced Lipofuscin Accumulation in the Hippocampus of Aged Per1−/−-Mice
4.2. Per1 Had No Influence on the Proliferation or Apoptosis of Granule Cells in Aged Mice
4.3. Enhanced Presenilin and Significant ß Accumulation in the Hippocampus of Aged Per1−/−-Mice
4.4. Loss of Synapses and Microglial Activation in the Hippocampus of Aged Per1−/−-Mice
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gibson, E.M.; Williams, W.P.; Kriegsfeld, L.J. Aging in the Circadian System: Considerations for Health, Disease Prevention, and Longevity. Exp. Gerontol. 2009, 44, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Harper, D.G.; Volicer, L.; Stopa, E.G.; McKee, A.C.; Nitta, M.; Satlin, A. Disturbance of endogenous circadian rhythm in aging and Alzheimer disease. Am. J. Geriatr. Psychiatry 2005, 13, 359–368. [Google Scholar] [CrossRef]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 2006, 22, 375–382. [Google Scholar] [CrossRef]
- Chen, Z.; McKnight, S.L. A conserved DNA damage response pathway responsible for coupling the cell division cycle to the circadian and metabolic cycles. Cell Cycle 2007, 6, 2906–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musiek, E.S.; Lim, M.M.; Yang, G.; Bauer, A.Q.; Qi, L.; Lee, Y.; Roh, J.H.; Ortiz-Gonzalez, X.; Dearborn, J.T.; Culver, J.P.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Investig. 2013, 123, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Rawashdeh, O.; Jilg, A.; Jedlicka, P.; Slawska, J.; Thomas, L.; Saade, A.; Schwarzacher, S.W.; Stehle, J.H. PERIOD1 coordinates hippocampal rhythms and memory processing with daytime. Hippocampus 2014, 24, 712–723. [Google Scholar] [CrossRef]
- Morioka, N.; Fujii, S.; Kondo, S.; Zhang, F.F.; Miyauchi, K.; Nakamura, Y.; Hisaoka-Nakashima, K.; Nakata, Y. Downregulation of spinal astrocytic connexin43 leads to upregulation of interleukin-6 and cyclooxygenase-2 and mechanical hypersensitivity in mice. Glia 2018, 66, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, S.M.; Phan, T.X.; Saraf, A.; Chen, X.; Storm, D.R. Genetic disruption of the core circadian clock impairs hippocampus-dependent memory. Learn. Mem. 2014, 21, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Bang, J.; Chang, H.W.; Jung, H.-R.; Cho, C.-H.; Hur, J.-A.; Lee, S.-I.; Choi, T.H.; Kim, S.-H.; Ha, E. Melatonin attenuates clock gene cryptochrome1, which may aggravate mouse anti-type II collagen antibody-induced arthritis. Rheumatol. Int. 2012, 32, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Wiebking, N.; Maronde, E.; Rami, A. Increased neuronal injury in clock gene Per-1 deficient-mice after cerebral ischemia. Curr. Neurovascular Res. 2013, 10, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Fekadu, J.; Rawashdeh, O. The Hippocampal Autophagic Machinery is Depressed in the Absence of the Circadian Clock Protein PER1 that may Lead to Vulnerability During Cerebral Ischemia. Curr. Neurovascular Res. 2017, 14, 207–214. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.; Klionsky, D.J.; Toit, A.D.; Hofmeyr, J.-H.S. On the relevance of precision autophagy flux control in vivo—Points of departure for clinical translation. Autophagy 2020, 16, 750–762. [Google Scholar] [CrossRef]
- Yoshimori, T. Autophagy: A regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004, 313, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagy in the eukaryotic cell. Eukaryot. Cell 2002, 1, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Ojo, B.; Rezaie, P.; Gabbott, P.L.; Davies, H.; Colyer, F.; Cowley, T.R.; Lynch, M.; Stewart, M.G. Age-related changes in the hippocampus (loss of synaptophysin and glial-synaptic interaction) are modified by systemic treatment with an NCAM-derived peptide, FGL. Brain Behav. Immun. 2012, 26, 778–788. [Google Scholar] [CrossRef]
- Kalfalah, F.; Janke, L.; Schiavi, A.; Tigges, J.; Ix, A.; Ventura, N.; Boege, F.; Reinke, H. Crosstalk of clock gene expression and autophagy in aging. Aging 2016, 8, 1876–1890. [Google Scholar] [CrossRef] [Green Version]
- Lefta, M.; Campbell, K.S.; Feng, H.-Z.; Jin, J.-P.; Esser, K.A. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H475–H485. [Google Scholar] [CrossRef] [Green Version]
- Lananna, B.V.; Nadarajah, C.J.; Izumo, M.; Cedeño, M.R.; Xiong, D.D.; Dimitry, J.; Tso, C.F.; McKee, C.A.; Griffin, P.; Sheehan, P.W.; et al. Cell-Autonomous Regulation of Astrocyte Activation by the Circadian Clock Protein BMAL1. Cell Rep. 2018, 25, 1–9.e5. [Google Scholar] [CrossRef] [Green Version]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Barca-Mayo, O.; Boender, A.J.; Armirotti, A.; De Pietri Tonelli, D. Deletion of astrocytic BMAL1 results in metabolic imbalance and shorter lifespan in mice. Glia 2020, 68, 1131–1147. [Google Scholar] [CrossRef] [Green Version]
- Nakazato, R.; Kawabe, K.; Yamada, D.; Ikeno, S.; Mieda, M.; Shimba, S.; Hinoi, E.; Yoneda, Y.; Takarada, T. Disruption of Bmal1 Impairs Blood–Brain Barrier Integrity via Pericyte Dysfunction. J. Neurosci. 2017, 37, 10052–10062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2016, 441, 880–884. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Steiger-Barraissoul, S.; Rami, A. Serum deprivation induced autophagy and predominantly an AIF-dependent apoptosis in hippocampal HT22 neurons. Apoptosis 2009, 14, 1274–1288. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Kögel, D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: Two sides of the same coin? Autophagy 2008, 4, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rami, A. Review: Autophagy in neurodegeneration: Firefighter and/or incendiarist? Neuropathol. Appl. Neurobiol. 2009, 35, 449–461. [Google Scholar] [CrossRef]
- Kim, M.; Fekadu, J.; Maronde, E.; Rami, A. Alleviation of autophagy by knockdown of Beclin-1 enhances susceptibility of hippocampal neurons to proapoptotic signals induced by amino acid starvation. Histochem. Cell Biol. 2013, 139, 99–108. [Google Scholar] [CrossRef]
- Fekadu, J.; Rami, A. Beclin-1 Deficiency Alters Autophagosome Formation, Lysosome Biogenesis and Enhances Neuronal Vulnerability of HT22 Hippocampal Cells. Mol. Neurobiol. 2016, 53, 5500–5509. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial Turnover and Aging of Long-Lived Postmitotic Cells: The Mitochondrial–Lysosomal Axis Theory of Aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Aging as an Epigenetic Phenomenon. Curr. Genom. 2017, 18, 385–407. [Google Scholar] [CrossRef] [Green Version]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Bouchard-Cannon, P.; Mendoza-Viveros, L.; Yuen, A.; Kærn, M.; Cheng, H.-Y.M. The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 2013, 5, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.A.H.; Schwarz-Herzke, B.; Mir, S.; Sahlender, B.; Victor, M.; Görg, B.; Schmuck, M.; Dach, K.; Fritsche, E.; Kremer, A.; et al. Deficiency of the clock gene Bmal1 affects neural progenitor cell migration. Brain Struct. Funct. 2019, 224, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Thinakaran, G.; Parent, A.T. Identification of the role of presenilins beyond Alzheimer’s disease. Pharmacol. Res. 2004, 50, 411–418. [Google Scholar] [CrossRef]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Moir, R.D.; Romano, D.M.; Tesco, G.; Kovacs, D.M.; Tanzi, R.E. Hypocapnia induces caspase-3 activation and increases Abeta production. Neurodegener. Dis. 2004, 1, 29–37. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Willis, E.F.; MacDonald, K.P.A.; Nguyen, Q.H.; Garrido, A.L.; Gillespie, E.R.; Harley, S.B.R.; Bartlett, P.F.; Schroder, W.A.; Yates, A.G.; Anthony, D.C.; et al. Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell 2020, 180, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Investig. 2012, 122, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Wujek, J.R.; Criste, G.A.; Jalabi, W.; Yin, X.; Kidd, G.J.; Stohlman, S.; Ransohoff, R. Evidence for synaptic stripping by cortical microglia. Glia 2007, 55, 360–368. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.-G.; Ding, J.-Q.; Chen, S.-D. Microglia in the aging brain: Relevance to neurodegeneration. Mol. Neurodegener. 2010, 5, 12. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Börner, J.H.; Rawashdeh, O.; Rami, A. Exacerbated Age-Related Hippocampal Alterations of Microglia Morphology, β-Amyloid and Lipofuscin Deposition and Presenilin Overexpression in Per1−/−-Mice. Antioxidants 2021, 10, 1330. https://doi.org/10.3390/antiox10091330
Börner JH, Rawashdeh O, Rami A. Exacerbated Age-Related Hippocampal Alterations of Microglia Morphology, β-Amyloid and Lipofuscin Deposition and Presenilin Overexpression in Per1−/−-Mice. Antioxidants. 2021; 10(9):1330. https://doi.org/10.3390/antiox10091330
Chicago/Turabian StyleBörner, Jan Hendrik, Oliver Rawashdeh, and Abdelhaq Rami. 2021. "Exacerbated Age-Related Hippocampal Alterations of Microglia Morphology, β-Amyloid and Lipofuscin Deposition and Presenilin Overexpression in Per1−/−-Mice" Antioxidants 10, no. 9: 1330. https://doi.org/10.3390/antiox10091330
APA StyleBörner, J. H., Rawashdeh, O., & Rami, A. (2021). Exacerbated Age-Related Hippocampal Alterations of Microglia Morphology, β-Amyloid and Lipofuscin Deposition and Presenilin Overexpression in Per1−/−-Mice. Antioxidants, 10(9), 1330. https://doi.org/10.3390/antiox10091330