
Tumor-Induced Cardiac Dysfunction: A Potential Role of ROS

 , ,
, ,  and
and
Abstract
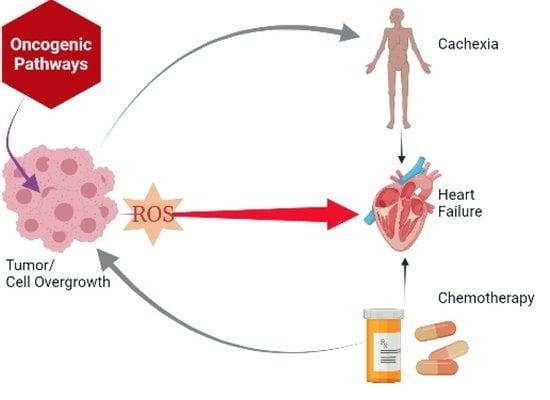
:
1. Introduction
2. Methods
2.1. Fly Stocks
2.2. Tumor Induction and Heart Function in Mice
2.3. Optical Coherence Tomography (OCT) Recordings
2.4. Cardiac Function Analysis
2.5. Measurement of Pericardin Fiber Thickness
2.6. ROS Staining
2.7. Hemolymph ROS Measurement
2.8. Antioxidant Feeding
2.9. Statistical Analysis
3. Results
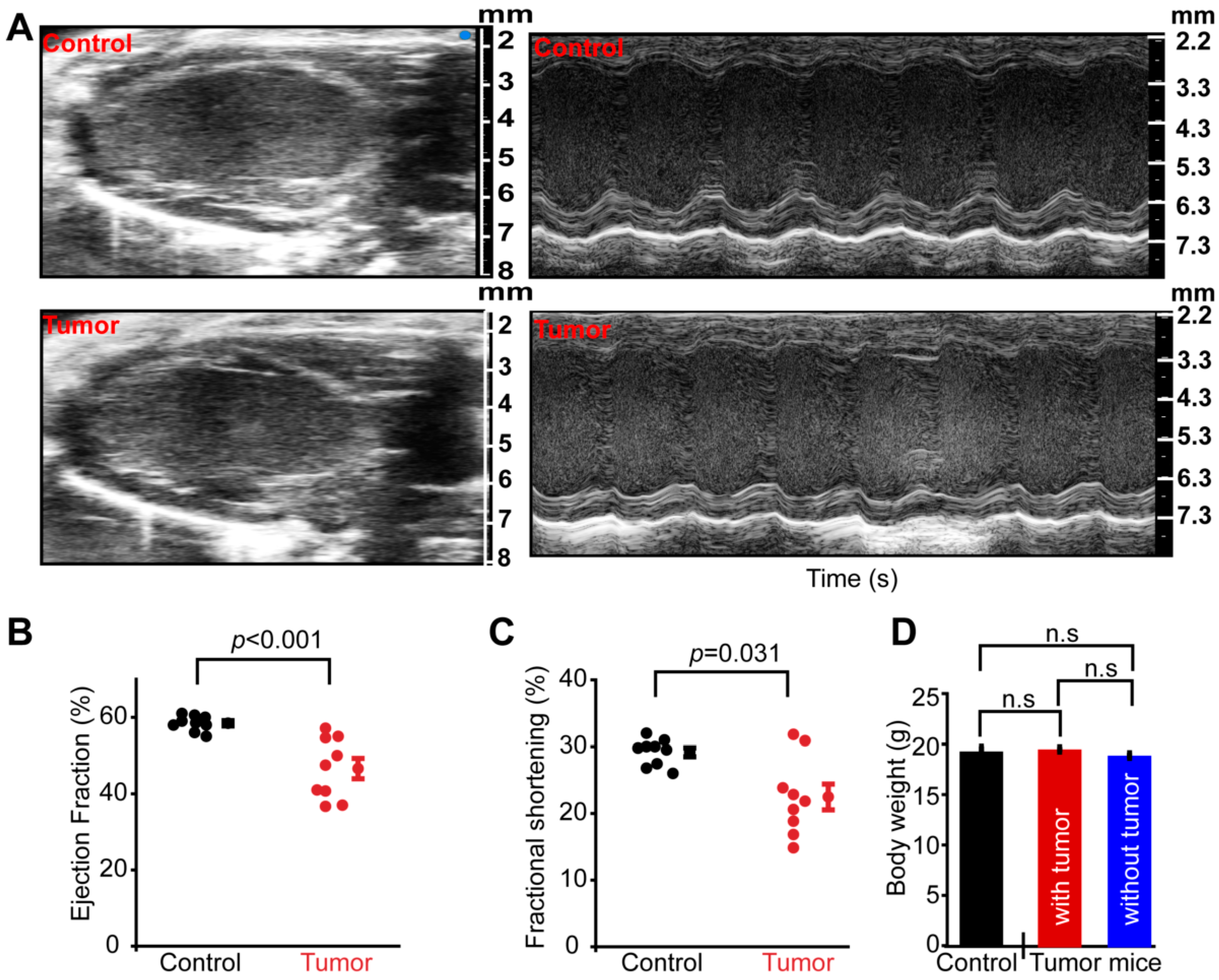
3.1. Tumorigenesis Causes Systemic Cardiac Dysfunction
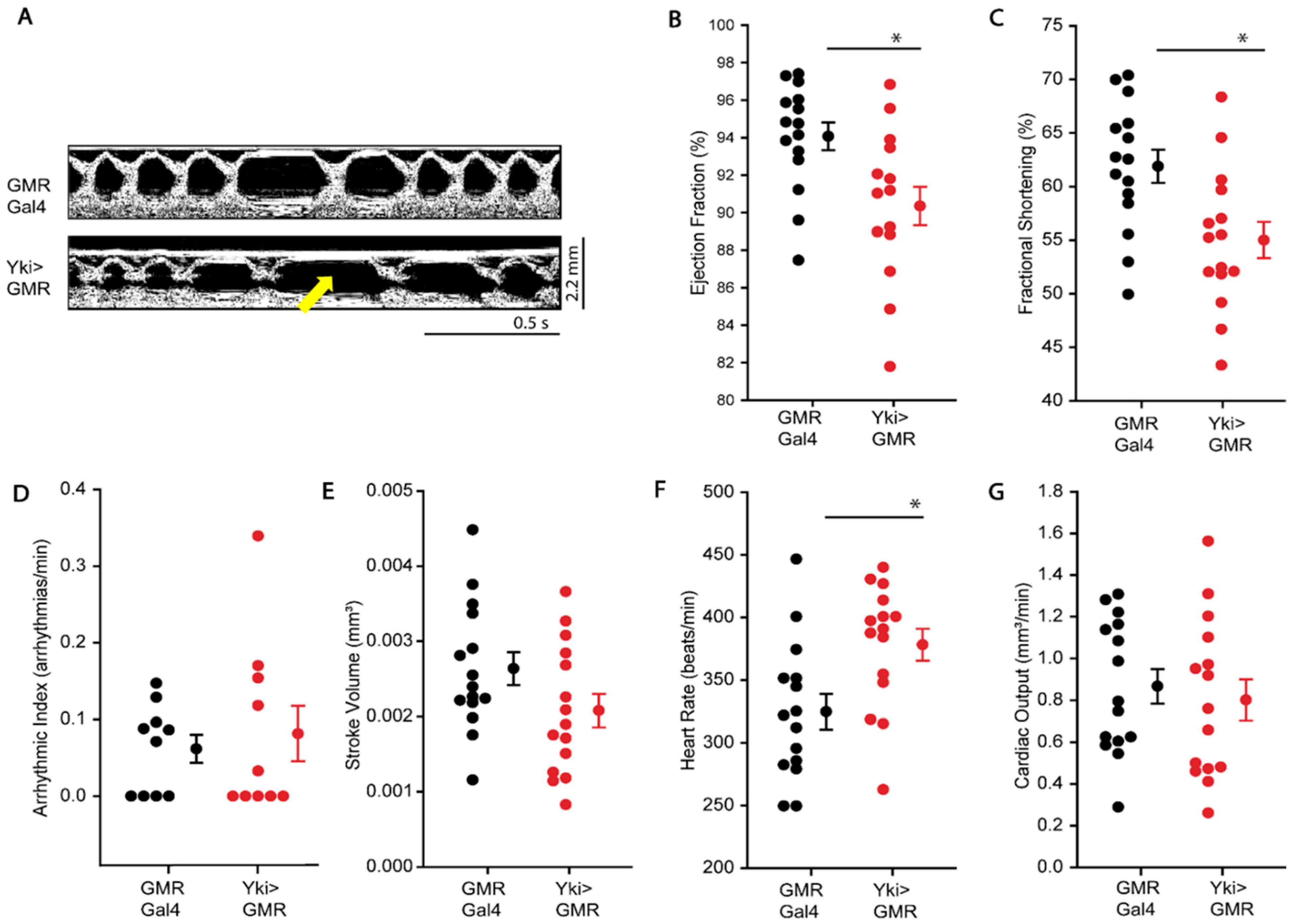
3.2. Overgrowth Causes Systemic Cardiac Dysfunction in Drosophila
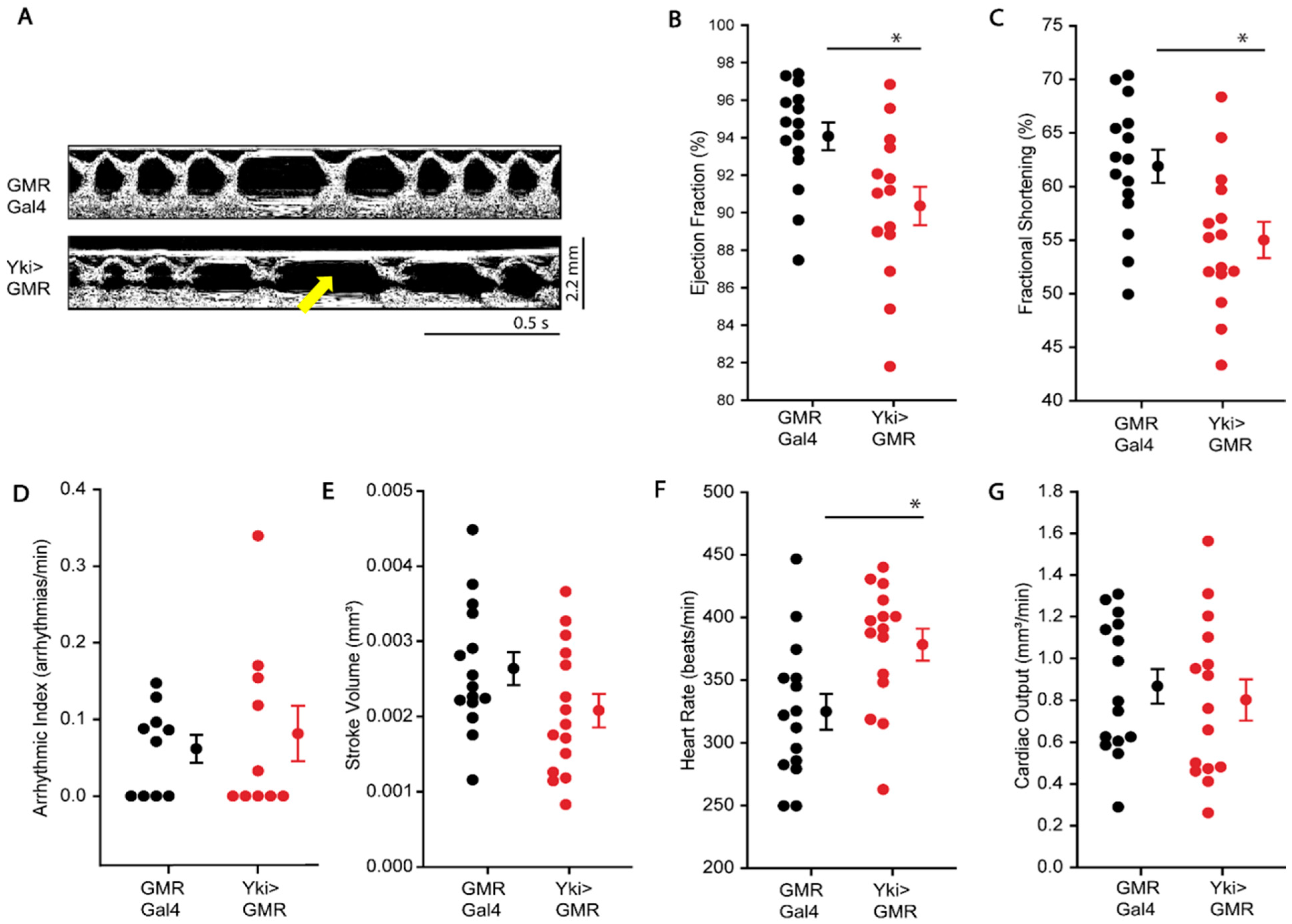
3.3. Effect of Overexpression of Yorkie-Induced Overgrowth on Cardiac Function
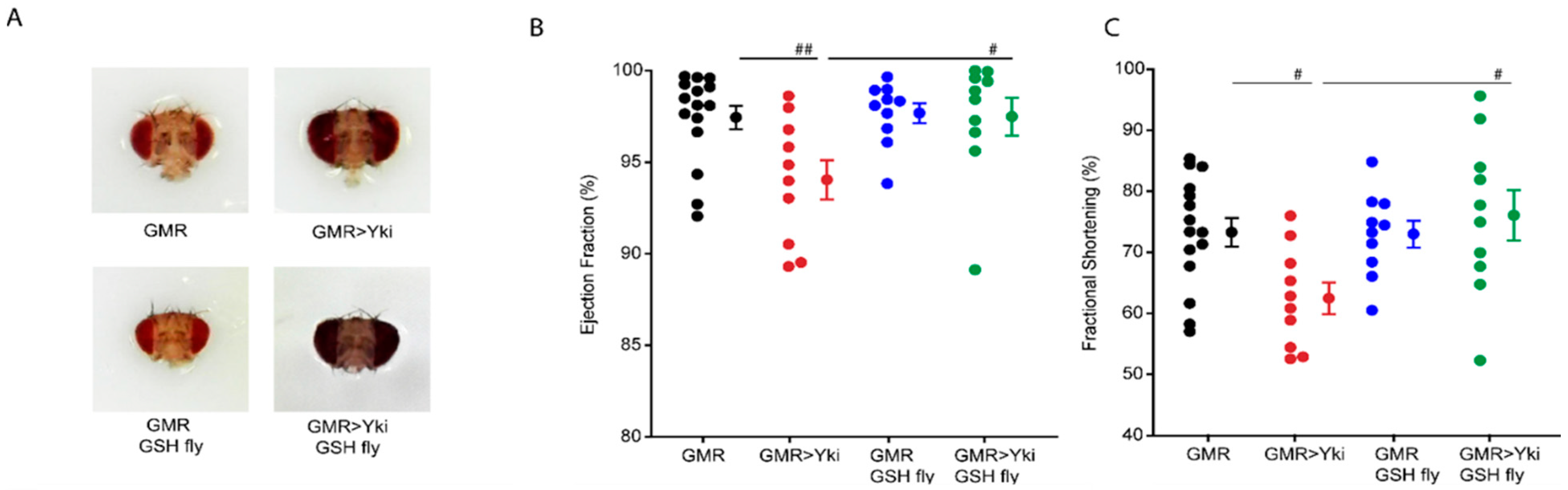
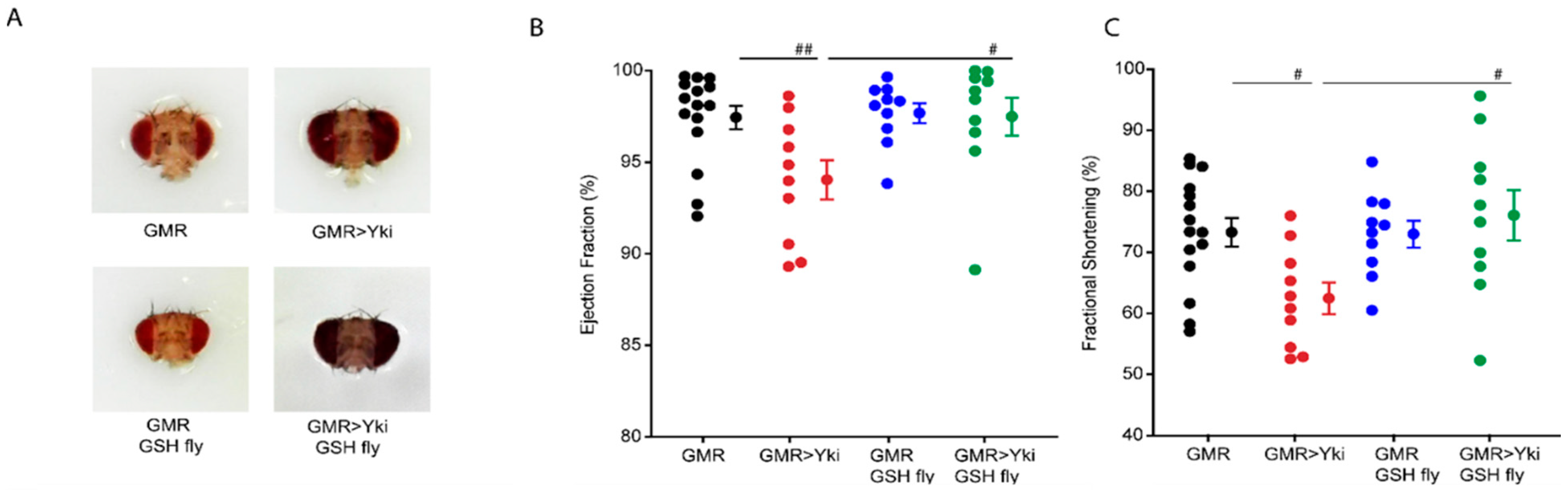
3.4. Quenching Reactive Oxygen Species Rescues Cardiac Phenotypes
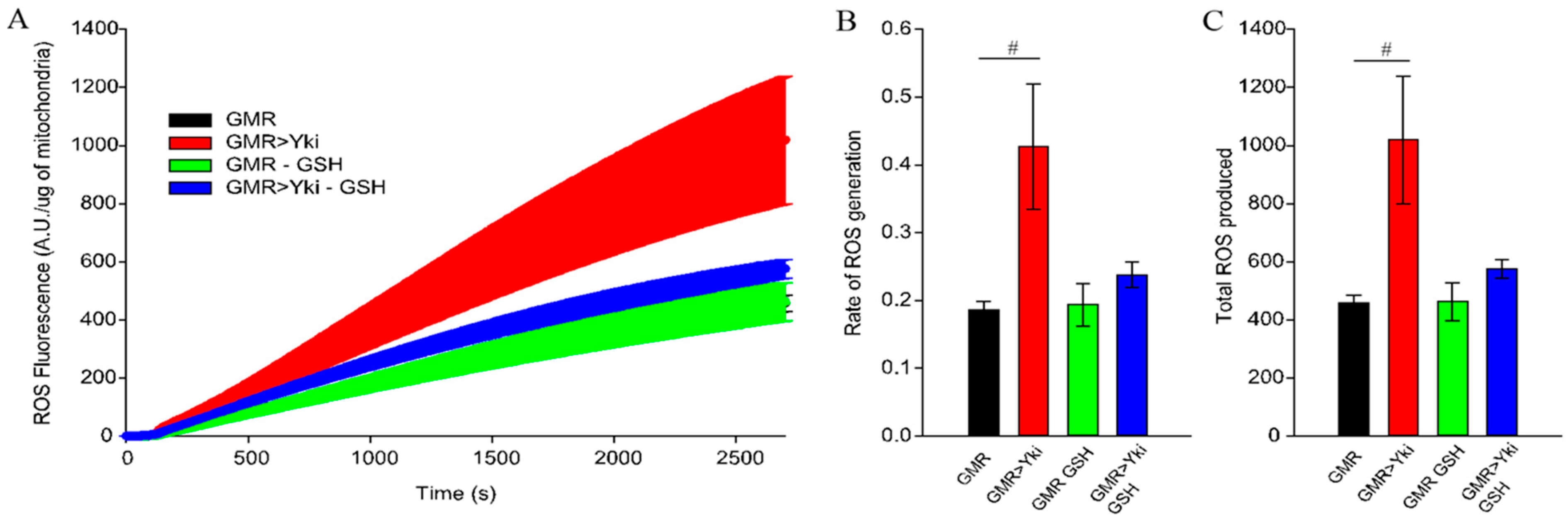
3.5. Systemic Reactive Oxygen Species Increase in Yki-Overexpressing Flies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.L.; Xu, J.; Kochanek, K.D.; Arias, E. Mortality in the United States, 2017; NCHS Data Brief: Hyattsville, MD, USA, 2018; pp. 1–8.
- von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. Cachexia Sarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: Facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Perez, I.E.; Taveras Alam, S.; Hernandez, G.A.; Sancassani, R. Cancer Therapy-Related Cardiac Dysfunction: An Overview for the Clinician. Clin. Med. Insights Cardiol. 2019, 13, 1179546819866445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamo, C.E.; Bloom, M.W. Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers 2019, 11, 571. [Google Scholar]
- Hamo, C.E.; Bloom, M.W. Getting to the Heart of the Matter: An Overview of Cardiac Toxicity Related to Cancer Therapy. Clin. Med. Insights Cardiol. 2015, 9 (Suppl. 2), 47–51. [Google Scholar] [CrossRef]
- Kazemi-Bajestani, S.M.R.; Becher, H.; Butts, C.; Basappa, N.S.; Smylie, M.; Joy, A.A.; Sangha, R.; Gallivan, A.; Chu, Q.; Baracos, V.E. Undiagnosed cardiac deficits in non-small cell carcinoma patients in the candidate population for anti-cachexia clinical trials. Support. Care Cancer 2019, 27, 1551–1561. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, H.; Li, X.; Sun, Y. Pay attention to cardiac remodeling in cancer cachexia. Support. Care Cancer 2016, 24, 3253–3259. [Google Scholar] [CrossRef]
- Kazemi-Bajestani, S.M.; Becher, H.; Fassbender, K.; Chu, Q.; Baracos, V.E. Concurrent evolution of cancer cachexia and heart failure: Bilateral effects exist. J. Cachexia Sarcopenia Muscle 2014, 5, 95–104. [Google Scholar] [CrossRef]
- Barkhudaryan, A.; Scherbakov, N.; Springer, J.; Doehner, W. Cardiac muscle wasting in individuals with cancer cachexia. ESC Heart Fail. 2017, 4, 458–467. [Google Scholar] [CrossRef]
- Devine, R.D.; Bicer, S.; Reiser, P.J.; Velten, M.; Wold, L.E. Metalloproteinase expression is altered in cardiac and skeletal muscle in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H685–H691. [Google Scholar] [CrossRef] [Green Version]
- Hinch, E.C.; Sullivan-Gunn, M.J.; Vaughan, V.C.; McGlynn, M.A.; Lewandowski, P.A. Disruption of pro-oxidant and antioxidant systems with elevated expression of the ubiquitin proteosome system in the cachectic heart muscle of nude mice. J. Cachexia Sarcopenia Muscle 2013, 4, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietzsch, S.; Ricke-Hoch, M.; Stapel, B.; Hilfiker-Kleiner, D. Modulation of cardiac AKT and STAT3 signalling in preclinical cancer models and their impact on the heart. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1867, 118519. [Google Scholar] [CrossRef] [PubMed]
- Caygill, E.E.; Brand, A.H. The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods Mol. Biol. 2016, 1478, 33–52. [Google Scholar]
- Pyter, L.M.; Suarez-Kelly, L.P.; Carson, W.E., 3rd; Kaur, J.; Bellisario, J.; Bever, S.R. Novel rodent model of breast cancer survival with persistent anxiety-like behavior and inflammation. Behav. Brain Res. 2017, 330, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H.; Johannesen, J.; Shah, K.; Goswami, S.K.; Patel, N.J.; Ponnalagu, D.; Kohut, A.R.; Singh, H. Inhibition of BKCa negatively alters cardiovascular function. Physiol. Rep. 2018, 6, e13748. [Google Scholar] [CrossRef]
- Chaudhury, A.; Wanek, A.; Ponnalagu, D.; Singh, H.; Kohut, A. Use of Speckle Tracking Echocardiography to Detect Induced Regional Strain Changes in the Murine Myocardium by Acoustic Radiation Force. J. Cardiovasc. Imaging 2021, 29, 147–157. [Google Scholar] [CrossRef]
- Kohut, A.; Patel, N.; Singh, H. Comprehensive echocardiography assessment of the right ventricle in murine models. J. Cardiovasc. Ultrasound 2016, 24, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.; Karekar, P.; Shah, K.; Hariharan, G.; Fleyshman, M.; Kaur, H.; Singh, H.; Gururaja Rao, S. Drosophila Voltage-Gated Calcium Channel alpha1-Subunits Regulate Cardiac Function in the Aging Heart. Sci. Rep. 2018, 8, 6910. [Google Scholar] [CrossRef]
- Goswami, S.K.; Ponnalagu, D.; Hussain, A.T.; Shah, K.; Karekar, P.; Gururaja Rao, S.; Meredith, A.L.; Khan, M.; Singh, H. Expression and Activation of BKCa Channels in Mice Protects against Ischemia-Reperfusion Injury of Isolated Hearts by Modulating Mitochondrial Function. Front. Cardiovasc. Med. 2018, 5, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, B.; Dougherty, J.A.; Ponnalagu, D.; Singh, H.; Angelos, M.; Chen, C.A.; Khan, M. Measurement of Oxidative Stress Markers In Vitro Using Commercially Available Kits. In Measuring Oxidants and Oxidative Stress in Biological Systems; Berliner, L.J., Parinandi, N.L., Eds.; Springer: Cham, Switzerland, 2020; Volume 34, pp. 39–60. [Google Scholar]
- Singh, H.; Lu, R.; Rodriguez, P.F.; Wu, Y.; Bopassa, J.C.; Stefani, E.; Toro, L. Visualization and quantification of cardiac mitochondrial protein clusters with STED microscopy. Mitochondrion 2012, 12, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Ponnalagu, D.; Gururaja Rao, S.; Farber, J.; Xin, W.; Hussain, A.T.; Shah, K.; Tanda, S.; Berryman, M.; Edwards, J.C.; Singh, H. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion 2016, 27, 6–14. [Google Scholar] [CrossRef]
- Lee, A.; Lin, A.; Shah, K.; Singh, H.; Miller, V.; Gururaja Rao, S. Optimization of Non-Thermal Plasma Treatment in an In Vivo Model Organism. PLoS ONE 2016, 11, e0160676. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Asp, M.L.; Nishijima, Y.; Belury, M.A. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int. J. Oncol. 2011, 39, 1321–1326. [Google Scholar]
- Saavedra, P.; Perrimon, N. Drosophila as a Model for Tumor-Induced Organ Wasting. Adv. Exp. Med. Biol. 2019, 1167, 191–205. [Google Scholar]
- Oh, H.; Irvine, K.D. Yorkie: The final destination of Hippo signaling. Trends Cell Biol. 2010, 20, 410–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YApp. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, R.; Gururaja-Rao, S.; Jones, K.T.; Slattery, M.; Negre, N.; Braas, D.; Christofk, H.; White, K.P.; Mann, R.; Banerjee, U. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev. 2012, 26, 2027–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, C.N.; Smith, Y.E.; Cao, Y.; Burrows, A.D.; Cross, R.S.; Ling, X.; Redvers, R.P.; Doherty, J.P.; Eckhardt, B.L.; Natoli, A.L.; et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Models Mech. 2015, 8, 237–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thackeray, J.T.; Pietzsch, S.; Stapel, B.; Ricke-Hoch, M.; Lee, C.W.; Bankstahl, J.P.; Scherr, M.; Heineke, J.; Scharf, G.; Haghikia, A.; et al. Insulin supplementation attenuates cancer-induced cardiomyopathy and slows tumor disease progression. JCI Insight 2017, 2, e93098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi-Bajestani, S.M.R.; Becher, H.; Butts, C.; Basappa, N.S.; Smylie, M.; Joy, A.A.; Sangha, R.; Gallivan, A.; Kavsak, P.; Chu, Q.; et al. Rapid atrophy of cardiac left ventricular mass in patients with non-small cell carcinoma of the lung. J. Cachexia Sarcopenia Muscle 2019, 10, 1070–1082. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Marcondes, M.C.; Tisdale, M.J. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002, 180, 69–74. [Google Scholar] [CrossRef]
- Razeghi, P.; Baskin, K.K.; Sharma, S.; Young, M.E.; Stepkowski, S.; Essop, M.F.; Taegtmeyer, H. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem. Biophys. Res. Commun. 2006, 342, 361–364. [Google Scholar] [CrossRef]
- Bjorklund, G.; Dadar, M.; Aaseth, J.; Chirumbolo, S.; Pen, J.J. Cancer-associated cachexia, reactive oxygen species, and nutrition therapy. Curr. Med. Chem. 2019, 26, 5728–5744. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ejection Fraction | Fractional Shortening | Stroke Volume | Heart Rate | Cardiac Output | |
|---|---|---|---|---|---|
| Yki |  |  | NC |  | NC |
| RasV12 | 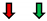 | 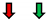 | NC |  | NC |
| Pi3K | 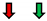 | 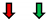 |  |  |  |
| Hep Act |  |  |  |  |  |
 Dpp Gal4;
Dpp Gal4;  GMR Gal4;
GMR Gal4;  Eyless Gal4; NC: No Change.
Eyless Gal4; NC: No Change.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karekar, P.; Jensen, H.N.; Russart, K.L.G.; Ponnalagu, D.; Seeley, S.; Sanghvi, S.; Smith, S.A.; Pyter, L.M.; Singh, H.; Gururaja Rao, S. Tumor-Induced Cardiac Dysfunction: A Potential Role of ROS. Antioxidants 2021, 10, 1299. https://doi.org/10.3390/antiox10081299
Karekar P, Jensen HN, Russart KLG, Ponnalagu D, Seeley S, Sanghvi S, Smith SA, Pyter LM, Singh H, Gururaja Rao S. Tumor-Induced Cardiac Dysfunction: A Potential Role of ROS. Antioxidants. 2021; 10(8):1299. https://doi.org/10.3390/antiox10081299
Chicago/Turabian StyleKarekar, Priyanka, Haley N. Jensen, Kathryn L. G. Russart, Devasena Ponnalagu, Sarah Seeley, Shridhar Sanghvi, Sakima A. Smith, Leah M. Pyter, Harpreet Singh, and Shubha Gururaja Rao. 2021. "Tumor-Induced Cardiac Dysfunction: A Potential Role of ROS" Antioxidants 10, no. 8: 1299. https://doi.org/10.3390/antiox10081299
APA StyleKarekar, P., Jensen, H. N., Russart, K. L. G., Ponnalagu, D., Seeley, S., Sanghvi, S., Smith, S. A., Pyter, L. M., Singh, H., & Gururaja Rao, S. (2021). Tumor-Induced Cardiac Dysfunction: A Potential Role of ROS. Antioxidants, 10(8), 1299. https://doi.org/10.3390/antiox10081299