
Pterostilbene Changes Epigenetic Marks at Enhancer Regions of Oncogenes in Breast Cancer Cells

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment with Pterostilbene (PTS)
2.2. Chromatin Immunoprecipitation (ChIP) Sequencing (ChIP-Seq)
2.3. Analysis of ChIP-Seq Data
2.4. DNA Isolation and Pyrosequencing
2.5. RNA Isolation, cDNA Synthesis and QPCR
2.6. Statistical Analysis
3. Results
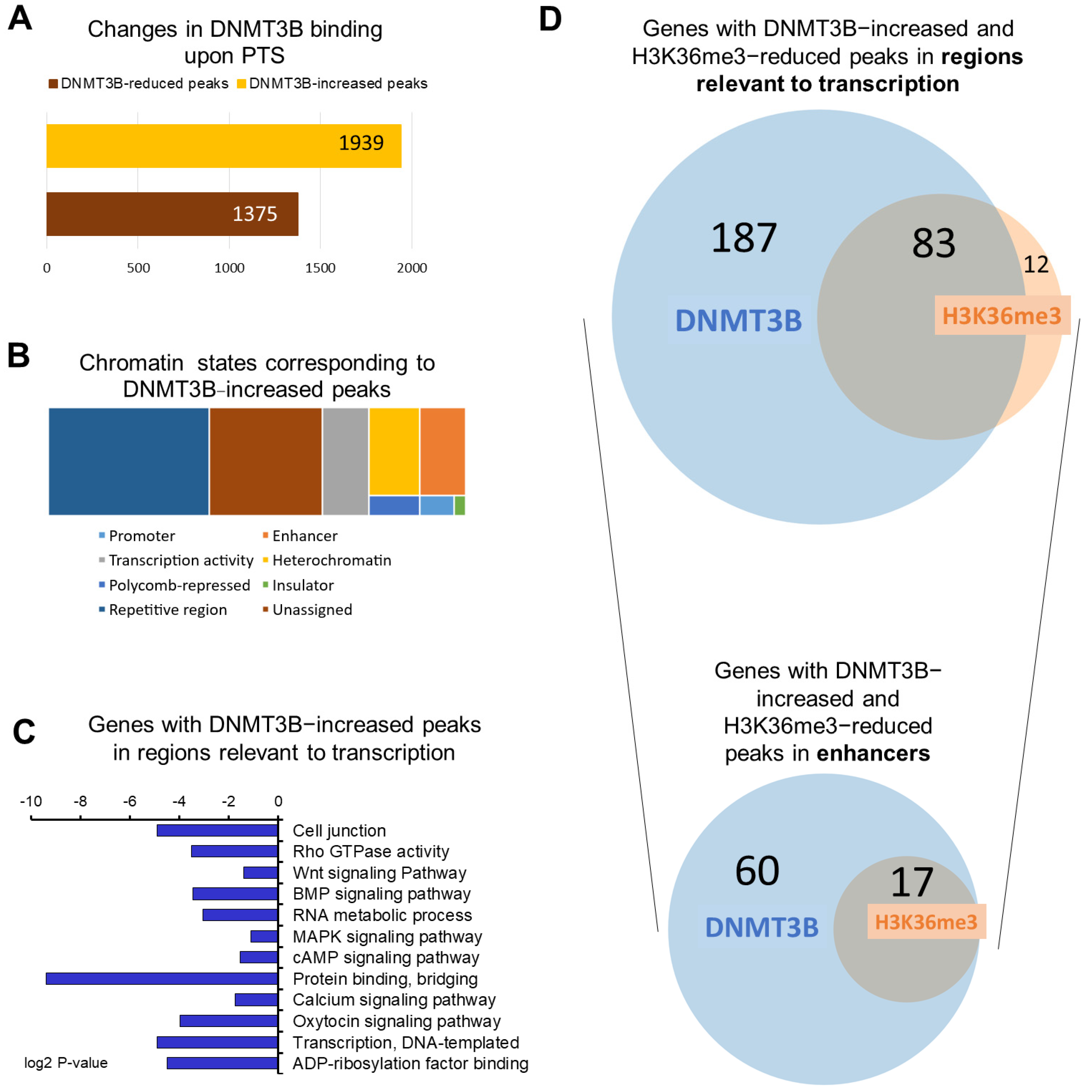
3.1. Overview of DNMT3B Binding in Highly Invasive MCF10CA1a Breast Cancer Cells in Response to Pterostilbene (PTS)
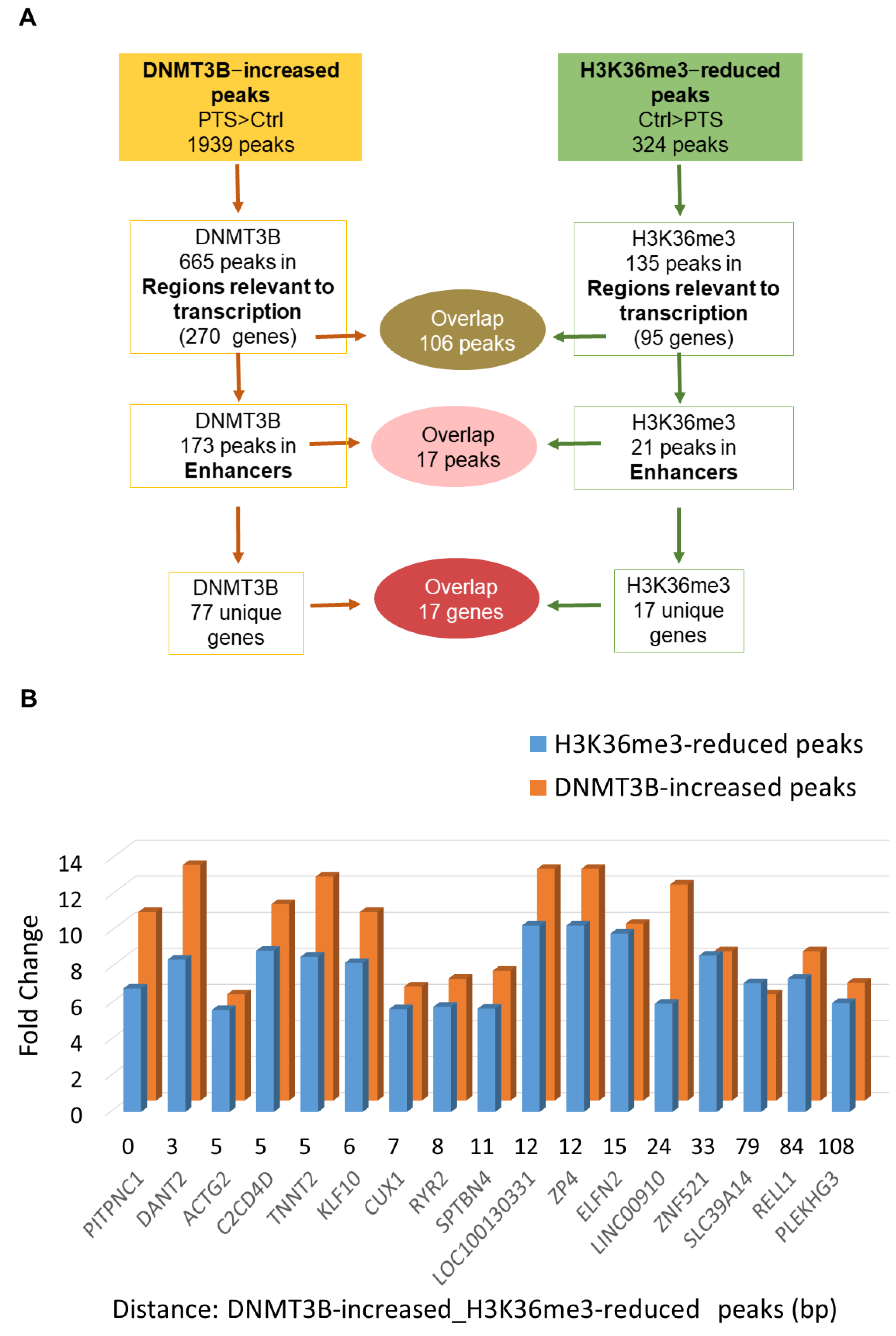
3.2. H3K36me3 Occupancy Is Reduced at Regions Enriched with DNMT3B in Response to PTS
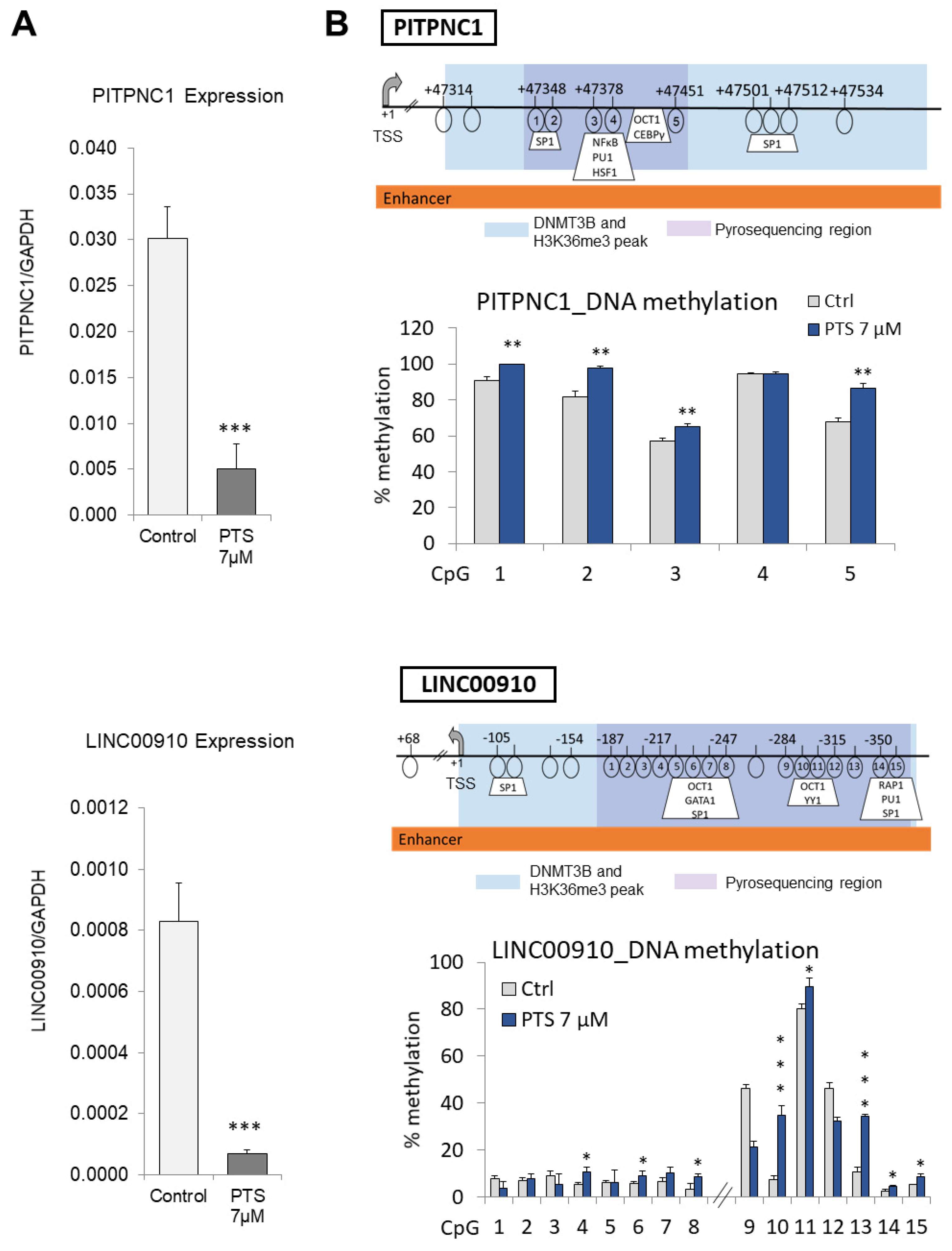
3.3. Candidate Genes with Enhancers That Are Characterized by Increased DNMT3B and Reduced H3K36me3 Occupancy Are Hypermethylated and Downregulated upon Exposure to PTS
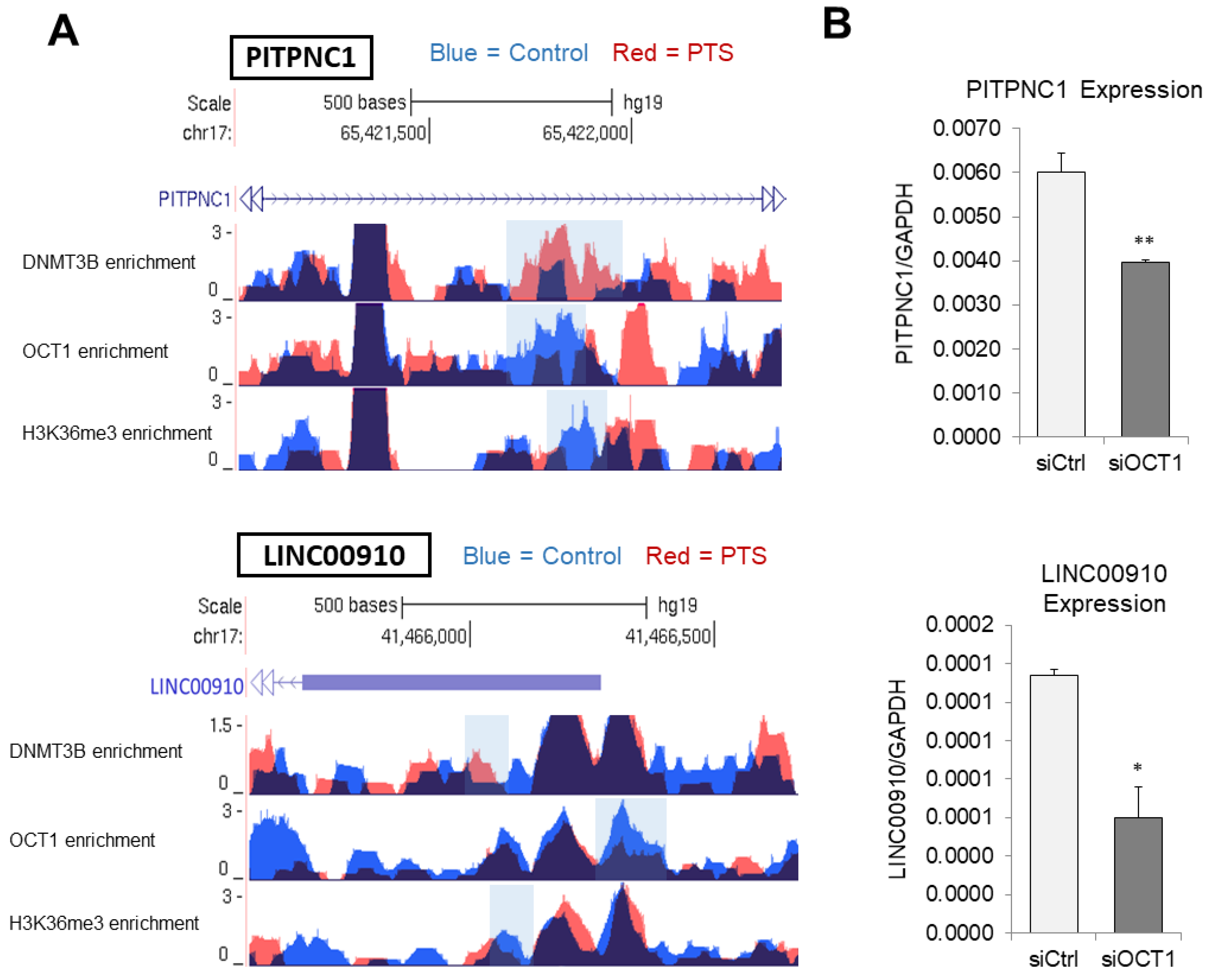
3.4. DNMT3B Enrichment and H3K36me3 Reduced Occupancy Coincide with a Decrease in OCT1 Binding at Target Enhancers in Response to PTS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, O.A.; Esteller, M. Epigenetic Modifications in Breast Cancer and Their Role in Personalized Medicine. Am. J. Pathol. 2013, 183, 1052–1063. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [Green Version]
- Xiang, T.-X.; Yuan, Y.; Li, L.; Wang, Z.-H.; Dan, L.-Y.; Chen, Y.; Ren, G.-S.; Tao, Q. Aberrant promoter CpG methylation and its translational applications in breast cancer. Chin. J. Cancer 2013, 32, 12–20. [Google Scholar] [CrossRef]
- Kolovos, P.; Knoch, T.A.; Grosveld, F.G.; Cook, P.R.; Papantonis, A. Enhancers and silencers: An integrated and simple model for their function. Epigenet. Chrom. 2012, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Angeloni, A.; Bogdanovic, O. Enhancer DNA methylation: Implications for gene regulation. Essays Biochem. 2019, 63, 707–715. [Google Scholar] [CrossRef]
- Sur, I.; Taipale, I.S.J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Ding, M.; Liu, Y.; Liao, X.; Zhan, H.; Liu, Y.; Huang, W. Enhancer RNAs (eRNAs): New Insights into Gene Transcription and Disease Treatment. J. Cancer 2018, 9, 2334–2340. [Google Scholar] [CrossRef] [Green Version]
- Zentner, G.E.; Tesar, P.J.; Scacheri, P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011, 21, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Lin, C. Enhancer, epigenetics, and human disease. Curr. Opin. Genet. Dev. 2016, 36, 27–33. [Google Scholar] [CrossRef]
- Pulakanti, K.; Pinello, L.; Stelloh, C.; Blinka, S.; Allred, J.; Milanovich, S.; Kiblawi, S.; Peterson, J.; Wang, A.; Yuan, G.-C.; et al. Enhancer transcribed RNAs arise from hypomethylated, Tet-occupied genomic regions. Epigenetics 2013, 8, 1303–1320. [Google Scholar] [CrossRef] [Green Version]
- Stefanska, B.; Rudnicka, K.; Bednarek, A.; Fabianowska-Majewska, K. Hypomethylation and induction of retinoic acid receptor beta 2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. Eur. J. Pharmacol. 2010, 638, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, B.; Salamé, P.; Bednarek, A.; Fabianowska-Majewska, K. Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. Br. J. Nutr. 2011, 107, 781–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papoutsis, A.J.; Borg, J.L.; Selmin, O.I.; Romagnolo, D.F. BRCA-1 promoter hypermethylation and silencing induced by the aromatic hydrocarbon receptor-ligand TCDD are prevented by resveratrol in MCF-7 Cells. J. Nutr. Biochem. 2012, 23, 1324–1332. [Google Scholar] [CrossRef]
- Gracia, A.; Elcoroaristizabal, X.; Fernández-Quintela, A.; Miranda, J.; Bediaga, N.G.; De Pancorbo, M.M.; Rimando, A.M.; Portillo, M.P. Fatty acid synthase methylation levels in adipose tissue: Effects of an obesogenic diet and phenol compounds. Genes Nutr. 2014, 9, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogens 2016, 37, 656–668. [Google Scholar] [CrossRef]
- Medina-Aguilar, R.; Perez-Plasencia, C.; Marchat, L.; Gariglio, P.; Garcia-Mena, J.; Cuevas, S.R.; Ruiz-Garcia, E.; La Vega, H.A.-D.; Juárez, J.H.; Flores-Pérez, A.; et al. Methylation Landscape of Human Breast Cancer Cells in Response to Dietary Compound Resveratrol. PLoS ONE 2016, 11, e0157866. [Google Scholar] [CrossRef] [PubMed]
- Beetch, M.; Harandi-Zadeh, S.; Shen, K.; Lubecka, K.; Kitts, D.D.; O’Hagan, H.; Stefanska, B. Dietary antioxidants remodel DNA methylation patterns in chronic disease. Br. J. Pharmacol. 2019, 177, 1382–1408. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yan, X.; Duan, W.; Yan, J.; Yi, W.; Liang, Z.; Wang, N.; Li, Y.; Chen, W.; Yu, S.; et al. Pterostilbene Exerts Antitumor Activity via the Notch1 Signaling Pathway in Human Lung Adenocarcinoma Cells. PLoS ONE 2013, 8, e62652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratyuk, T.P.; Park, E.-J.; Marler, L.E.; Ahn, S.; Yuan, Y.; Choi, Y.; Yu, R.; van Breemen, R.; Sun, B.; Hoshino, J.; et al. Resveratrol derivatives as promising chemopreventive agents with improved potency and selectivity. Mol. Nutr. Food Res. 2011, 55, 1249–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, G.; Buscemi, S.; Galvano, F.; Mistretta, A.; Marventano, S.; Vela, V.L.; Drago, F.; Gangi, S.; Basile, F.; Biondi, A. Mediterranean diet and cancer: Epidemiological evidence and mechanism of selected aspects. BMC Surg. 2013, 13, S14. [Google Scholar] [CrossRef] [Green Version]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [Green Version]
- Williamson, G.; Manach, C. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am. J. Clin. Nutr. 2005, 81, 243S–255S. [Google Scholar] [CrossRef] [PubMed]
- Beetch, M.; Lubecka, K.; Shen, K.; Flower, K.; Harandi-Zadeh, S.; Suderman, M.; Flanagan, J.; Stefanska, B. Stilbenoid-Mediated Epigenetic Activation of Semaphorin 3A in Breast Cancer Cells Involves Changes in Dynamic Interactions of DNA with DNMT3A and NF1C Transcription Factor. Mol. Nutr. Food Res. 2019, 63, e1801386. [Google Scholar] [CrossRef]
- Beetch, M.; Boycott, C.; Harandi-Zadeh, S.; Yang, T.; Martin, B.; Dixon-McDougall, T.; Ren, K.; Gacad, A.; Dupuis, J.H.; Ullmer, M.; et al. Pterostilbene leads to DNMT3B-mediated DNA methylation and silencing of OCT1-targeted oncogenes in breast cancer cells. J. Nutr. Biochem. 2021, 108815. [Google Scholar] [CrossRef]
- Brown, S.E.; Suderman, M.; Hallett, M.; Szyf, M. DNA demethylation induced by the methyl-CpG-binding domain protein MBD3. Gene 2008, 420, 99–106. [Google Scholar] [CrossRef]
- Ciernia, A.V.; Laufer, B.I.; Hwang, H.; Dunaway, K.W.; Mordaunt, C.; Coulson, R.; Yasui, D.H.; LaSalle, J.M. Epigenomic Convergence of Neural-Immune Risk Factors in Neurodevelopmental Disorder Cortex. Cereb. Cortex 2019, 30, 640–655. [Google Scholar] [CrossRef]
- Laufer, B.I.; Hwang, H.; Ciernia, A.V.; Mordaunt, C.E.; LaSalle, J.M. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics 2019, 14, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformation 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137–R139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.; Thomas, S.; Holloway, A.K.; Pollard, K.S. Features that define the best ChIP-seq peak calling algorithms. Briefings Bioinform. 2016, 18, 441–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nat. Cell Biol. 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef]
- Gagliardi, M.; Strazzullo, M.; Matarazzo, M.R. DNMT3B Functions: Novel Insights from Human Disease. Front. Cell Dev. Biol. 2018, 6, 140. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Gacquer, D.; Van Heurck, R.; Kumar, D.; Wojno, M.; Bilheu, A.; Herpoel, A.; Lambert, N.; Cheron, J.; Polleux, F.; et al. Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell 2018, 173, 1370–1384.e16. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Liu, C.; Yang, Y.; Song, L.; Liu, X.; Wang, C.; Peng, Z.; Li, H.; Zhong, L. Panx1 promotes invasion-metastasis cascade in hepatocellular carcinoma. J. Cancer 2019, 10, 5681–5688. [Google Scholar] [CrossRef] [Green Version]
- Jalaleddine, N.; El-Hajjar, L.; Dakik, H.; Shaito, A.; Saliba, J.; Safi, R.; Zibara, K.; El-Sabban, M. Pannexin1 Is Associated with Enhanced Epithelial-To-Mesenchymal Transition in Human Patient Breast Cancer Tissues and in Breast Cancer Cell Lines. Cancers 2019, 11, 1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, J.; Wang, Z.; Wang, P.; Li, S. Upregulation of SOX2 activated LncRNA PVT1 expression promotes breast cancer cell growth and invasion. Biochem. Biophys. Res. Commun. 2017, 493, 429–436. [Google Scholar] [CrossRef]
- Sarver, A.L.; Murray, C.D.; Temiz, N.A.; Tseng, Y.-Y.; Bagchi, A. MYCandPVT1synergize to regulate RSPO1 levels in breast cancer. Cell Cycle 2016, 15, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Halberg, N.; Sengelaub, C.A.; Navrazhina, K.; Molina, H.; Uryu, K.; Tavazoie, S.F. PITPNC1 Recruits RAB1B to the Golgi Network to Drive Malignant Secretion. Cancer Cell 2016, 29, 339–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.R.; Chase, P.B.; Pinto, J.R. Troponin through the looking-glass: Emerging roles beyond regulation of striated muscle contraction. Oncotarget 2017, 9, 1461–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, J.; Pereira, R.; Oliveira, J.; Alves, Â.; Marques-Magalhães, A.; Frutuoso, A.; Leal, C.; Barros, N.; Fernandes, R.; Almeida, D.Q.; et al. Structural and molecular analysis of the cancer prostate cell line PC3: Oocyte zona pellucida glycoproteins. Tissue Cell 2018, 55, 91–106. [Google Scholar] [CrossRef]
- Guerrero-Martínez, J.A.; Reyes, J.C. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci. Rep. 2018, 8, 2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daams, R.; Massoumi, R. Nemo-Like Kinase in Development and Diseases: Insights from Mouse Studies. Int. J. Mol. Sci. 2020, 21, 9203. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Liangpunsakul, S.; Wang, L. Long non-coding RNA in liver metabolism and disease: Current status. Liver Res. 2017, 1, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Satpathy, A.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef]
- Perry, R.B.-T.; Ulitsky, I. The functions of long noncoding RNAs in development and stem cells. Development 2016, 143, 3882–3894. [Google Scholar] [CrossRef] [Green Version]
- Kedinger, V.; Sansregret, L.; Harada, R.; Vadnais, C.; Cadieux, C.; Fathers, K.; Park, M.; Nepveu, A. p110 CUX1 Homeodomain Protein Stimulates Cell Migration and Invasion in Part through a Regulatory Cascade Culminating in the Repression of E-cadherin and Occludin. J. Biol. Chem. 2009, 284, 27701–27711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, L.J.; Hawsawi, O.; Sweeney, J.; Bowen, N.; Hudson, T.; Odero-Marah, V. CCAAT-displacement protein/cut homeobox transcription factor (CUX1) represses estrogen receptor-alpha (ER-α) in triple-negative breast cancer cells and can be antagonized by muscadine grape skin extract (MSKE). PLoS ONE 2019, 14, e0214844. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qin, K.; Zhang, Y.; Gong, J.; Li, N.; Lv, D.; Xiang, R.; Tan, X. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 411, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.-O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-Stimulated, Agonist-Stimulated and Store-Operated Ca2+ Influx in MDA-MB-468 Breast Cancer Cells and the Effect of EGF-Induced EMT on Calcium Entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.-Q.; Wang, J.; Chen, S.-Y. Methylation modification in gastric cancer and approaches to targeted epigenetic therapy (Review). Int. J. Oncol. 2017, 50, 1921–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlady, R.A.; Novakova, S.; Opavska, J.; Klinkebiel, D.; Peters, S.L.; Bies, J.; Hannah, J.; Iqbal, J.; Anderson, K.M.; Siebler, H.M.; et al. Loss of Dnmt3b function upregulates the tumor modifier Ment and accelerates mouse lymphomagenesis. J. Clin. Investig. 2012, 122, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhang, H.; Wang, Y.; Li, X.; Lu, P.; Dong, F.; Pang, Y.; Ma, S.; Cheng, H.; Hao, S.; et al. Loss of Dnmt3b accelerates MLL-AF9 leukemia progression. Leukemia 2016, 30, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Arrieta, I.; Hernández-Sotelo, D.; Castro-Coronel, Y.; Leyva-Vazquez, M.A.; Illades-Aguiar, B. DNMT3B modulates the expression of cancer-related genes and downregulates the expression of the gene VAV3 via methylation. Am. J. Cancer Res. 2017, 7, 77–87. [Google Scholar] [PubMed]
- Roll, J.D.; Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer 2008, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.-F.; Zhang, Y.-J.; Lei, Y.; Wu, D.-W.; Ma, T.-H.; Liu, X.-Q. Hypermethylation of the CHRDL1 promoter induces proliferation and metastasis by activating Akt and Erk in gastric cancer. Oncotarget 2017, 8, 23155–23166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-L.; Choi, H.S.; Kim, J.-H.; Jeong, D.K.; Kim, K.-S.; Lee, D.-S. Dihydrotanshinone-Induced NOX5 Activation Inhibits Breast Cancer Stem Cell through the ROS/Stat3 Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 9296439. [Google Scholar] [CrossRef]
- Shikauchi, Y.; Saiura, A.; Kubo, T.; Niwa, Y.; Yamamoto, J.; Murase, Y.; Yoshikawa, H. SALL3 Interacts with DNMT3A and Shows the Ability To Inhibit CpG Island Methylation in Hepatocellular Carcinoma. Mol. Cell. Biol. 2009, 29, 1944–1958. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan, H.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, A.R.; Peng, M.; Podicheti, R.; Sriramkumar, S.; Kamplain, C.M.; Rusch, D.B.; Shields, C.E.D.; Sears, C.L.; O’Hagan, H.M. Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis. Cancer Res. 2017, 77, 3467–3478. [Google Scholar] [CrossRef] [Green Version]
- Eräsalo, H.; Hämäläinen, M.; Leppänen, T.; Mäki-Opas, I.; Laavola, M.; Haavikko, R.; Yli-Kauhaluoma, J.; Moilanen, E. Natural Stilbenoids Have Anti-Inflammatory Properties in Vivo and Down-Regulate the Production of Inflammatory Mediators NO, IL6, and MCP1 Possibly in a PI3K/Akt-Dependent Manner. J. Nat. Prod. 2018, 81, 1131–1142. [Google Scholar] [CrossRef]
- Liu, Y.; You, Y.; Lu, J.; Chen, X.; Yang, Z. Recent Advances in Synthesis, Bioactivity, and Pharmacokinetics of Pterostilbene, an Important Analog of Resveratrol. Molecules 2020, 25, 5166. [Google Scholar] [CrossRef] [PubMed]
- Levenson, A.S.; Kumar, A. Pterostilbene as a Potent Chemopreventive Agent in Cancer. In Natural Products for Cancer Chemoprevention; Single Compounds and Combinations; Pezzuto, J.M., Vang, O., Eds.; Springer Nature Switzerland AG: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Bu, J.; Chen, A.; Yan, X.; He, F.; Dong, Y.; Zhou, Y.; He, J.; Zhan, D.; Lin, P.; Hayashi, Y.; et al. SETD2-mediated crosstalk between H3K36me3 and H3K79me2 in MLL-rearranged leukemia. Leukemia 2017, 32, 890–899. [Google Scholar] [CrossRef]
- Lien, H.-C.; Jeng, Y.-M.; Jhuang, Y.-L.; Yuan, R.-H. Increased Trimethylation of histone H3K36 associates with biliary differentiation and predicts poor prognosis in resectable hepatocellular carcinoma. PLoS ONE 2018, 13, e0206261. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Guo, L.; Duan, Z.J.; Tepper, C.G.; Xue, L.; Chen, X.; Kung, H.-J.; Gao, A.C.; Zou, J.X.; Chen, H.-W. Histone Methyltransferase NSD2/MMSET Mediates Constitutive NF-κB Signaling for Cancer Cell Proliferation, Survival, and Tumor Growth via a Feed-Forward Loop. Mol. Cell. Biol. 2012, 32, 3121–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivenbark, A.G.; Coleman, W.B.; Stahl, B.D. Histone methylation patterns in human breast cancer. FASEB J. 2009, 23, 38.1. [Google Scholar] [CrossRef]
- Zhang, L.-Q.; Li, Q.-Z.; Jin, W.; Zuo, Y.; Guo, S.-C. Genome-wide analysis of H3K36me3 and its regulations to cancer-related genes expression in human cell lines. Biosystems 2018, 171, 59–65. [Google Scholar] [CrossRef]
- Tiedemann, R.L.; Hlady, R.A.; Hanavan, P.D.; Lake, D.F.; Tibes, R.; Lee, J.-H.; Choi, J.-H.; Ho, T.H.; Robertson, K.D. Dynamic reprogramming of DNA methylation in SETD2-deregulated renal cell carcinoma. Oncotarget 2015, 7, 1927–1946. [Google Scholar] [CrossRef] [Green Version]
- Finogenova, K.; Bonnet, J.; Poepsel, S.; Schäfer, I.B.; Finkl, K.; Schmid, K.; Litz, C.; Strauss, M.; Benda, C.; Müller, J. Structural basis for PRC2 decoding of active histone methylation marks H3K36me2/3. eLife 2020, 9, e61964. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 Methylation Antagonizes PRC2-mediated H3K27 Methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alajem, A.; Roth, H.; Ratgauzer, S.; Bavli, D.; Motzik, A.; Lahav, S.; Peled, I.; Ram, O. DNA methylation patterns expose variations in enhancer-chromatin modifications during embryonic stem cell differentiation. PLoS Genet. 2021, 17, e1009498. [Google Scholar] [CrossRef]
- Vázquez-Arreguín, K.; Tantin, D. The Oct1 transcription factor and epithelial malignancies: Old protein learns new tricks. Biochim. Biophys. Acta Bioenerg. 2016, 1859, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakya, A.; Cooksey, R.; Cox, J.E.; Wang, V.; McClain, D.; Tantin, D. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat. Cell Biol. 2009, 11, 320–327. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 2018, 10, 17. [Google Scholar] [CrossRef]
- Baubec, T.; Colombo, D.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nat. Cell Biol. 2015, 520, 243–247. [Google Scholar] [CrossRef]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.L.; Seagroves, T.N. Fluorescence-Activated Cell Sorting of Murine Mammary Cancer Stem-Like Cell Subpopulations with HIF Activity. Methods Mol. Biol. 2018, 247–263. [Google Scholar] [CrossRef]
- Assi, M.; Dauguet, N.; Jacquemin, P. DIE-RNA: A Reproducible Strategy for the Digestion of Normal and Injured Pancreas, Isolation of Pancreatic Cells from Genetically Engineered Mouse Models and Extraction of High Quality RNA. Front. Physiol. 2018, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Porter, W.; Snowden, E.; Hahn, F.; Ferguson, M.; Tong, F.; Dillmore, W.S.; Blaesius, R. High accuracy gene expression profiling of sorted cell subpopulations from breast cancer PDX model tissue. PLoS ONE 2020, 15, e0238594. [Google Scholar] [CrossRef]
- Ischenko, I.; D’Amico, S.; Rao, M.; Li, J.; Hayman, M.J.; Powers, S.; Petrenko, O.; Reich, N.C. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 2021, 12, 1482. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harandi-Zadeh, S.; Boycott, C.; Beetch, M.; Yang, T.; Martin, B.J.E.; Ren, K.; Kwasniak, A.; Dupuis, J.H.; Lubecka, K.; Yada, R.Y.; et al. Pterostilbene Changes Epigenetic Marks at Enhancer Regions of Oncogenes in Breast Cancer Cells. Antioxidants 2021, 10, 1232. https://doi.org/10.3390/antiox10081232
Harandi-Zadeh S, Boycott C, Beetch M, Yang T, Martin BJE, Ren K, Kwasniak A, Dupuis JH, Lubecka K, Yada RY, et al. Pterostilbene Changes Epigenetic Marks at Enhancer Regions of Oncogenes in Breast Cancer Cells. Antioxidants. 2021; 10(8):1232. https://doi.org/10.3390/antiox10081232
Chicago/Turabian StyleHarandi-Zadeh, Sadaf, Cayla Boycott, Megan Beetch, Tony Yang, Benjamin J. E. Martin, Kevin Ren, Anna Kwasniak, John H. Dupuis, Katarzyna Lubecka, Rickey Y. Yada, and et al. 2021. "Pterostilbene Changes Epigenetic Marks at Enhancer Regions of Oncogenes in Breast Cancer Cells" Antioxidants 10, no. 8: 1232. https://doi.org/10.3390/antiox10081232
APA StyleHarandi-Zadeh, S., Boycott, C., Beetch, M., Yang, T., Martin, B. J. E., Ren, K., Kwasniak, A., Dupuis, J. H., Lubecka, K., Yada, R. Y., Howe, L. J., & Stefanska, B. (2021). Pterostilbene Changes Epigenetic Marks at Enhancer Regions of Oncogenes in Breast Cancer Cells. Antioxidants, 10(8), 1232. https://doi.org/10.3390/antiox10081232