
Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease



Abstract
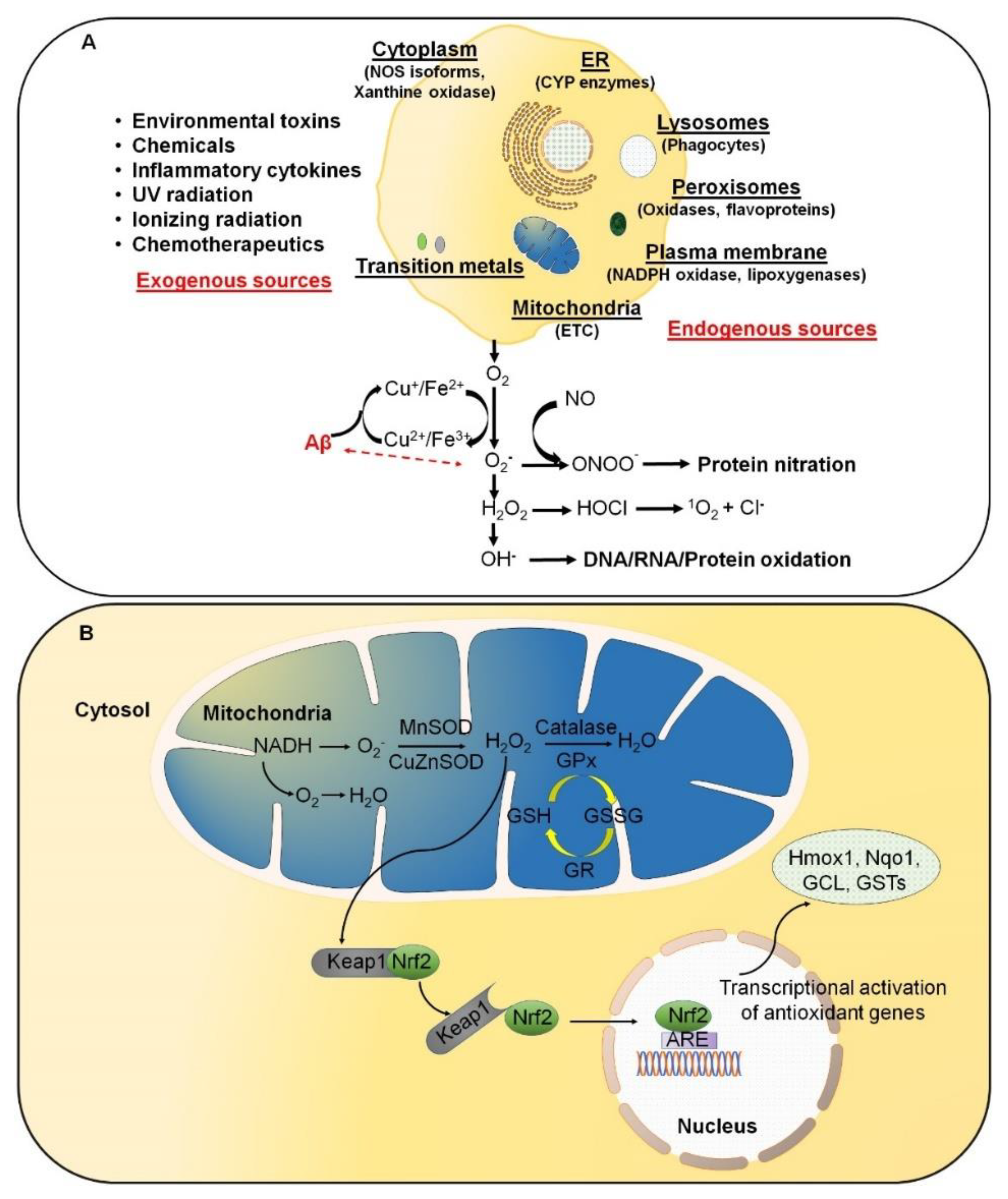
:1. Introduction
2. Markers of Oxidative Stress
3. Linking OS and Proteinopathy in AD
3.1. Oxidative Stress and Aβ Proteinopathy
3.2. Oxidative Stress and Tau Proteinopathy
4. Linking Microbiota with Oxidative Stress and AD
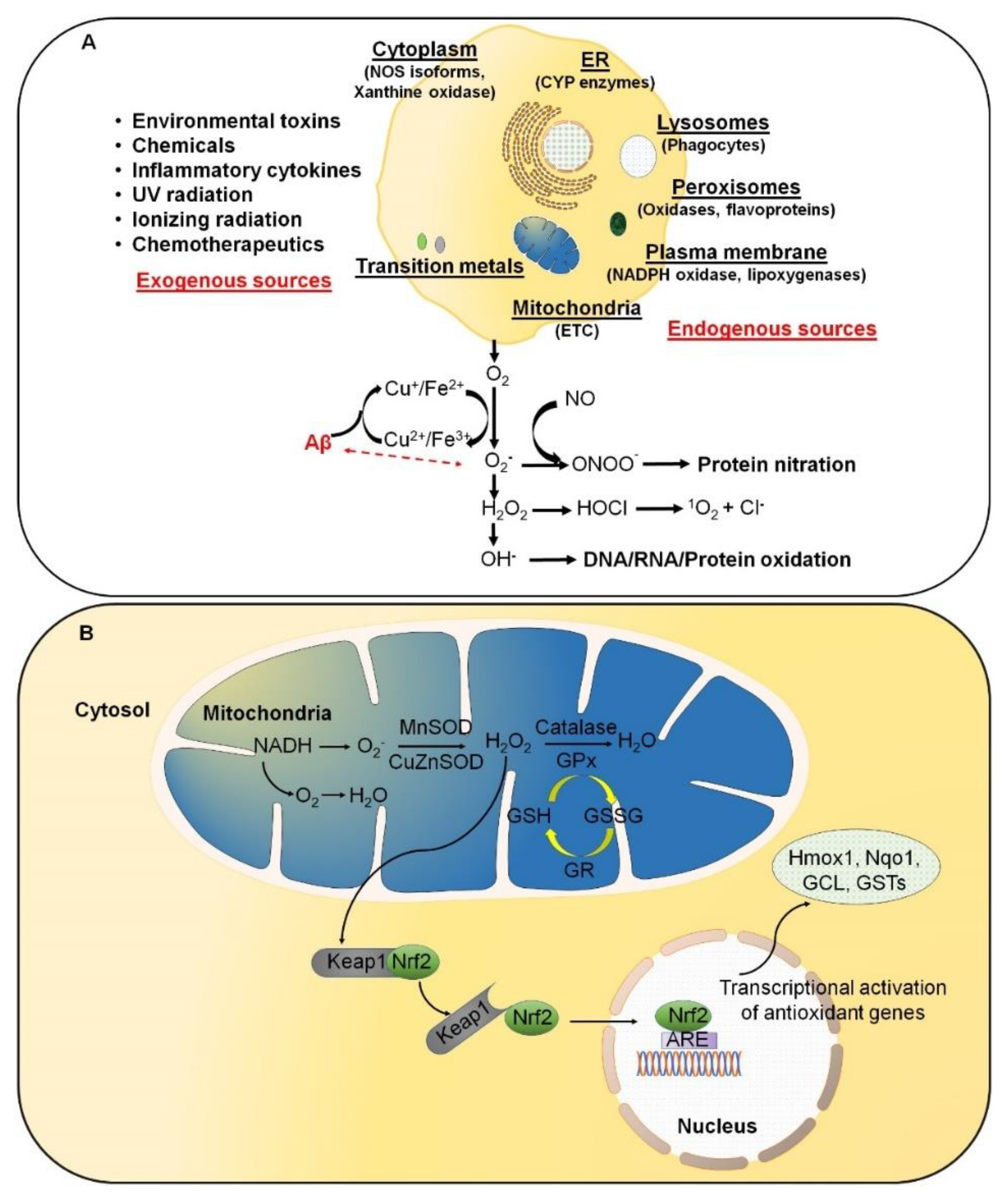
5. Antioxidants and AD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Kiley, P.J.; Storz, G. Exploiting Thiol Modifications. PLoS Biol. 2004, 2, e400. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [Green Version]
- Holland, R.; Fishbein, J.C. Chemistry of the cysteine sensors in kelch-like ECH-associated protein. Antioxid. Redox Signal. 2010, 13, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Niforou, K.; Cheimonidou, C.; Trougakos, I.P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014, 2, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010, 647, 37–74. [Google Scholar]
- Copple, I.M.; Goldring, C.E.; Kitteringham, N.R.; Park, B.K. The Keap1-Nrf2 cellular defense pathway: Mechanisms of regulation and role in protection against drug-induced toxicity. Handb. Exp. Pharmacol. 2010, 196, 233–266. [Google Scholar]
- Hensen, S.M.M.; Heldens, L.; van Enckevort, C.M.W.; Van Genesen, S.T.; Pruijn, G.J.M.; Lubsen, N.H. Activation of the antioxidant response in methionine deprived human cells results in an HSF1-independent increase in HSPA1A mRNA levels. Biochimie 2013, 95, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, E.N.; Iliaki, K.K.; Höhn, A.; Grimm, S.; Papassideri, I.S.; Grune, T.; Trougakos, I.P. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic. Biol. Med. 2013, 65, 1155–1163. [Google Scholar] [CrossRef]
- Tsakiri, E.N.; Sykiotis, G.P.; Papassideri, I.S.; Terpos, E.; Dimopoulos, M.A.; Gorgoulis, V.G.; Bohmann, D.; Trougakos, I.P. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 2013, 12, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Tabner, B.J.; El-Agnaf, O.M.A.; German, M.J.; Fullwood, N.J.; Allsop, D. Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 2005, 33, 1082–1086. [Google Scholar] [CrossRef]
- Pérez, V.I.; Buffenstein, R.; Masamsetti, V.; Leonard, S.; Salmon, A.B.; Mele, J.; Andziak, B.; Yang, T.; Edrey, Y.; Friguet, B.; et al. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl. Acad. Sci. USA 2009, 106, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Nedić, O.; Rattan, S.I.S.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47, 28–38. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A.; et al. β-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Lai, A.Y. Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimers Dis. 2011, 2011, 548380. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Jeandel, C.; Nicolas, M.B.; Dubois, F.; Nabet-Belleville, F.; Penin, F.; Cuny, G. Lipid peroxidation and free radical scavengers in Alzheimer’s disease. Gerontology 1989, 35, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy Samples of Alzheimer’s Cortex Show Increased Peroxidation In Vitro. J. Neurochem. 1990, 55, 342–345. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’ s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [CrossRef]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. Rev. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef] [Green Version]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Du Yan, S.; Yan, S.F.; Stern, D.M. The Biology of the Receptor for Advanced Glycation end Products and Its Ligands. Biochim. Biophys Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Gkogkolou, P.; Böhm, M. Advanced glycation end products: Keyplayers in skin aging? Dermatoendocrinology 2012, 4, 259. [Google Scholar] [CrossRef] [Green Version]
- Maillard, L.C. Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compte-Rendu L’academie Sci. 1912, 154, 66–68. [Google Scholar]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [Green Version]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanbari, H.A.; Ghanbari, K.; Harris, P.L.R.; Jones, P.K.; Kubat, Z.; Castellani, R.J.; Wolozin, B.L.; Smith, M.A.; Perry, G. Oxidative damage in cultured human olfactory neurons from Alzheimer’s disease patients. Aging Cell 2004, 3, 41–44. [Google Scholar] [CrossRef] [Green Version]
- Migliore, L.; Fontana, I.; Trippi, F.; Colognato, R.; Coppedè, F.; Tognoni, G.; Nucciarone, B.; Siciliano, G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol. Aging 2005, 26, 567–573. [Google Scholar] [CrossRef]
- Moreira, P.I.; Harris, P.L.R.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimer Dis. 2007, 12, 195–206. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative Stress-Mediated Regulation of Proteasome Complexes. Mol. Cell. Proteom. 2011, 10, R110.006924. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; Dias, I.H.K.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Rajendran, L.; Annaert, W. Membrane Trafficking Pathways in Alzheimer’s Disease. Traffic 2012, 13, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Zhang, G.; Morrison, C.M.; Bosch, M.E.; Weir, R.A.; Lu, Y.; Dong, W.; Gendelman, H.E. AAV2/1 CD74 Gene Transfer Reduces β-amyloidosis and Improves Learning and Memory in a Mouse Model of Alzheimer’s Disease. Mol. Ther. 2015, 23, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Pimplikar, S.W.; Nixon, R.A.; Robakis, N.K.; Shen, J.; Tsai, L.H. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 2010, 30, 14946–14954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco-Quinto, J.; Eckman, E.A. Endothelin-converting enzymes degrade intracellular β-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J. Biol. Chem. 2013, 288, 5606–5615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Li, Y.; Zhang, X.; Bu, G.; Xu, H.; Zhang, Y.W. Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Kolaj, I.; Imindu Liyanage, S.; Weaver, D.F. Phenylpropanoids and Alzheimer’s disease: A potential therapeutic platform. Neurochem. Int. 2018, 120, 99–111. [Google Scholar] [CrossRef]
- Armstrong, R.A. The pathogenesis of Alzheimer’s disease: A reevaluation of the “amyloid cascade hypothesis”. Int. J. Alzheimers. Dis. 2011, 2011, 630865. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, M.Y.; Aksenova, M.V.; Harris, M.E.; Hensley, K.; Butterfield, D.A.; Carney, J.M. Enhancement of β-Amyloid Peptide Aβ(1–40)-Mediated Neurotoxicity by Glutamine Synthetase. J. Neurochem. 1995, 65, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef] [Green Version]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Hemachandra Reddy, P.; Flint Beal, M. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev. 2005, 49, 618–632. [Google Scholar] [CrossRef]
- Mhatre, M.; Floyd, R.A.; Hensley, K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: Common links and potential therapeutic targets. J. Alzheimers Dis. 2004, 6, 147–157. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, J.H.; Lee, J.P.; Kim, E.M.; Chang, K.A.; Park, C.H.; Jeong, S.J.; Wittendorp, M.C.; Seo, J.H.; Choi, S.H.; et al. Amyloid β peptide induces cytochrome c release from isolated mitochondria. Neuroreport 2002, 13, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Torres, L.L.; Quaglio, N.B.; De Souza, G.T.; Garcia, R.T.; Dati, L.M.M.; Moreira, W.L.; De Melo Loureiro, A.P.; De Souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M.Y. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwon, A.R.; Park, J.S.; Arumugam, T.V.; Kwon, Y.K.; Chan, S.L.; Kim, S.H.; Baik, S.H.; Yang, S.; Yun, Y.K.; Choi, Y.; et al. Oxidative lipid modification of nicastrin enhances amyloidogenic γ-secretase activity in Alzheimer’s disease. Aging Cell 2012, 11, 559–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The Aβ peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Opazo, C.; Huang, X.; Cherny, R.A.; Moir, R.D.; Roher, A.E.; White, A.R.; Cappai, R.; Masters, C.L.; Tanzi, R.E.; Inestrosa, N.C.; et al. Metalloenzyme-like activity of Alzheimer’s disease β-amyloid: Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O. J. Biol. Chem. 2002, 277, 40302–40308. [Google Scholar] [CrossRef] [Green Version]
- Nelson, T.J.; Alkon, D.L. Oxidation of cholesterol by amyloid precursor protein and β-amyloid peptide. J. Biol. Chem. 2005, 280, 7377–7387. [Google Scholar] [CrossRef] [Green Version]
- Ortwerth, B.J.; Olesen, P.R. Ascorbic acid-induced crosslinking of lens proteins: Evidence supporting a Maillard reaction. Biochim. Biophys. Acta BBA Protein Struct. Mol. 1988, 956, 10–22. [Google Scholar] [CrossRef]
- Prabhakaram, M.; Ortwerth, B.J. Determination of glycation crosslinking by the sugar-dependent incorporation of [14C]lysine into protein. Anal. Biochem. 1994, 216, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Cunningham, A.M.; Riederer, P.; Braak, E. Advanced glycation endproducts are associated with Hirano bodies in Alzheimer’s disease. Brain Res. 1998, 796, 307–310. [Google Scholar] [CrossRef]
- Li, Y.M.; Dickson, D.W. Enhanced binding of advanced glycation endproducts (AGE) by the ApoE4 isoform links the mechanism of plaque deposition in Alzheimer’s disease. Neurosci. Lett. 1997, 226, 155–158. [Google Scholar] [CrossRef]
- Kim, D.E.; Priefer, R. Therapeutic Potential of Direct Clearance of the Amyloid-β in Alzheimer’s Disease. Brain Sci. 2020, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Jianyi, M.; Ann, Y.; Brewer, B.H., Jr.; Saumya, D.; Huntington, P. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef]
- Kloske, C.M.; Wilcock, D.M. The Important Interface between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2020, 11, 754. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mooldijk, S.S.; Licher, S.; Waqas, K.; Ikram, M.K.; Uitterlinden, A.G.; Zillikens, M.C.; Ikram, M.A. Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw. Open 2021, 4, e2033012. [Google Scholar] [CrossRef] [PubMed]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Eskici, G.; Axelsen, P.H. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 2012, 51, 6289–6311. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s Disease Amyloid-β Binds Copper and Zinc to Generate an Allosterically Ordered Membrane-penetrating Structure Containing Superoxide Dismutase-like Subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, I.F.; Dringen, R. Astrocyte functions in the copper homeostasis of the brain. Neurochem. Int. 2013, 62, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Nie, G.; Zhang, J.; Luo, Y.; Zhang, P.; Zhang, Z.; Zhao, B. β-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic. Biol. Med. 2011, 50, 122–129. [Google Scholar] [CrossRef]
- Honda, K.; Casadesus, G.; Petersen, R.B.; Perry, G.; Smith, M.A. Oxidative stress and redox-active iron in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 179–182. [Google Scholar] [CrossRef]
- Sensi, S.L.; Rapposelli, I.G.; Frazzini, V.; Mascetra, N. Altered oxidant-mediated intraneuronal zinc mobilization in a triple transgenic mouse model of Alzheimer’s disease. Exp. Gerontol. 2008, 43, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Drewes, G.; Ebneth, A.; Mandelkow, E.M. MAPs, MARKs and microtubule dynamics. Trends Biochem. Sci. 1998, 23, 307–311. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [Green Version]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Moloney, C.M.; Lowe, V.J.; Murray, M.E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimer’s Dement. 2021. [Google Scholar] [CrossRef]
- Kundel, F.; Hong, L.; Falcon, B.; McEwan, W.A.; Michaels, T.C.T.; Meisl, G.; Esteras, N.; Abramov, A.Y.; Knowles, T.J.P.; Goedert, M.; et al. Measurement of Tau Filament Fragmentation Provides Insights into Prion-like Spreading. ACS Chem. Neurosci. 2018, 9, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Terro, F. Tau protein phosphatases in Alzheimer’s disease: The leading role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; Van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T. Oxidative Stress in Alzheimer’s Disease: Molecular Hallmarks of Underlying Vulnerability. In Biological, Diagnostic and Therapeutic Advances in Alzheimer’s Disease: Non-Pharmacological Therapies for Alzheimer’s Disease; Springer: Singapore, 2019; p. 91. ISBN 9789811396366. [Google Scholar]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Petersen, J.D.; Kaech, S.; Banker, G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J. Neurosci. 2014, 34, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007, 2, 536. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Dubey, M.; Chaudhury, P.; Kabiru, H.; Shea, T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskelet. 2008, 65, 89–99. [Google Scholar] [CrossRef]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March separate, strike together—Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2021, 288, 127–141. [Google Scholar] [CrossRef]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, C.; Farías, G.; Maccioni, R.B. Glycation of tau to an Alzheimer’s type protein interferes with its interaction with microtubules. Cell. Mol. Boil. 1998, 44, 1117–1127. [Google Scholar]
- Ledesma, M.D.; Pérez, M.; Colaco, C.; Avila, J. Tau glycation is involved in aggregation of the protein but not in the formation of filaments. Cell. Mol. Biol. 1998, 44, 1111–1116. [Google Scholar]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef]
- Collins, S.M.; Bercik, P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology 2009, 136, 2003–2014. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.M.; Surette, M.G.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Mahony, S.M. The microbiome-gut-brain axis: From bowel to behavior. Neurogastroenterol. Motil. 2011, 23, 187–192. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T. Role of the Microbiota in Immunity and inflammation. Cell 2014, 157, 121. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. 2013, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Douglas-Escobar, M.; Elliott, E.; Neu, J. Effect of Intestinal Microbial Ecology on the Developing Brain. JAMA Pediatr. 2013, 167, 374–379. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Coy, K.D.M.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Reese, A.T.; Cho, E.H.; Klitzman, B.; Nichols, S.P.; A Wisniewski, N.; Villa, M.M.; Durand, H.K.; Jiang, S.; Midani, F.S.; Nimmagadda, S.N.; et al. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. eLife 2018, 7, e35987. [Google Scholar] [CrossRef]
- Mercante, J.W.; Neish, A.S. Reactive Oxygen Production Induced by the Gut Microbiota: Pharmacotherapeutic Implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef]
- Kahn, M.S.; Kranjac, D.; Alonzo, C.A.; Haase, J.H.; Cedillos, R.O.; McLinden, K.A.; Boehm, G.W.; Chumley, M.J. Prolonged elevation in hippocampal Aβ and cognitive deficits following repeated endotoxin exposure in the mouse. Behav. Brain Res. 2012, 229, 176–184. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Olsen, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Smith, D.; Leong, B.J.; Brännström, K.; Almqvist, F.; Chapman, M.R. Promiscuous Cross-seeding between Bacterial Amyloids Promotes Interspecies Biofilms. J. Biol. Chem. 2012, 287, 35092. [Google Scholar] [CrossRef] [Green Version]
- Mitew, S.; Kirkcaldie, M.T.; Dickson, T.; Vickers, J.C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 2013, 34, 2341–2351. [Google Scholar] [CrossRef]
- Brenner, S.R. Blue-green algae or cyanobacteria in the intestinal micro-flora may produce neurotoxins such as Beta-N-Methylamino-l-Alanine (BMAA) which may be related to development of amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson-Dementia-Complex in humans and Equine Motor Neuron Disease in Horses. Med. Hypotheses 2013, 80, 103. [Google Scholar] [CrossRef]
- Schwartz, K.; Boles, B.R. Microbial amyloids—Functions and interactions within the host. Curr. Opin. Microbiol. 2013, 16, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, M.J.; Lukiw, W.J.; Kammerman, E.M.; Hill, J.M. Intracerebral propagation of Alzheimer’s disease: Strengthening evidence of a herpes simplex virus etiology. Alzheimers Dement. 2013, 9, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Manuelidis, L. Infectious particles, stress, and induced prion amyloids: A unifying perspective. Virulence 2013, 4, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Köhler, C.; Maes, M.; Slyepchenko, A.; Berk, M.; Solmi, M.; Lanctôt, K.; Carvalho, A. The Gut-Brain Axis, Including the Microbiome, Leaky Gut and Bacterial Translocation: Mechanisms and Pathophysiological Role in Alzheimer’s Disease. Curr. Pharm. Des. 2016, 22, 6152–6166. [Google Scholar] [CrossRef]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-Q.; Zheng, Q.; Wang, Q.; Zeng, Q.-P. Biotic/Abiotic Stress-Driven Alzheimer’s Disease. Front. Cell. Neurosci. 2016, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ahn, E.H.; Kang, S.S.; Liu, X.; Alam, A.; Ye, K. Gut dysbiosis contributes to amyloid pathology, associated with C/EBPβ/AEP signaling activation in Alzheimer’s disease mouse model. Sci. Adv. 2020, 6, eaba0466. [Google Scholar] [CrossRef] [PubMed]
- Luca, M.; Di Mauro, M.; Di Mauro, M.; Luca, A. Gut Microbiota in Alzheimer’s Disease, Depression, and Type 2 Diabetes Mellitus: The Role of Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 4730539. [Google Scholar] [CrossRef] [PubMed]
- Akbari, E.; Asemi, Z.; Kakhaki, R.D.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [Green Version]
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.V.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum IgG Antibody Levels to Periodontal Microbiota Are Associated with Incident Alzheimer Disease. PLoS ONE 2014, 9, e114959. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Diling, C.; Jian, Y.; Ting, L.; Guoyan, H.; Hualun, L.; Xiaocui, T.; Guoxiao, L.; Ou, S.; Chaoqun, Z.; et al. Effects of Oligosaccharides from Morinda officinalis on Gut Microbiota and Metabolome of APP/PS1 Transgenic Mice. Front. Neurol. 2018, 9, 412. [Google Scholar] [CrossRef] [PubMed]
- Brandscheid, C.; Schuck, F.; Reinhardt, S.; Schäfer, K.-H.; Pietrzik, C.U.; Grimm, M.; Hartmann, T.; Schwiertz, A.; Endres, K. Altered Gut Microbiome Composition and Tryptic Activity of the 5xFAD Alzheimer’s Mouse Model. J. Alzheimers. Dis. 2017, 56, 775–788. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 1–21. [Google Scholar] [CrossRef]
- Du, X.T.; Wang, L.; Wang, Y.J.; Andreasen, M.; Zhan, D.W.; Feng, Y.; Li, M.; Zhao, M.; Otzen, D.; Xue, D.; et al. Aβ1-16 can aggregate and induce the production of reactive oxygen species, nitric oxide, and inflammatory cytokines. J. Alzheimer Dis. 2011, 27, 401–413. [Google Scholar] [CrossRef]
- Bhatti, A.B.; Usman, M.; Ali, F.; Satti, S.A. Vitamin supplementation as an adjuvant treatment for Alzheimer’s disease. J. Clin. Diagn. Res. 2016, 10, OE07–OE11. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Vitamin A and Alzheimer’s disease. Geriatr. Gerontol. Int. 2012, 12, 180–188. [Google Scholar] [CrossRef]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.Y.; Trojanowski, J.Q.; Praticò, D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004, 18, 323–325. [Google Scholar] [CrossRef]
- Conte, V.; Uryu, K.; Fujimoto, S.; Yao, Y.; Rokach, J.; Longhi, L.; Trojanowski, J.Q.; Lee, V.M.Y.; McIntosh, T.K.; Praticò, D. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J. Neurochem. 2004, 90, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s β-Amyloid Fibrils In Vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fiala, M.; Cashman, J.; Sayre, J.; Espinosa, A.; Mahanian, M.; Zaghi, J.; Badmaev, V.; Graves, M.C.; Bernard, G.; et al. Curcuminoids enhance amyloid-β uptake by macrophages of Alzheimer’s disease patients. J. Alzheimer Dis. 2006, 10, 1–7. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wruck, C.J.; Claussen, M.; Fuhrmann, G.; Römer, L.; Schulz, A.; Pufe, T.; Waetzig, V.; Peipp, M.; Herdegen, T.; Götz, M.E. Luteolin protects rat PC 12 and C6 cells against MPP+ induced toxicity via an ERK dependent Keapl-Nrf2-ARE pathway. J. Neural. Transm. Suppl. 2007, 72, 57–67. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Quinto, J.P.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef]
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed β-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2005, 336, 444–449. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [Green Version]
- Cornejo, A.; Aguilar Sandoval, F.; Caballero, L.; Machuca, L.; Muñoz, P.; Caballero, J.; Perry, G.; Ardiles, A.; Areche, C.; Melo, F. Rosmarinic acid prevents fibrillization and diminishes vibrational modes associated to β sheet in tau protein linked to Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2017, 32, 945–953. [Google Scholar] [CrossRef]
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates β-amyloid-induced oxidative stress via Akt/GSK-3β/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic. Biol. Med. 2018, 120, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Casanovas, B.; Camps, J.; Navarro, G.; Martínez-Pinilla, E. Antixoxidant Supplements versus Health Benefits of Brief/Intermittent Exposure to Potentially Toxic Physical or Chemical Agents. Curr. Issues Mol. Biol. 2021, 43, 47. [Google Scholar] [CrossRef]
- Cuttler, J.M. Application of Low Doses of Ionizing Radiation in Medical Therapies. Dose Response 2020, 18, 1559325819895739. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Ishihara, T.; Yokota, O.; Terada, S.; Trojanowski, J.Q.; Lee, V.M.Y.; Kuroda, S. Effects of α-tocopherol on an animal model of tauopathies. Free Radic. Biol. Med. 2004, 37, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef]
- Das, B.; Dasgupta, S.; Ray, S. Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural. Regen. Res. 2019, 14, 1880–1892. [Google Scholar]
- Yang, X.; Dai, G.; Li, G.; Yang, E.S. Coenzyme Q10 reduces β-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2010, 41, 110–113. [Google Scholar] [CrossRef]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J. Alzheimer Dis. 2011, 27, 211–223. [Google Scholar] [CrossRef]
- Mcmanus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant mitoq prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [Green Version]
- Atitlán-Gil, A.; Bretón-De La Loza, M.M.; Jiménez-Ortega, J.C.; Belefant-Miller, H.; Betanzos-Cabrera, G. Activated and Micronized Zeolite in the Modulation of Cellular Oxidative Stress in Mexican Smokers: A Randomized Clinical Trial. Revista de Investigación Clínica 2017, 69, 146–151. [Google Scholar] [CrossRef]
- Hira, S.; Saleem, U.; Anwar, F.; Sohail, M.F.; Raza, Z.; Ahmad, B. β-Carotene: A Natural Compound Improves Cognitive Impairment and Oxidative Stress in a Mouse Model of Streptozotocin-Induced Alzheimer’s Disease. Biomolecules 2019, 9, 441. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Su, C.; Li, R.; Wang, H.; Ren, Y.; Sun, H.; Yang, J.; Sun, J.; Shi, J.; Tian, J.; et al. Mechanisms and effects of curcumin on spatial learning and memory improvement in APPswe/PS1dE9 mice. J. Neurosci. Res. 2014, 92, 218–231. [Google Scholar] [CrossRef]
- Handattu, S.P.; Monroe, C.E.; Nayyar, G.; Palgunachari, M.N.; Kadish, I.; Van Groen, T.; Anantharamaiah, G.M.; Garber, D.W. In vivo and in vitro effects of an apolipoprotein E mimetic peptide on amyloid-β pathology. J. Alzheimer Dis. 2013, 36, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, A.; Bi, X.; Baudry, M. Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer’s disease: Protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J. Alzheimer Dis. 2012, 30, 183–208. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Y.; Lian, D.; Lai, J.; Wu, Y.; Wang, L.; Chen, Y.Y.; Zhang, Y.; Boini, K.M.M.; Huang, Y.; Chen, Y.Y. Cathepsin B-Mediated NLRP3 Inflammasome Formation and Activation in Angiotensin II -Induced Hypertensive Mice: Role of Macrophage Digestion Dysfunction. Cell. Physiol. Biochem. 2018, 50, 1585–1600. [Google Scholar] [CrossRef]
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.L.; Qu, Z.W.; Qin, C.; Zhang, J.T. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J. Pineal Res. 2004, 37, 129–136. [Google Scholar] [CrossRef]
- Dragicevic, N.; Copes, N.; O’Neal-Moffitt, G.; Jin, J.; Buzzeo, R.; Mamcarz, M.; Tan, J.; Cao, C.; Olcese, J.M.; Arendash, G.W.; et al. Melatonin treatment restores mitochondrial function in Alzheimer’s mice: A mitochondrial protective role of melatonin membrane receptor signaling. J. Pineal Res. 2011, 51, 75–86. [Google Scholar] [CrossRef]
- Otalora, B.B.; Popovic, N.; Gambini, J.; Popovic, M.; Viña, J.; Bonet-Costa, V.; Reiter, R.J.; Camello, P.J.; Rol, M.Á.; Madrid, J.A. Circadian system functionality, hippocampal oxidative stress, and spatial memory in the APPswePS1dE9 transgenic model of Alzheimer disease: Effects of melatonin or ramelteon. Chronobiol. Int. 2012, 29, 822–834. [Google Scholar] [CrossRef]
- Huang, Q.; Aluise, C.D.; Joshi, G.; Sultana, R.; Clair, S.D.K.; Markesbery, W.R.; Butterfield, D.A. Potential in vivo amelioration by N-Acetyl-l-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: Toward therapeutic modulation of mild cognitive impairment. J. Neurosci. Res. 2010, 88, 2618–2629. [Google Scholar] [CrossRef]

{kind=link}
| Antioxidant | Mechanism | Experimental Model | Reference |
|---|---|---|---|
| Vitamins | |||
| α-Tocopherol | Reduces Aβ and lipid peroxidation; delays development of tau pathology; reduction in learning deficits and motor weakness | Tg2576 mice | [156,157,167,168,169,170,173] |
| Ascorbic acid | Reduces Aβ oligomers, tau phosphorylation, and oxidative stress | hAPP-J20 mice | [174] |
| Retinol | Reduces MDA levels; upregulates SOD activity; reduces Aβ pathology | APPswe/ PS1M146V/ tauP301L (3 × Tg) mice; in vitro enzymatic assay and in silico modeling | [175] |
| Naturally present | |||
| CoQ10 | Reduces MDA levels; upregulates SOD activity; reduces Aβ pathology | Tg19959 mice; APP/PS1 Tg mice | [176,177] |
| Synthetic | |||
| Mito Q | Prevents cognitive decline, oxidative stress, Aβ accumulation, synaptic loss, and caspase activation | 3 × Tg mice | [178] |
| Plant-based | |||
| Zeolite | Increases endogenous SOD; reduces Aβ levels and plaque burden | Randomized clinical trial | [179] |
| β-Carotene | Improves cognitive impairment and oxidative stress | Streptozotocin-induced AD mice model | [180] |
| Curcumin | Inhibits Aβ fibrillization and oligomerization; clearance of Aβ by macrophages; reduces Aβ40 and 42 and Aβ-derived diffusible ligands; increases Aβ-degrading enzymes; promotes destabilization | Tg2576 mice; APPSw mice; APPswe/PS1dE9 mice; in vitro enzymatic assay | [160,161,162,163,181] |
| Ferulic acid | Inhibits the fibrillization and/or oligomerization of Aβ | In vitro enzymatic assay | [167,168] |
| Rosmarinic acid | Inhibits the fibrillization and/or oligomerization of Aβ | Molecular docking analysis; Tg2576 mice; PC12 neuroblastoma | [168,169,170] |
| Nordihydroguaiaretic acid (NDGA) | Inhibits the fibrillization and/or oligomerization of Aβ | Tg2576 mice | [168] |
| Mimetic | |||
| ApoE mimetic peptide Ac-hE18A-NH2 | Reduces oxidative stress and ApoE secretion; inhibits Aβ plaque deposition | APP/PS1ΔE9 mice and U251 human astrocyte cells | [182] |
| Catalase mimetic | Protects against oxidative stress, DNA, and protein oxidation; reduces Aβ and tau phosphorylation | 3 × Tg-AD mice | [183] |
| Drug | |||
| Melatonin | Inhibits time-dependent elevation of Aβ; reduces abnormal oxidation and nitration of proteins; increases survival; alleviates learning and memory deficits; decreases choline acetyltransferase activity and increases acetyltransferase activity; increases mitochondrial function | Tg2576 mice; APP 695 Tg mouse model; APP/PS1 mice; APPswePS1dE9 mice | [166,184,185,186,187] |
| N-Acetyl-l-cysteine | Reduces lipid peroxidation, oxidative stress, and glutathione peroxidase activity | APP/PS-1 knock-in mice | [188] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. https://doi.org/10.3390/antiox10081231
Sharma C, Kim SR. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants. 2021; 10(8):1231. https://doi.org/10.3390/antiox10081231
Chicago/Turabian StyleSharma, Chanchal, and Sang Ryong Kim. 2021. "Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease" Antioxidants 10, no. 8: 1231. https://doi.org/10.3390/antiox10081231
APA StyleSharma, C., & Kim, S. R. (2021). Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants, 10(8), 1231. https://doi.org/10.3390/antiox10081231