
Glutathione S-Transferases in Cancer


Abstract
1. Introduction
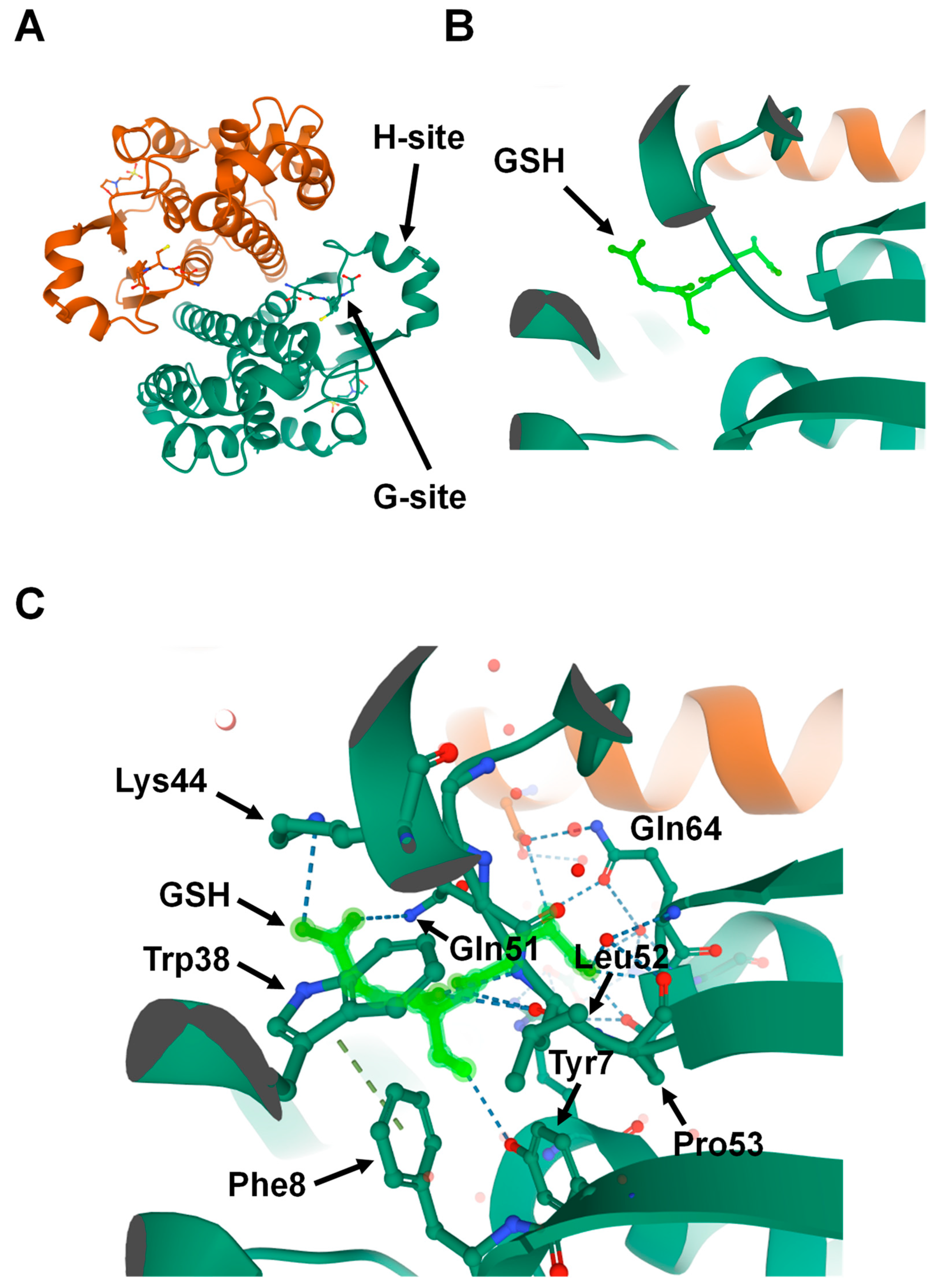
2. Structure
3. Metabolism of Xenobiotic Compounds
4. Cellular Signaling
5. Cellular Metabolism
6. Chemoresistance
7. GSTs Glutathionylate Various Proteins
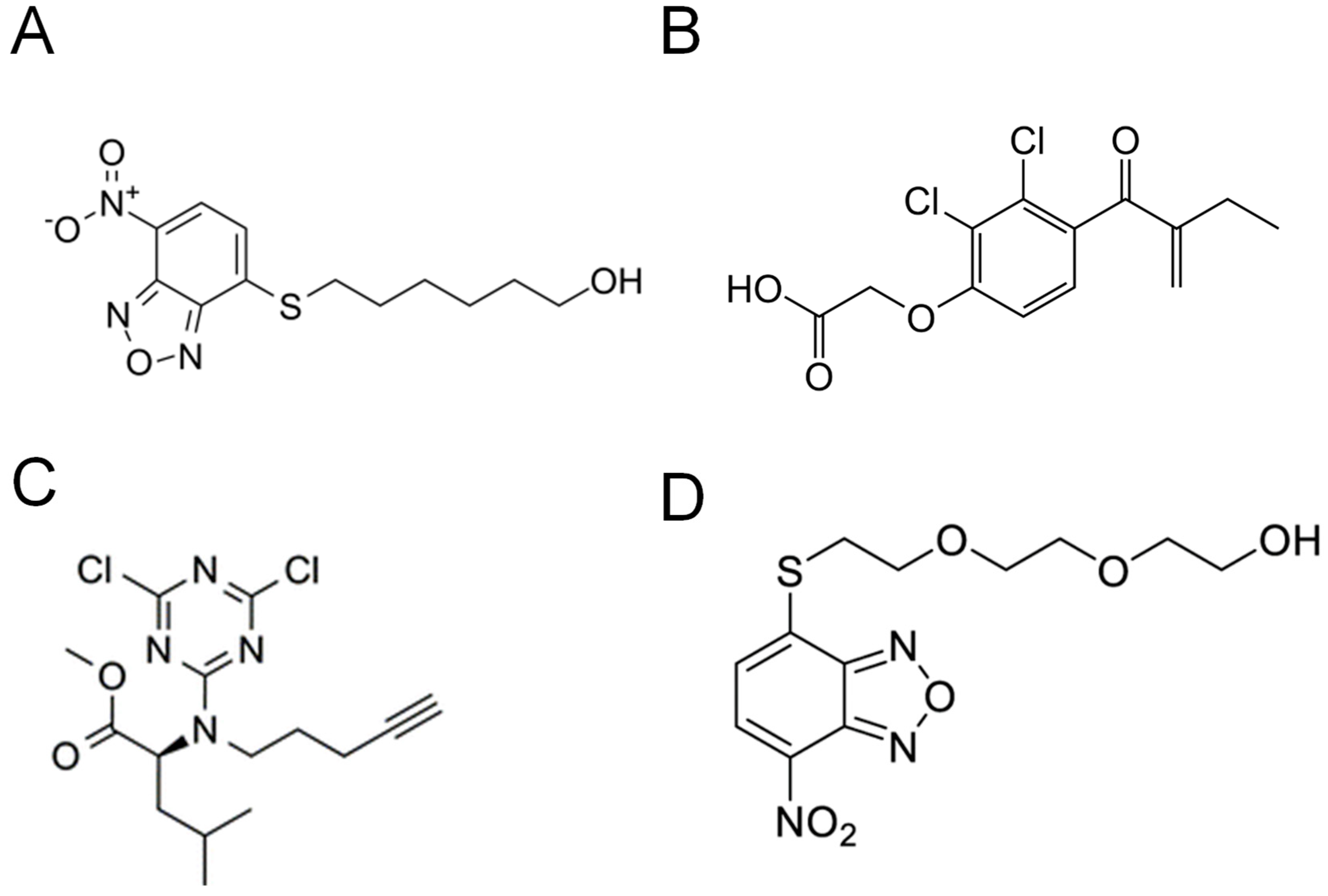
8. GST Inhibitors and Their Therapeutic Implications
8.1. Inhibitors That Bind to the G-Site
8.2. Inhibitors That Bind to the H-Site
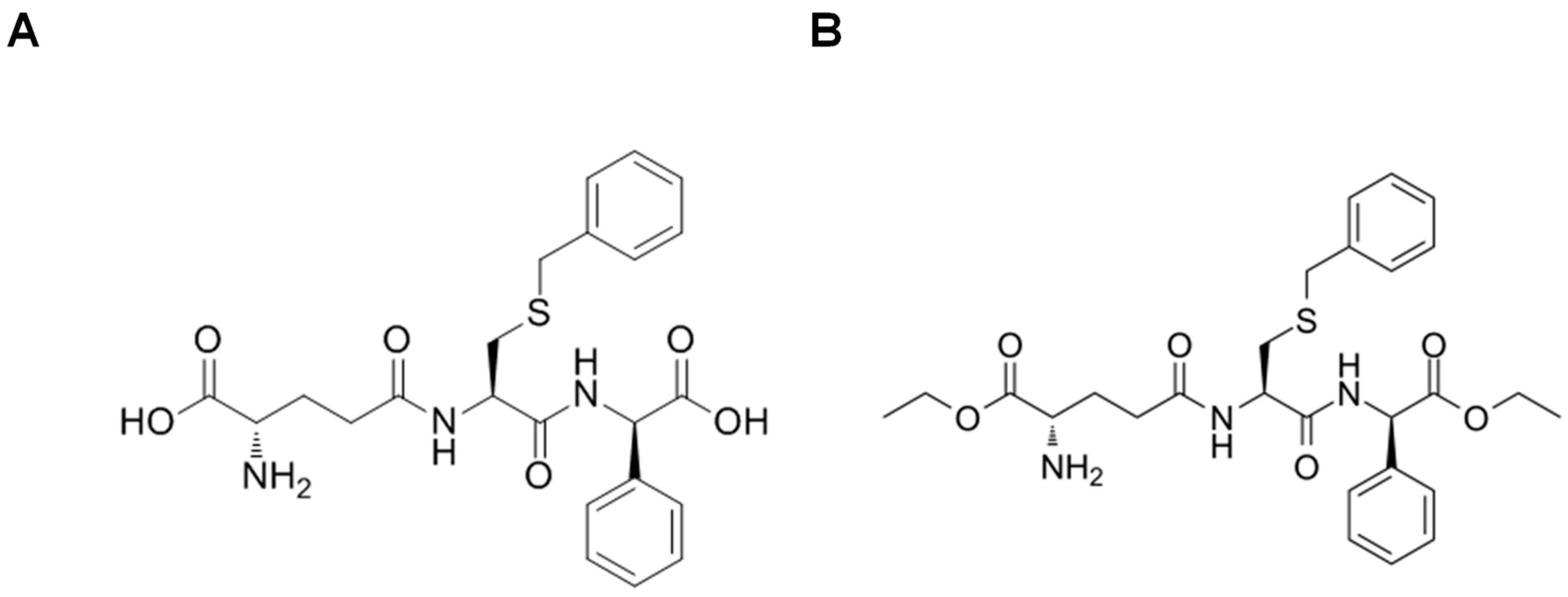
8.3. Glutathione Peptidomimetics
8.4. Natural Compounds
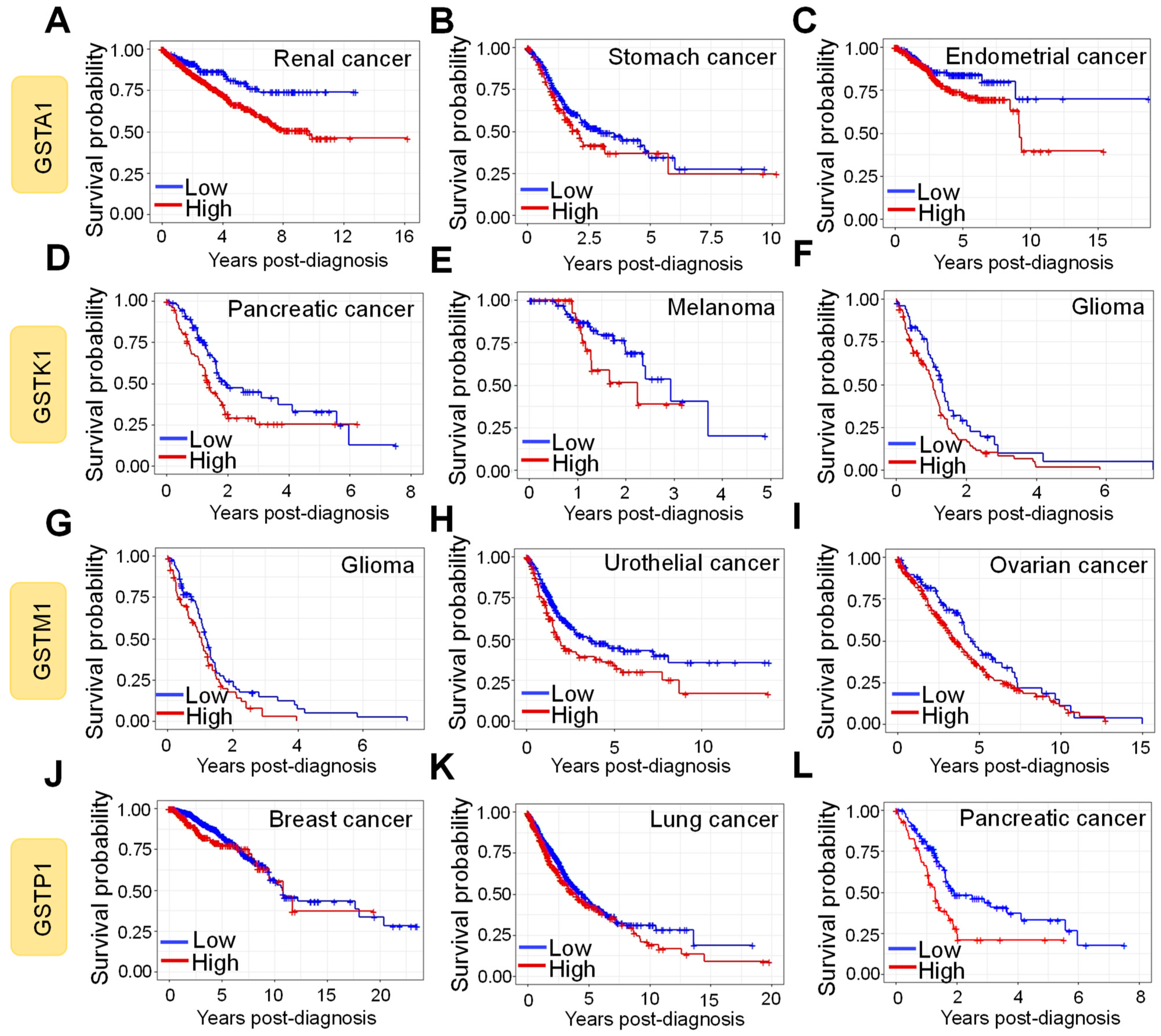
9. Prognostic Impact of GST Protein Expression
10. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grek, C.L.; Ye, Z.-W.; Manevich, Y.; Tew, K.D.; Townsend, D.M. Pleiotropic Functions of glutathione S-transferase P. Adv. Cancer Res. 2014, 122, 143–175. [Google Scholar] [CrossRef] [PubMed]
- Kural, C.; Kocdogan, A.K.; Şimşek, G.G.; Oguztuzun, S.; Kaygın, P.; Yılmaz, I.; Bayram, T.; Izci, Y. Glutathione S-transferases and cytochrome P450 enzyme expression in patients with intracranial tumors: Preliminary report of 55 patients. Med. Princ. Pr. 2018, 28, 56–62. [Google Scholar] [CrossRef]
- Soleo, L.; Strzelczyk, R. Xenobiotics and glutathione. G Ital. Med. Lav. Ergon. 1999, 21, 302–308. [Google Scholar]
- Tew, K.D.; Townsend, D.M. Regulatory functions of glutathioneS-transferase P1-1 unrelated to detoxification. Drug Metab. Rev. 2011, 43, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lym-phocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef]
- Júnior, P.I.D.B.; Senna, S.M.; Vidor, A.C.; Miyasaka, C.K.; Curi, R.; Williams, J.F. Glutathione metabolism and glutathione S-conjugate export ATPase (MRP1/GS-X pump) activity in cancer. II. Cell-to-cell variability, relation with cellular activation state and functional absence of GS-X pump in lymphocytes. Biochem. Mol. Boil. Int. 1998, 45, 1243–1254. [Google Scholar]
- Booth, J.; Boyland, E.; Sims, P. An enzyme from rat liver catalysing conjugations with glutathione. Biochem. J. 1961, 79, 516–524. [Google Scholar] [CrossRef]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 is a driver of triple-negative breast cancer cell metabolism and pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef]
- Checa-Rojas, A.; Delgadillo-Silva, L.F.; Velasco-Herrera, M.D.C.; Andrade-Domínguez, A.; Gil, J.; Santillán, O.; Lozano, L.; Toledo-Leyva, A.; Ramírez-Torres, A.; Talamas-Rohana, P.; et al. GSTM3 and GSTP1: Novel players driving tumor progression in cervical cancer. Oncotarget 2018, 9, 21696–21714. [Google Scholar] [CrossRef]
- Townsend, D.M.; Findlay, V.J.; Fazilev, F.; Ogle, M.; Fraser, J.; Saavedra, J.E.; Ji, X.; Keefer, L.K.; Tew, K.D. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol. Pharmacol. 2006, 69, 501–508. [Google Scholar] [CrossRef]
- Reinemer, P.; Dirr, H.W.; Ladenstein, R.; Schäffer, J.; Gallay, O.; Huber, R. The three-dimensional structure of class pi glutathione S-transferase in complex with glutathione sulfonate at 2.3 A resolution. EMBO J. 1991, 10, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef]
- Kilili, K.G.; Atanassova, N.; Vardanyan, A.; Clatot, N.; Al-Sabarna, K.; Kanellopoulos, P.N.; Makris, A.M.; Kampranis, S.C. Differential roles of tau class glutathione S-transferases in oxidative stress. J. Biol. Chem. 2004, 279, 24540–24551. [Google Scholar] [CrossRef]
- Ladner, J.E.; Parsons, J.F.; Rife, C.L.; Gilliland, G.L.; Armstrong, R.N. Parallel evolutionary pathways for glutathione transferases: Structure and mechanism of the mitochon-drial class kappa enzyme rGSTK1-1. Biochemistry 2004, 43, 352–361. [Google Scholar] [CrossRef]
- Li, J.; Xia, Z.; Ding, J. Thioredoxin-like domain of human kappa class glutathione transferase reveals sequence homology and structure similarity to the theta class enzyme. Protein Sci. 2005, 14, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.J.; Babbitt, P.C. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry 2009, 48, 11108–11116. [Google Scholar] [CrossRef]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef] [PubMed]
- Wilce, M.C.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzym. 1994, 1205, 1–18. [Google Scholar] [CrossRef]
- Cohen, L.; Jefferies, A. Environmental exposures and cancer: Using the precautionary principle. Ecancermedicalscience 2019, 13, ed91. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef]
- Bocedi, A.; Noce, A.; Marrone, G.; Noce, G.; Cattani, G.; Gambardella, G.; Di Lauro, M.; Di Daniele, N.; Ricci, G. Glutathione transferase P1-1 an enzyme useful in biomedicine and as biomarker in clinical practice and in environmental pollution. Nutrition 2019, 11, 1741. [Google Scholar] [CrossRef]
- Kim, K.-H.; Jahan, S.A.; Kabir, E.; Brown, R.J. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef]
- Kaisarevic, S.; Dakic, V.; Hrubik, J.; Glisic, B.; Varel, U.L.-V.; Pogrmic-Majkic, K.; Fa, S.; Teodorovic, I.; Brack, W.; Kovacevic, R. Differential expression of CYP1A1 and CYP1A2 genes in H4IIE rat hepatoma cells exposed to TCDD and PAHs. Environ. Toxicol. Pharmacol. 2015, 39, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Bakhiya, N.; Batke, M.; Laake, J.; Monien, B.H.; Frank, H.; Seidel, A.; Engst, W.; Glatt, H. Directing role of organic anion transporters in the excretion of mercapturic acids of alkylated polycyclic aromatic hydrocarbons. Drug Metab. Dispos. 2007, 35, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.E.; Anders, M.W. The mercapturic acid pathway. Crit. Rev. Toxicol. 2019, 49, 819–929. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef]
- Ballatori, N.; Rebbeor, J. Roles of MRP2 and oatp1 in hepatocellular export of reduced glutathione. Semin. Liver Dis. 1998, 18, 377–387. [Google Scholar] [CrossRef]
- Biswas, S.K.; Rahman, I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Mol. Asp. Med. 2009, 30, 60–76. [Google Scholar] [CrossRef]
- Meister, A. Biosynthesis and functions of glutathione, an essential biofactor. J. Nutr. Sci. Vitaminol. 1992, 38, 1–6. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Asp. Med. 2009, 30, 29–41. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Asp. Med. 2009, 30, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Storr, S.J.; Woolston, C.M.; Zhang, Y.; Martin, S.G. Redox environment, free radical, and oxidative DNA damage. Antioxid. Redox Signal. 2013, 18, 2399–2408. [Google Scholar] [CrossRef]
- Ning, J.; Grant, M.H. The role of reduced glutathione and glutathione reductase in the cytotoxicity of chromium (VI) in osteoblasts. Toxicol. Vitr. 2000, 14, 329–335. [Google Scholar] [CrossRef]
- Kang, Y.J.; Enger, M.D. Effect of cellular glutathione depletion on cadmium-induced cytotoxicity in human lung carcinoma cells. Cell Biol. Toxicol. 1987, 3, 347–360. [Google Scholar] [CrossRef]
- Shimizu, M.; Hochadel, J.F.; Fulmer, B.A.; Waalkes, M.P. Effect of glutathione depletion and metallothionein gene expression on arsenic-induced cytotoxicity and c-myc expression in vitro. Toxicol. Sci. 1998, 45, 204–211. [Google Scholar] [CrossRef]
- Russo, A.; Mitchell, J.B.; McPherson, S.; Friedman, N. Alteration of bleomycin cytotoxicity by glutathione depletion or elevation. Int. J. Radiat. Oncol. 1984, 10, 1675–1678. [Google Scholar] [CrossRef]
- Perry, R.R.; Greaves, B.R.; Rasberry, U.; Barranco, S.C. Effect of treatment duration and glutathione depletion on mitomycin C cytotoxicity in vitro. Cancer Res. 1992, 52, 4608–4612. [Google Scholar] [PubMed]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox signaling during hypoxia in mammalian cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Li, L.-C.; Wang, Z.-W.; Hu, X.-P.; Wu, Z.-Y.; Hu, Z.-P.; Ruan, Y.-L. MDG-1 inhibits H2O2-induced apoptosis and inflammation in human umbilical vein endothelial cells. Mol. Med. Rep. 2017, 16, 3673–3679. [Google Scholar] [CrossRef]
- Riou, C.; Remy, C.; Rabilloud, R.; Rousset, B.; Fonlupt, P. H2O2 induces apoptosis of pig thyrocytes in culture. J. Endocrinol. 1998, 156, 315–322. [Google Scholar] [CrossRef][Green Version]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef]
- Dekant, W.; Vamvakas, S. Glutathione-dependent bioactivation of xenobiotics. Xenobiotica 1993, 23, 873–887. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Tew, K.D.; Ronai, Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18, 6104–6111. [Google Scholar] [CrossRef] [PubMed]
- Gate, L.; Majumdar, R.S.; Lunk, A.; Tew, K.D. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J. Biol. Chem. 2004, 279, 8608–8616. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Arifoglu, P.; Ronai, Z.; Tew, K.D. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through inter-action with the C terminus. J. Biol. Chem. 2001, 276, 20999–21003. [Google Scholar] [CrossRef]
- De Luca, A.; Mei, G.; Rosato, N.; Nicolai, E.; Federici, L.; Palumbo, C.; Pastore, A.; Serra, M.; Caccuri, A.M. The fine-tuning of TRAF2-GSTP1-1 interaction: Effect of ligand binding and in situ detection of the com-plex. Cell Death Dis. 2014, 5, e1015. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fan, Y.; Xue, B.; Luo, L.; Shen, J.; Zhang, S.; Jiang, Y.; Yin, Z. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2–ASK1 signals. Oncogene 2006, 25, 5787–5800. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Mohammad, J.; Orr, M.; Reindl, K.M. Glutathione S-transferase pi-1 knockdown reduces pancreatic ductal adenocarcinoma growth by Ac-tivating oxidative stress response pathways. Cancers 2020, 12, 1501. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin–dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef]
- Liu, X.; Sui, X.; Zhang, C.; Wei, K.; Bao, Y.; Xiong, J.; Zhou, Z.; Chen, Z.; Wang, C.; Zhu, H.; et al. Glutathione S-transferase A1 suppresses tumor progression and indicates better prognosis of human primary hepatocellular carcinoma. J. Cancer 2020, 11, 83–91. [Google Scholar] [CrossRef]
- Saisawang, C.; Wongsantichon, J.; Robinson, R.C.; Ketterman, A.J. Glutathione transferase Omega 1-1 (GSTO1-1) modulates Akt and MEK1/2 signaling in human neuro-blastoma cell SH-SY5Y. Proteins 2019, 87, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cravatt, B.F. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 2013, 341, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, R.; Xiao, F.; Zhang, S.-X.; Gong, M.; Wang, X.-L.; Zhang, Y.; Huang, J.-Y. Expressions of glutathione S-transferase P1 and 4- hydroxynonenal and the progression of prostate cancer. Zhonghua nan ke xue Natl. J. Androl. 2017, 23, 412–416. [Google Scholar]
- Barbati, S.; Bonnefoy, A.; Botta, A.; Chiron, S. Secondary oxidation of cyclic 1,N2-propano and 1,N2-etheno-2′-deoxyguanosine DNA adducts. Conse-quences in oxidative stress biomarker development. Chemosphere 2010, 80, 1081–1087. [Google Scholar] [CrossRef]
- Bartsch, H.; Arab, K.; Nair, J. Biomarkers for hazard identification in humans. Environ. Health 2011, 10, S11. [Google Scholar] [CrossRef] [PubMed]
- Koo, M.M.; Swann, R.; McPhail, S.; Abel, G.A.; Elliss-Brookes, L.; Rubin, G.P.; Lyratzopoulos, G. Presenting symptoms of cancer and stage at diagnosis: Evidence from a cross-sectional, population-based study. Lancet Oncol. 2020, 21, 73–79. [Google Scholar] [CrossRef]
- Sabater, L.; Muñoz, E.; Roselló, S.; Dorcaratto, D.; Garcés-Albir, M.; Huerta, M.; Roda, D.; Gómez-Mateo, M.C.; Ferrández-Izquierdo, A.; Darder, A.; et al. Borderline resectable pancreatic cancer. Challenges and controversies. Cancer Treat. Rev. 2018, 68, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergia, N.; Shah, P.C.; Denlinger, C.S. Survivorship in non-small cell lung cancer: Challenges faced and steps forward. J. Natl. Compr. Cancer Netw. 2015, 13, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA A Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Mehrling, T. Chemotherapy is getting ‘smarter’. Future Oncol. 2015, 11, 549–552. [Google Scholar] [CrossRef]
- Gupta, R.; Amanam, I.; Chung, V. Current and future therapies for advanced pancreatic cancer. J. Surg. Oncol. 2017, 116, 25–34. [Google Scholar] [CrossRef]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Salfity, S.; Seshacharyulu, P.; Rachagani, S.; Thomas, A.; Das, S.; Majhi, P.D.; Nimmakayala, R.K.; Vengoji, R.; Lele, S.M.; et al. MUC16 regulates TSPYL5 for lung cancer cell growth and chemoresistance by suppressing p53. Clin. Cancer Res. 2017, 23, 3906–3917. [Google Scholar] [CrossRef]
- Tang, J.; Guo, F.; Du, Y.; Liu, X.; Qin, Q.; Liu, X.; Yin, T.; Jiang, L.; Wang, Y. Continuous exposure of non-small cell lung cancer cells with wild-type EGFR to an inhibitor of EGFR tyrosine kinase induces chemoresistance by activating STAT3. Int. J. Oncol. 2015, 46, 2083–2095. [Google Scholar] [CrossRef]
- Kuroda, H.; Takeno, M.; Murakami, S.; Miyazawa, N.; Kaneko, T.; Ishigatsubo, Y. Inhibition of heme oxygenase-1 with an epidermal growth factor receptor inhibitor and cisplatin decreases proliferation of lung cancer A549 cells. Lung Cancer 2010, 67, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zheng, W.; Li, N.; Su, Z.; Zhao, L.; Zhou, H.; Jia, L. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 41942. [Google Scholar] [CrossRef] [PubMed]
- Deeley, R.G.; Cole, S.P. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett. 2005, 580, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kauvar, L.; Tew, K.D. Importance of glutathione and associated enzymes in drug response. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1997, 9, 295–302. [Google Scholar]
- Huang, J.; Gu, M.; Chen, C. Expression of glutathione S-transferase-pi in operative specimens as marker of chemo-resistance in patients with ovarian cancer. Zhonghua Fu Chan Ke Za Zhi 1997, 32, 458–461. [Google Scholar]
- Zhang, F.; Qi, L.; Chen, H. Value of P-glycoprotein and glutathione S-transferase-pi as chemo-resistant indicators in ovarian cancers. Zhonghua Zhong Liu za Zhi J. Oncol. 2001, 23, 313–316. [Google Scholar]
- Satoh, T.; Nishida, M.; Miyazaki, Y.; Sugita, M.; Arisawa, Y.; Oki, A.; Nishide, K.; Kono, K.; Tsunoda, H.; Kubo, T. An immunohistological study on expression of glutathione S-transferase pi (form) in human ovarian carcinoma. Nihon Sanka Fujinka Gakkai Zasshi 1995, 47, 931–938. [Google Scholar]
- Mousseau, M.; Chauvin, C.; Nissou, M.; Chaffanet, M.; Plantaz, D.; Pasquier, B.; Schaerer, R.; Benabid, A. A study of the expression of four chemoresistance-related genes in human primary and metastatic brain tumours. Eur. J. Cancer 1993, 29, 753–759. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Brem, H.; Brem, S.; Sloan, A.; Barger, G.; Huang, W.; Parker, R. In vitro drug response and molecular markers associated with drug resistance in malignant gliomas. Clin. Cancer Res. 2006, 12, 4523–4532. [Google Scholar] [CrossRef]
- Geng, M.; Wang, L.; Chen, X.; Cao, R.; Li, P. The association between chemosensitivity and Pgp, GST-π and Topo II expression in gastric cancer. Diagn. Pathol. 2013, 8, 198. [Google Scholar] [CrossRef]
- Yu, D.-S.; Hsieh, D.S.; Chang, S.Y. Increasing expression of GST-pi MIF, and ID1 genes in chemoresistant prostate cancer cells. Arch. Androl. 2006, 52, 275–281. [Google Scholar] [CrossRef]
- Wang, Z.; Liang, S.; Lian, X.; Liu, L.; Zhao, S.; Xuan, Q.; Guo, L.; Liu, H.; Yang, Y.; Dong, T.; et al. Identification of proteins responsible for adriamycin resistance in breast cancer cells using proteomics analysis. Sci. Rep. 2015, 5, 9301. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, Y.; Shen, X.; Ruan, Y.; Lu, Y.; Jin, X.; Song, P.; Guo, Y.; Zhang, X.; Qu, H.; et al. CLDN6 promotes chemoresistance through GSTP1 in human breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 157. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Konishi, H.; Ichikawa, D.; Matsubara, D.; Shoda, K.; Arita, T.; Kosuga, T.; Komatsu, S.; Shiozaki, A.; Okamoto, K.; et al. Glutathione S-transferase Pi 1 is a valuable predictor for cancer drug resistance in esophageal squamous cell carcinoma. Cancer Sci. 2019, 110, 795–804. [Google Scholar] [CrossRef]
- Li, J.; Ye, T.; Liu, Y.; Kong, L.; Sun, Z.; Liu, D.; Wang, J.; Xing, H.R. Transcriptional activation of Gstp1 by MEK/ERK signaling confers chemo-resistance to cisplatin in lung cancer stem cells. Front. Oncol. 2019, 9, 476. [Google Scholar] [CrossRef]
- Ishii, T.; Teramoto, S.; Matsuse, T. GSTP1 affects chemoresistance against camptothecin in human lung adenocarcinoma cells. Cancer Lett. 2004, 216, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Nandy, S.B.; Lakshmanaswamy, R. Cancer stem cells and metastasis. Prog. Mol. Biol. Transl. Sci. 2017, 151, 137–176. [Google Scholar] [PubMed]
- Liu, T.; Xu, F.; Du, X.; Lai, D.; Liu, T.; Zhao, Y.; Huang, Q.; Jiang, L.; Huang, W.; Cheng, W.; et al. Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem-like cells from human prostate cancer cell lines 22RV1. Mol. Cell Biochem. 2010, 340, 265–273. [Google Scholar] [CrossRef]
- Shafee, N.; Smith, C.R.; Wei, S.; Kim, Y.; Mills, G.B.; Hortobagyi, G.N.; Stanbridge, E.J.; Lee, E.Y.-H.P. Cancer stem cells contribute to cisplatin resistance in Brca1/p53–mediated mouse mammary tumors. Cancer Res. 2008, 68, 3243–3250. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M. Understanding the causes of multidrug resistance in cancer: A comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Inoue, K.; Shimizu, T.; Akimoto, K.; Kubota, K. Dual pharmacological inhibition of glutathione and thioredoxin systems synergizes to kill colorectal carcinoma stem cells. Cancer Med. 2016, 5, 2544–2557. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Xie, L.; Lu, Y.; Hu, Z.; Chang, J. miR-133b reverses cisplatin resistance by targeting GSTP1 in cisplatin-resistant lung cancer cells. Int. J. Mol. Med. 2018, 41, 2050–2058. [Google Scholar] [CrossRef]
- Chen, J.; Solomides, C.; Simpkins, H. Sensitization of mesothelioma cells to platinum-based chemotherapy by GSTpi knockdown. Biochem. Biophys. Res. Commun. 2014, 447, 77–82. [Google Scholar] [CrossRef]
- Hour, T.C.; Chen, J.; Huang, C.Y.; Guan, J.Y.; Lu, S.H.; Hsieh, C.Y.; Pu, Y.S. Characterization of chemoresistance mechanisms in a series of cisplatin-resistant transitional carcinoma cell lines. Anticancer. Res. 2000, 20, 3221–3225. [Google Scholar]
- Tew, K.D.; Manevich, Y.; Grek, C.; Xiong, Y.; Uys, J.; Townsend, D.M. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free. Radic. Biol. Med. 2011, 51, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Robin, S.K.D.; Ansari, M.; Uppugunduri, C.R.S. Spectrophotometric screening for potential inhibitors of cytosolic glu-tathione s-transferases. J. Vis. Exp. 2020, 10, 164. [Google Scholar]
- Lee, K.G.Z.; Babak, M.V.; Weiss, A.; Dyson, P.J.; Nowak-Sliwinska, P.; Montagner, D.; Ang, W.H.; Novak-Slowinska, P. Development of an efficient dual-action GST-inhibiting anticancer platinum (IV) prodrug. ChemMedChem 2018, 13, 1210–1217. [Google Scholar] [CrossRef]
- El-Karim, S.S.A.; Anwar, M.M.; Zaki, E.R.; Elseginy, S.A.; Nofal, Z.M. Synthesis and molecular modeling of new benzimidazoles as glutathioneS-transferase inhibitors and anticancer agents. Future Med. Chem. 2018, 10, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Brozovic, A.; Fritz, G.; Christmann, M.; Zisowsky, J.; Jaehde, U.; Osmak, M.; Kaina, B. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int. J. Cancer 2004, 112, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Ridgway, L.D.; Korapati, A.L.; Zhang, Q.; Tian, L.; Wang, Y.; Siddik, Z.H.; Mills, G.B.; Claret, F.X. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to fas ligand induction and cell death in ovarian carcinoma cells. J. Biol. Chem. 2003, 278, 19245–19256. [Google Scholar] [CrossRef]
- Besirli, C.G.; Johnson, E.M. JNK-independent activation of c-Jun during neuronal apoptosis induced by multiple DNA-damaging agents. J. Biol. Chem. 2003, 278, 22357–22366. [Google Scholar] [CrossRef]
- Huang, X.-L.; Zhang, H.; Yang, X.-Y.; Dong, X.-Y.; Xie, X.-Y.; Yin, H.-B.; Gou, X.; Lin, Y.; He, W.-Y. Activation of a c-Jun N-terminal kinase-mediated autophagy pathway attenuates the anticancer activity of gemcitabine in human bladder cancer cells. Anti-Cancer Drugs 2017, 28, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, X.; Yang, X. Ursolic acid inhibits growth and induces apoptosis in gemcitabine-resistant human pancreatic cancer via the JNK and PI3K/Akt/NF-kappaB pathways. Oncol. Rep. 2012, 28, 501–510. [Google Scholar] [CrossRef]
- Dominko, K.; Đikić, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arch. Ind. Hyg. Toxicol. 2018, 69, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.F. Redox control of enzyme activities by thiol/disulfide exchange. Methods Enzymol. 1984, 107, 330–351. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase P-mediated Protein S-glutathionylation of resident endoplasmic reticulum proteins influences sensitivity to drug-induced unfolded protein response. Antioxid. Redox Signal. 2017, 26, 247–261. [Google Scholar] [CrossRef]
- Chae, H.Z.; Oubrahim, H.; Park, J.W.; Rhee, S.G.; Chock, P.B. Protein glutathionylation in the regulation of peroxiredoxins: A family of thiol-specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antioxid. Redox Signal. 2012, 16, 506–523. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free. Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Hofmann, B.; Hecht, H.-J.; Flohé, L. Peroxiredoxins. Biol. Chem. 2002, 383, 347–364. [Google Scholar] [CrossRef]
- Noguera-Mazon, V.; Lemoine, J.; Walker, O.; Rouhier, N.; Salvador, A.; Jacquot, J.; Lancelin, J.; Krimm, I. Glutathionylation induces the dissociation of 1-Cys D-peroxiredoxin non-covalent homodimer. J. Biol. Chem. 2006, 281, 31736–31742. [Google Scholar] [CrossRef]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathi-onylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef] [PubMed]
- Arriga, R.; Pacifici, F.; Capuani, B.; Coppola, A.; Orlandi, A.; Scioli, M.G.; Pastore, D.; Andreadi, A.; Sbraccia, P.; Tesauro, M.; et al. Peroxiredoxin 6 is a key antioxidant enzyme in modulating the link between glycemic and lipogenic metabolism. Oxid. Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Fisher, A.B. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospho-lipid metabolism. Free Radic. Biol. Med. 2005, 38, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Hutchens, S.; Tew, K.; Townsend, D. Allelic variants of glutathione S-transferase P1-1 differentially mediate the peroxidase function of peroxiredoxin VI and alter membrane lipid peroxidation. Free. Radic. Biol. Med. 2013, 54, 62–70. [Google Scholar] [CrossRef]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.H.; Chen, Y.-R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nat. Cell Biol. 2010, 468, 1115–1118. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, J.G.R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Vignoli, B.; Ramesh, N.; Polanco, M.J.; Coutelier, M.; Stephen, C.D.; Canossa, M.; Monin, M.-L.; Aeschlimann, P.; Turberville, S.; et al. Mutations in TGM6 induce the unfolded protein response in SCA35. Hum. Mol. Genet. 2017, 26, 3749–3762. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv. 2007, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.; Gilbert, H.F. Protein disulfide isomerase. Biochim. Biophys. Acta BBA Proteins Proteom. 2004, 1699, 35–44. [Google Scholar] [CrossRef]
- Townsend, D.M.; Manevich, Y.; He, L.; Xiong, Y.; Bowers, R.R.; Hutchens, S.; Tew, K.D. Nitrosative stress–Induced S-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009, 69, 7626–7634. [Google Scholar] [CrossRef]
- Stournaras, C.; Drewes, G.; Blackholm, H.; Merkler, I.; Faulstich, H. Glutathionyl(cysteine-374) actin forms filaments of low mechanical stability. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzym. 1990, 1037, 86–91. [Google Scholar] [CrossRef]
- Rokutan, K.; Johnston, R.B., Jr.; Kawai, K. Oxidative stress induces S-thiolation of specific proteins in cultured gastric mucosal cells. Am. J. Physiol. 1994, 266, G247–G254. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Rossi, R.; Colombo, R.; Milzani, A. Reversible S-glutathionylation of Cys374 regulates actin filament formation by inducing structural changes in the actin molecule. Free. Radic. Biol. Med. 2003, 34, 23–32. [Google Scholar] [CrossRef]
- Lind, C.; Gerdes, R.; Hamnell, Y.; Schuppe-Koistinen, I.; Von Löwenhielm, H.B.; Holmgren, A.; Cotgreave, I.A. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch. Biochem. Biophys. 2002, 406, 229–240. [Google Scholar] [CrossRef]
- Chen, W.; Wen, K.-K.; Sens, A.E.; Rubenstein, P.A. Differential interaction of cardiac, skeletal muscle, and yeast tropomyosins with fluorescent (Pyrene235) yeast actin. Biophys. J. 2006, 90, 1308–1318. [Google Scholar] [CrossRef]
- Kaus-Drobek, M.; Mücke, N.; Szczepanowski, R.H.; Wedig, T.; Czarnocki-Cieciura, M.; Polakowska, M.; Herrmann, H.; Wysłouch-Cieszyńska, A.; Dadlez, M. Vimentin S-glutathionylation at Cys328 inhibits filament elongation and induces severing of mature filaments in vitro. FEBS J. 2020, 287, 5304–5322. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Rainwater, R.; Parks, D.; Anderson, M.E.; Tegtmeyer, P.; Mann, K. Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 1995, 15, 3892–3903. [Google Scholar] [CrossRef]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 Is Inhibited by Glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef]
- Yusuf, M.A.; Chuang, T.; Bhat, G.J.; Srivenugopal, K.S. Cys-141 glutathionylation of human p53: Studies using specific polyclonal antibodies in cancer samples and cell lines. Free. Radic. Biol. Med. 2010, 49, 908–917. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive oxygen and nitrogen species-induced protein modifications: Implication in carcinogenesis and anticancer therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: Refining the toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [CrossRef]
- Benavides, F.; Blando, J.; Perez, C.J.; Garg, R.; Conti, C.J.; John DiGiovanni, J.; Kazanietz, M.G. Transgenic overexpression of PKC-epsilon in the mouse prostate induces preneoplastic lesions. Cell Cycle 2011, 10, 268–277. [Google Scholar] [CrossRef]
- Wang, H.; Gutierrez-Uzquiza, A.; Garg, R.; Barrio-Real, L.; Abera, M.B.; Lopez-Haber, C.; Rosemblit, C.; Lu, H.; Abba, M.; Kazanietz, M.G. Transcriptional regulation of oncogenic protein kinase C (PKC) by STAT1 and Sp1 proteins. J. Biol. Chem. 2014, 289, 19823–19838. [Google Scholar] [CrossRef]
- Ward, N.E.; Stewart, J.R.; Ioannides, A.C.G.; O’Brian, C.A. Oxidant-induced S-glutathiolation inactivates protein kinase C-α (PKC-α): A potential mechanism of PKC isozyme regulation. Biochemistry 2000, 39, 10319–10329. [Google Scholar] [CrossRef]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.-K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; Pérez-Sala, D.; Lamas, S. Novel application of S-nitrosoglutathione‒Sepharose to identify proteins that are potential targets for S-nitrosoglutathione-induced mixed-disulphide formation. Biochem. J. 2000, 349, 567–578. [Google Scholar] [CrossRef]
- Odin, J.A.; Huebert, R.C.; Casciola-Rosen, L.; LaRusso, N.F.; Rosen, A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J. Clin. Investig. 2001, 108, 223–232. [Google Scholar] [CrossRef]
- Ralat, L.A.; Manevich, Y.; Fisher, A.B.; Colman, R.F. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry 2006, 45, 360–372. [Google Scholar] [CrossRef]
- Kruyer, A.; Ball, L.E.; Townsend, D.M.; Kalivas, P.W.; Uys, J.D. Post-translational S-glutathionylation of cofilin increases actin cycling during cocaine seeking. PLoS ONE 2019, 14, e0223037. [Google Scholar] [CrossRef]
- Patel, B.G.; Wilder, T.; Solaro, R.J. Novel control of cardiac myofilament response to calcium by S-glutathionylation at specific sites of myosin binding protein C. Front. Physiol. 2013, 4, 336. [Google Scholar] [CrossRef] [PubMed]
- Carletti, B.; Passarelli, C.; Sparaco, M.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. Effect of protein glutathionylation on neuronal cytoskeleton: A potential link to neurodegeneration. Neuroscience 2011, 192, 285–294. [Google Scholar] [CrossRef]
- Yu, C.-X.; Li, S.; Whorton, A.R. Redox regulation of PTEN by S-nitrosothiols. Mol. Pharmacol. 2005, 68, 847–854. [Google Scholar] [CrossRef]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J. Biol. Chem. 2007, 282, 18427–18436. [Google Scholar] [CrossRef]
- Goch, G.; Vdovenko, S.; Kozłowska, H.; Bierzynski, A. Affinity of S100A1 protein for calcium increases dramatically upon glutathionylation. FEBS J. 2005, 272, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Zhang, L.; Yeh, A.; Chen, C.; Green-Church, K.B.; Zweier, J.L.; Chen, Y. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry 2007, 46, 5754–5765. [Google Scholar] [CrossRef]
- Reynaert, N.L.; Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.; Matthews, D.E. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 loss-of-function associated with cerebral cavernous malformation disease leads to enhanced S-glutathionylation of distinct structural and regulatory proteins. Antioxidants 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Townsend, D.M.; Uys, J.D.; Manevich, Y.; Coker, W.J.; Pazoles, C.J.; Tew, K.D. S-glutathionylated serine proteinase inhibitors as plasma biomarkers in assessing response to re-dox-modulating drugs. Cancer Res. 2012, 72, 2383–2393. [Google Scholar] [CrossRef]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2008, 150, 1122–1131. [Google Scholar] [CrossRef]
- Rinna, A.; Torres, M.; Forman, H.J. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free. Radic. Biol. Med. 2006, 41, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J. Thioredoxin-1 and posttranslational modifications. Antioxid. Redox Signal. 2006, 8, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, G.; Talcott, K.E.; Bhattacharya, S.K.; Crabb, J.W.; Sears, J.E. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp. Cell Res. 2006, 312, 3526–3538. [Google Scholar] [CrossRef]
- O’Brian, C.A.; Chu, F. ReviewPost-translational disulfide modifications in cell signaling—Role of inter-protein, intra-protein, S-glutathionyl, and S-cysteaminyl disulfide modifications in signal transmission. Free. Radic. Res. 2005, 39, 471–480. [Google Scholar] [CrossRef]
- Musdal, Y.; Hegazy, U.M.; Aksoy, Y.; Mannervik, B. FDA-approved drugs and other compounds tested as inhibitors of human glutathione transferase P1-1. Chem. Interact. 2013, 205, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Dong, D. Human cytosolic glutathione transferases: Structure, function, and drug discovery. Trends Pharmacol. Sci. 2012, 33, 656–668. [Google Scholar] [CrossRef]
- Shishido, Y.; Tomoike, F.; Kimura, Y.; Kuwata, K.; Yano, T.; Fukui, K.; Fujikawa, H.; Sekido, Y.; Murakami-Tonami, Y.; Kameda, T.; et al. A covalent G-site inhibitor for glutathione S-transferase Pi (GSTP1-1). Chem. Commun. 2017, 53, 11138–11141. [Google Scholar] [CrossRef]
- Shishido, Y.; Tomoike, F.; Kuwata, K.; Fujikawa, H.; Sekido, Y.; Murakami-Tonami, Y.; Kameda, T.; Abe, N.; Kimura, Y.; Shuto, S.; et al. A covalent inhibitor for glutathione S-transferase Pi (GSTP1-1) in human cells. Chembiochemistry 2019, 20, 900–905. [Google Scholar] [CrossRef]
- Kulaksiz-Erkmen, G.; Dalmizrak, O.; Dincsoy-Tuna, G.; Dogan, A.; Ogus, I.H.; Ozer, N. Amitriptyline may have a supportive role in cancer treatment by inhibiting glutathione S-transferase pi (GST-π) and alpha (GST-α). J. Enzym. Inhib. Med. Chem. 2011, 28, 131–136. [Google Scholar] [CrossRef]
- Bachovchin, D.A.; Brown, S.J.; Rosen, H.; Cravatt, B.F. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat. Biotechnol. 2009, 27, 387–394. [Google Scholar] [CrossRef]
- Tsuboi, K.; Bachovchin, D.A.; Speers, A.E.; Spicer, T.P.; Fernández-Vega, V.; Hodder, P.; Rosen, H.; Cravatt, B.F. Potent and selective inhibitors of glutathiones-transferase omega 1 that impair cancer drug resistance. J. Am. Chem. Soc. 2011, 133, 16605–16616. [Google Scholar] [CrossRef]
- Urzúa, U.; Roby, K.F.; Gangi, L.M.; Cherry, J.M.; Powell, J.I.; Munroe, D.J. Transcriptomic analysis of an in vitro murine model of ovarian carcinoma: Functional similarity to the human disease and identification of prospective tumoral markers and targets. J. Cell. Physiol. 2005, 206, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-D.; Pan, L.-Y.; Yuan, Y.; Lang, J.-H.; Mao, N. Identification of platinum-resistance associated proteins through proteomic analysis of human ovarian cancer cells and their platinum-resistant sublines. J. Proteome Res. 2007, 6, 772–780. [Google Scholar] [CrossRef]
- Turella, P.; Cerella, C.; Filomeni, G.; Bullo, A.; De Maria, F.; Ghibelli, L.; Ciriolo, M.R.; Cianfriglia, M.; Mattei, M.; Federici, G.; et al. Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Res. 2005, 65, 3751–3761. [Google Scholar] [CrossRef] [PubMed]
- Turella, P.; Filomeni, G.; Dupuis, M.L.; Ciriolo, M.R.; Molinari, A.; De Maria, F.; Tombesi, M.; Cianfriglia, M.; Federici, G.; Ricci, G.; et al. A strong glutathione S-transferase inhibitor overcomes the P-glycoprotein-mediated resistance in tumor cells. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) triggers a caspase-dependent apoptosis in MDR1-expressing leukemia cells. J. Biol. Chem. 2006, 281, 23725–23732. [Google Scholar] [CrossRef] [PubMed]
- Federici, L.; Sterzo, C.L.; Pezzola, S.; Di Matteo, A.; Scaloni, F.; Federici, G.; Caccuri, A.M. Structural basis for the binding of the anticancer compound 6-(7-Nitro-2,1,3-Benzoxadiazol-4-Ylthio)hexanol to human glutathione S-transferases. Cancer Res. 2009, 69, 8025–8034. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Dorio, A.S.; Mazzon, E.; Muzi, A.; Sau, A.; Cuzzocrea, S.; Vernole, P.; Federici, G.; Caccuri, A.M.; Graziani, G. The glutathione transferase inhibitor 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) increases temozolomide efficacy against malignant melanoma. Eur. J. Cancer 2011, 47, 1219–1230. [Google Scholar] [CrossRef]
- Awasthi, S.; Srivastava, S.K.; Ahmad, F.; Ahmad, H.; Ansari, G.A. Interactions of glutathione S-transferase-pi with ethacrynic acid and its glutathione conjugate. Biochim. Biophys. Acta 1993, 1164, 173–178. [Google Scholar] [CrossRef]
- Li, T.; Liu, G.; Li, H.; Yang, X.; Jing, Y.; Zhao, G. The synthesis of ethacrynic acid thiazole derivatives as glutathione S-transferase pi inhibitors. Bioorg. Med. Chem. 2012, 20, 2316–2322. [Google Scholar] [CrossRef]
- Crawford, L.A.; Weerapana, E. A tyrosine-reactive irreversible inhibitor for glutathione S-transferase Pi (GSTP1). Mol. BioSyst. 2016, 12, 1768–1771. [Google Scholar] [CrossRef]
- O’Brien, M.L.; Vulevic, B.; Freer, S.; Boyd, J.; Shen, H.; Tew, K.D. Glutathione peptidomimetic drug modulator of multidrug resistance-associated protein. J. Pharmacol. Exp. Ther. 1999, 291, 1348–1355. [Google Scholar] [PubMed]
- McMillan, D.H.; Velden, J.L.J.; Lahue, K.G.; Qian, X.; Schneider, R.W.; Iberg, M.S.; Nolin, J.D.; Abdalla, S.; Casey, D.T.; Tew, K.D.; et al. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase pi. JCI Insight 2016, 1, e85717. [Google Scholar] [CrossRef]
- Raza, A.; Galili, N.; Callander, N.; Ochoa, L.; Piro, L.; Emanuel, P.; Williams, S.; Burrisiii, H.; Faderl, S.; Estrov, Z.; et al. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra®, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J. Hematol. Oncol. 2009, 2, 20. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.-W.; Janssen-Heininger, Y.; Townsend, D.M.; Tew, K.D. Development of telintra as an inhibitor of glutathione s-transferase P. React. Oxyg. Species 2020, 264, 71–91. [Google Scholar]
- Miyanaga, N.; Akaza, H.; Hinotsu, S.; Fujioka, T.; Naito, S.; Namiki, M.; Takahashi, S.; Hirao, Y.; Horie, S.; Tsukamoto, T.; et al. Prostate cancer chemoprevention study: An investigative randomized control study using purified isoflavones in men with rising prostate-specific antigen. Cancer Sci. 2011, 103, 125–130. [Google Scholar] [CrossRef]
- Nagata, C.; Mizoue, T.; Tanaka, K.; Tsuji, I.; Tamakoshi, A.; Matsuo, K.; Wakai, K.; Inoue, M.; Tsugane, S.; Sasazuki, S.; et al. Soy intake and breast cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn. J. Clin. Oncol. 2014, 44, 282–295. [Google Scholar] [CrossRef]
- Jian, L.; Xie, L.P.; Lee, A.H.; Binns, C.W. Protective effect of green tea against prostate cancer: A case-control study in southeast China. Int. J. Cancer 2003, 108, 130–135. [Google Scholar] [CrossRef]
- Roh, J.-L.; Kim, E.H.; Park, J.Y.; Kim, J.W.; Kwon, M.; Lee, B.-H. Piperlongumine selectively kills cancer cells and increases cisplatin antitumor activity in head and neck cancer. Oncotarget 2014, 5, 9227–9238. [Google Scholar] [CrossRef] [PubMed]
- Harshbarger, W.; Gondi, S.; Ficarro, S.B.; Hunter, J.; Udayakumar, D.; Gurbani, D.; Singer, W.D.; Liu, Y.; Li, L.; Marto, J.A.; et al. Structural and biochemical analyses reveal the mechanism of glutathione s-transferase Pi 1 inhibition by the anti-cancer compound piperlongumine. J. Biol. Chem. 2017, 292, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, J.; Dhillon, H.; Chikara, S.; Mamidi, S.; Sreedasyam, A.; Chittem, K.; Orr, M.; Wilkinson, J.C.; Reindl, K.M. Piperlongumine potentiates the effects of gemcitabine in in vitro and in vivo human pancreatic cancer models. Oncotarget 2017, 9, 10457–10469. [Google Scholar] [CrossRef][Green Version]
- Hang, W.; Yin, Z.-X.; Liu, G.; Zeng, Q.; Shen, X.-F.; Sun, Q.-H.; Li, D.-D.; Jian, Y.-P.; Zhang, Y.-H.; Wang, Y.-S.; et al. Piperlongumine and p53-reactivator APR-246 selectively induce cell death in HNSCC by targeting GSTP1. Oncogene 2018, 37, 3384–3398. [Google Scholar] [CrossRef]
- Tanaka, T.; Makita, H.; Ohnishi, M.; Hirose, Y.; Wang, A.; Mori, H.; Satoh, K.; Hara, A.; Ogawa, H. Chemoprevention of 4-nitroquinoline 1-oxide-induced oral carcinogenesis by dietary curcumin and hes-peridin: Comparison with the protective effect of beta-carotene. Cancer Res. 1994, 54, 4653–4659. [Google Scholar] [PubMed]
- Huang, M.T.; Lou, Y.R.; Ma, W.; Newmark, H.L.; Reuhl, K.R.; Conney, A.H. Inhibitory effects of dietary curcumin on forestomach, duodenal, and colon carcinogenesis in mice. Cancer Res. 1994, 54, 5841–5847. [Google Scholar]
- Duvoix, A.; Morceau, F.; Delhalle, S.; Schmitz, M.; Schnekenburger, M.; Galteau, M.-M.; Dicato, M.; Diederich, M. Induction of apoptosis by curcumin: Mediation by glutathione S-transferase P1-1 inhibition. Biochem. Pharmacol. 2003, 66, 1475–1483. [Google Scholar] [CrossRef]
- De Luca, A.; Rotili, D.; Carpanese, D.; Lenoci, A.; Calderan, L.; Scimeca, M.; Mai, A.; Bonanno, E.; Rosato, A.; Geroni, C.; et al. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget 2015, 6, 4126–4143. [Google Scholar] [CrossRef] [PubMed]
- Luisi, G.; Mollica, A.; Carradori, S.; Lenoci, A.; De Luca, A.; Caccuri, A.M. Nitrobenzoxadiazole-based GSTP1-1 inhibitors containing the full peptidyl moiety of (pseudo)glutathione. J. Enzym. Inhib. Med. Chem. 2015, 31, 924–930. [Google Scholar] [CrossRef]
- De Luca, A.; Carpanese, D.; Rapanotti, M.C.; Viguria, T.M.S.; Forgione, M.A.; Rotili, D.; Fulci, C.; Iorio, E.; Quintieri, L.; Chimenti, S.; et al. The nitrobenzoxadiazole derivative MC3181 blocks melanoma invasion and metastasis. Oncotarget 2017, 8, 15520–15538. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Economopoulos, K.P.; Sergentanis, T.N. GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: A comprehensive meta-analysis. Eur. J. Cancer 2010, 46, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, J.; Zhang, Y.; Mu, P.; Wang, X. Expression levels of MRP1, GST-pi, and GSK3beta in ovarian cancer and the relationship with drug resistance and prognosis of patients. Oncol. Lett. 2019, 18, 22–28. [Google Scholar] [PubMed]
- Xu, L.; Cai, J.; Yang, Q.; Ding, H.; Wu, L.; Li, T.; Wang, Z. Prognostic significance of several biomarkers in epithelial ovarian cancer: A meta-analysis of published studies. J. Cancer Res. Clin. Oncol. 2013, 139, 1257–1277. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.-F.; Xie, R.-H.; Yang, A.-K.; Chen, Z.-H.; Wu, Q.-L.; Liu, W.-W.; Ou, S.-M.; Xia, L.-P.; Chen, M.-Y.; Zhang, J.-X. Correlation of GST-pi and PCNA expression to prognosis of advanced maxillary sinus squamous cell carcinoma. Ai zheng Aizheng Chin. J. Cancer 2005, 24, 1267–1271. [Google Scholar]
- Gong, M.; Dong, W.; Shi, Z.; Xu, Y.; Ni, W.; An, R. Genetic polymorphisms of GSTM1, GSTT1, and GSTP1 with prostate cancer risk: A meta-analysis of 57 studies. PLoS ONE 2012, 7, e50587. [Google Scholar] [CrossRef]
- Harpole, D.H.; Moore, M.B.; Herndon, J.E.; Aloia, T.; D’Amico, T.A.; Sporn, T.; Parr, A.; Linoila, I.; Allegra, C. The prognostic value of molecular marker analysis in patients treated with trimodality therapy for esophageal cancer. Clin. Cancer Res. 2001, 7, 562–569. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Model | Anti-Neoplastic Agent | Outcome | Reference |
|---|---|---|---|
| Ovarian cancer | Cisplatin, doxorubicin | Response rate lower in GSTP1-positive patients | [85,86,87] |
| Glioma | Cisplatin, irinotecan | GSTP1 is overexpressed in resistant tumors | [88,89] |
| Gastric cancer | Fluorouracil (5-FU), cisplatin | GSTP1 is overexpressed in resistant tumors | [90] |
| Prostate cancer | Doxorubicin | GSTP1 is overexpressed in resistant tumors | [91] |
| Breast cancer | Adriamycin | GSTP1 is overexpressed in resistant tumors | [92] |
| Breast cancer | Adriamycin | Increased apoptosis and DNA damage upon GSTP1 knockdown | [93] |
| Esophageal squamous cancer cells | Cisplatin | Synergistic effect of GSTP1 knockdown and cisplatin treatment | [94] |
| Lung cancer stem cells | Cisplatin | Synergistic effect of GSTP1 inhibition and cisplatin treatment | [95] |
| Lung cancer | Camptothecin | Increased apoptosis upon GSTP1 knockdown and camptothecin treatment | [96] |
| Lung cancer stem cells | Cisplatin | miRNA-mediated inhibition of GSTP1 reverses cisplatin resistance | [105] |
| Protein | Impact of Glutathionylation | Reference |
|---|---|---|
| 1-cys Prx (Prdx VI) | restores peroxidase activity | [121,157] |
| 2-cys Prx | restores peroxidase activity | [121,127,128] |
| NOS | inhibits activity | [129] |
| PDI | inhibits isomerase activity | [134,136] |
| Actin | inhibits polymerization | [138,139,140] |
| Vimentin | inhibits elongation | [8] |
| Cofilin | reduces depolymerization activity | [8,158] |
| Myosin | increases Ca2+ sensitivity | [8,159] |
| β-tubulin | inhibits polymerization | [140,160] |
| p53 | reduces DNA binding | [146,147] |
| PKC | inhibits activity | [153] |
| Complex-I | inhibits activity | [154] |
| Cytochrome oxidase | inhibits activity | [8] |
| adenosine triphosphate (ATP)-ase | inhibits activity | [14] |
| Carbonic anhydrase | inhibits activity | [155] |
| Pyruvate dehydrogenase | inhibits activity | [156] |
| ERK | inhibits activity | [153] |
| protein-tyrosine phosphatase (PTP1B) | inhibits activity | [14] |
| Phosphatase and tensin homolog (PTEN) | inhibits activity | [161] |
| Aldolase | inhibits activity | [140] |
| Adenylate kinase 2 | inhibits activity | [8] |
| Vimentin | inhibits activity | [8] |
| c-Jun | inhibits activity | [155] |
| NF-κB subunits 65 and 50 | inhibits activity | [162] |
| HSP60 | inhibits activity | [8] |
| HSP70 | inhibits activity | [8] |
| S100 A1, S100 A4, S100 B | increases activity | [163] |
| Nicotinamide adenine dinucleotide hydrogen (NADH) ubiquinone reductase | inhibits activity | [164] |
| Inhibitor of nuclear factor kappa B kinase (IKK) β-subunit | inhibits activity | [165] |
| GAPDH | inhibits activity | [166] |
| Caspase 3 | inhibits activity | [155,167] |
| SerpinA1 and A3 | inhibits activity | [168] |
| TRAF2 | inhibits activity | [134] |
| STAT3 | inhibits activity | [169] |
| Src homology region 2 domain-containing phosphatase 1 and 2 (SHP-1, SHP-2) | inhibits activity | [170] |
| Thioredoxin (Trx) | inhibits activity | [171] |
| p12 | inhibits activity | [167] |
| p17 | inhibits activity | [167] |
| Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) | increases activity | [172] |
| CCAAT/enhancer-binding homologous protein (CHOP) | inhibits activity | [134] |
| Protein kinase B (Akt) | increases activity | [62] |
| Calreticulin | inhibits activity | [166] |
| Enolase 1 (Eno1) | inhibits activity | [166] |
| High mobility group box 1 (HMGB1) | inhibits activity | [173] |
| Ras | increases activity | [174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. https://doi.org/10.3390/antiox10050701
Singh RR, Reindl KM. Glutathione S-Transferases in Cancer. Antioxidants. 2021; 10(5):701. https://doi.org/10.3390/antiox10050701
Chicago/Turabian StyleSingh, Rahul Raj, and Katie M. Reindl. 2021. "Glutathione S-Transferases in Cancer" Antioxidants 10, no. 5: 701. https://doi.org/10.3390/antiox10050701
APA StyleSingh, R. R., & Reindl, K. M. (2021). Glutathione S-Transferases in Cancer. Antioxidants, 10(5), 701. https://doi.org/10.3390/antiox10050701