
Intestinal and Hepatic Uptake of Dietary Peroxidized Lipids and Their Decomposition Products, and Their Subsequent Effects on Apolipoprotein A1 and Paraoxonase1

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Thiobarbituric Acid Reactive Substances (TBARS) Assay
2.3. Cell Culture
2.4. Preparation of 13-HPODE and 13-HODE
2.5. Synthesis of 9-ONA
2.6. Cell Treatment 13-HPODE and Decomposition Products
2.7. Analyses of 13-HPODE and 13-HODE HPLC
2.8. TLC and Radioautography for Cell [14C]-13-HPODE Decomposition Products
2.9. Extraction and LC-HRMS Analysis of 13-HPODE Decomposition Products
2.10. Gene Expression
2.11. Western Blot Analysis
2.12. ApoA1 and ApoB ELISAs
2.13. PON1 Activity
2.14. Statistics
3. Results
3.1. Lipid Peroxides and Decomposition Products Detected in Dietary Cooking Oils
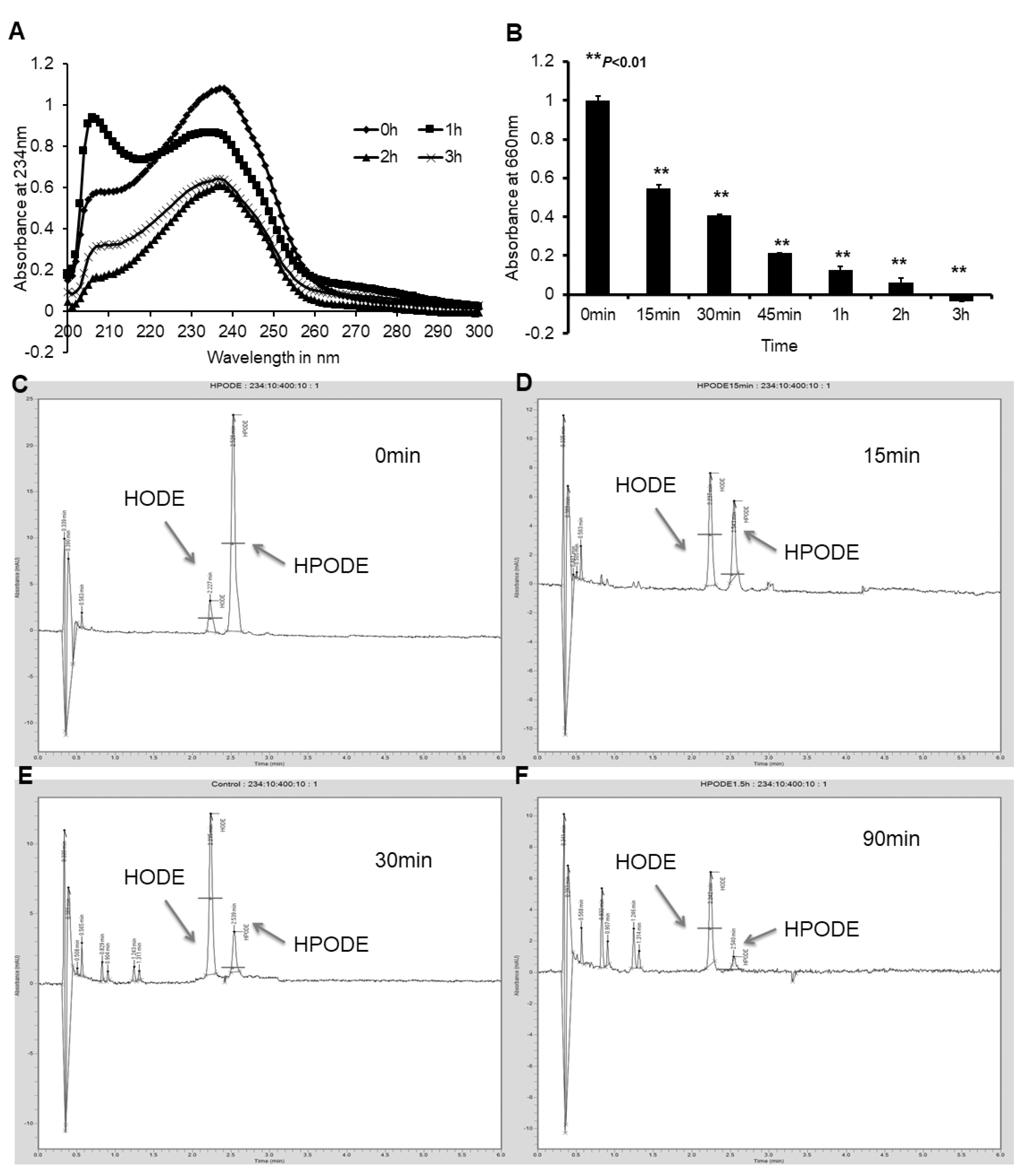
3.2. Caco-2 and HepG2 Cells Efficiently Reduce Lipid Hydroperoxides to Corresponding Hydroxides
3.3. Cellular Uptake of Lipid Peroxide and Corresponding Hydroxide
3.4. TLC Radioautography of [14C]-13-HPODE Decomposition by Cells
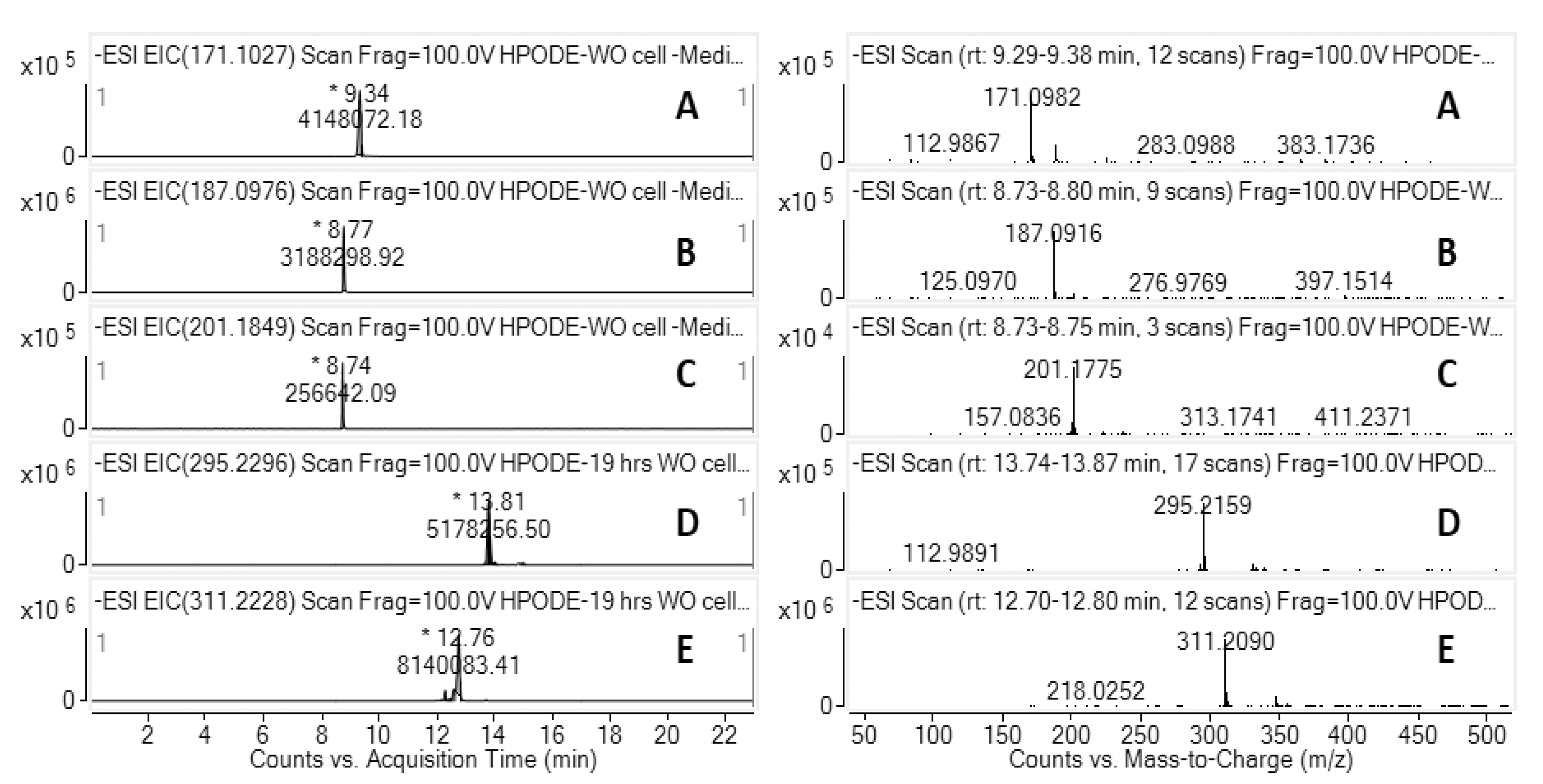
3.5. LC-HRMS Analysis of 13-HPODE Decomposition Products by Caco-2 Cells
3.6. Oxidation of 9-ONA to AzA, and Uptake of AzA in Differentiated Caco-2 Cells
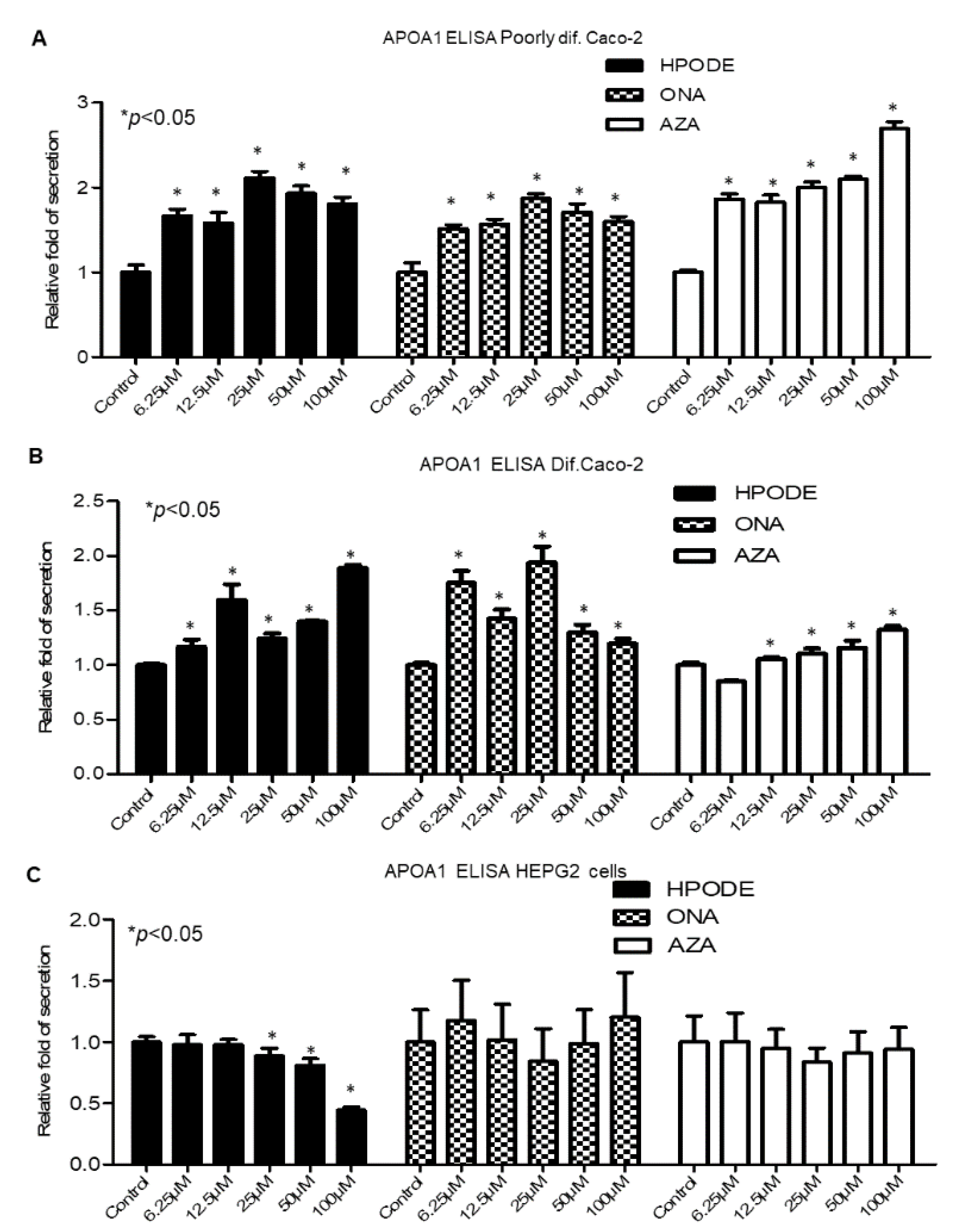
3.7. Intestinal and Hepatic 13-HPODE-, 9-ONA-, and AzA-Induced ApoA1 Secretion
3.8. Intestinal and Hepatic 13-HPODE-, 9-ONA-, and Aza-Induced PON1 Activity
3.9. Suppression of ApoB48 Secretion by AzA in Differentiated Caco-2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef]
- Kuller, L.H. Nutrition, lipids, and cardiovascular disease. Nutr. Rev. 2006, 64, S15–S26. [Google Scholar] [CrossRef]
- Chahoud, G.; Aude, Y.W.; Mehta, J.L. Dietary recommendations in the prevention and treatment of coronary heart disease: Do we have the ideal diet yet? Am. J. Cardiol. 2004, 94, 1260–1267. [Google Scholar] [CrossRef]
- Lichtenstein, A.H. Dietary fat and cardiovascular disease risk: Quantity or quality? J. Women’s Health 2003, 12, 109–114. [Google Scholar] [CrossRef]
- Lemieux, H.; Bulteau, A.L.; Friguet, B.; Tardif, J.C.; Blier, P.U. Dietary fatty acids and oxidative stress in the heart mitochondria. Mitochondrion 2011, 11, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Behniwal, P.K.; Soni, G.L.; Vadhera, S.; Singh, R. In vitro absorption of nutrients from small intestine of rats fed peroxidized oil. Indian J. Exp. Biol. 1993, 31, 658–659. [Google Scholar]
- Grootveld, M.; Percival, B.C.; Grootveld, K.L. Chronic non-communicable disease risks presented by lipid oxidation products in fried foods. Hepatobiliary Surg. Nutr. 2018, 7, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Glavind, J.; Hartmann, S. The occurrence of peroxidized lipids in atheromatous human aortas. Experientia 1951, 7, 464. [Google Scholar] [CrossRef]
- Desai, I.D.; Tappel, A.L. Damage to proteins by peroxidized lipids. J. Lipid Res. 1963, 4, 204–207. [Google Scholar] [CrossRef]
- Nielsen, H. Reaction between peroxidized phospholipid and protein: I. Covalent binding of peroxidized cardiolipin to albumin. Lipids 1978, 13, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H. Reaction between peroxidized phospholipid and protein: II. Molecular weight and phosphorus content of albumin after reaction with peroxidized cardiolipin. Lipids 1979, 14, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Barsacchi, R.; Camici, P.; Bottigli, U.; Salvadori, P.A.; Pelosi, G.; Maiorino, M.; Ursini, F. Correlation between hydroperoxide-induced chemiluminescence of the heart and its function. Biochim. Biophys. Acta 1983, 762, 241–247. [Google Scholar] [CrossRef]
- Gulati, J.; Gupta, P.P.; Soni, G.L.; Vadhera, S.; Singh, R. Effects of peroxidized oil on the development of experimental atherosclerosis in rabbits. Indian Heart J. 1992, 44, 235–239. [Google Scholar] [PubMed]
- Mickel, H.S.; Horbar, J. The effect of peroxidized arachidonic acid upon human platelet aggregation. Lipids 1974, 9, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Mukai, F.H.; Goldstein, B.D. Mutagenicity of malonaldehyde, a decomposition product of peroxidized polyunsaturated fatty acids. Science 1976, 191, 868–869. [Google Scholar] [CrossRef] [PubMed]
- Hegstad, A.C.; Strand, H.; Ytrehus, K. Phospholipid peroxidation after 60 min of global ischaemia and 10 min of reperfusion. A study in the isolated rat heart. J. Mol. Cell. Cardiol. 1994, 26, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Ytrehus, K.; Aspang, E.M. Phospholipid peroxidation in isolated perfused rat hearts subjected to hypothermia followed by rewarming: Inverse relation to loss of function. Cryobiology 1994, 31, 263–271. [Google Scholar] [CrossRef]
- Goheen, S.C.; O’Rourke, L.; Larkin, E.C. Ozone and the peroxidation of polyunsaturated fatty acids in vivo. Environ. Res. 1986, 40, 47–57. [Google Scholar] [CrossRef]
- Zalejska-Fiolka, J.; Wielkoszyński, T.; Kasperczyk, S.; Kasperczyk, A.; Birkner, E. Effects of oxidized cooking oil and α-lipoic acid on blood antioxidants: Enzyme activities and lipid peroxidation in rats fed a high-fat diet. Biol. Trace Elem. Res. 2012, 145, 217–221. [Google Scholar] [CrossRef]
- Yin, H.; Porter, N.A. New insights regarding the autoxidation of polyunsaturated fatty acids. Antioxid. Redox Signal. 2005, 7, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, K.L. Linoleic acid, vegetable oils & inflammation. MO Med. 2014, 111, 41–43. [Google Scholar]
- Whelan, J.; Fritsche, K. Linoleic acid. Adv. Nutr. 2013, 4, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Raghavamenon, A.; Garelnabi, M.; Babu, S.; Aldrich, A.; Litvnov, D.; Parthasarathy, S. a-Tocopherol Is Ineffective in Preventing the Decomposition of Preformed Lipid Peroxides and May Promote the Accumulation of Toxic Aldehydes: A Potential Explanation for the Failure of Antioxidants to Affect Human Atherosclerosis. Antioxid. Redox Signal. 2009, 11, 11. [Google Scholar] [CrossRef]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Biasi, F.; Leonarduzzi, G. 4-Hydroxynonenal-protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol. Asp. Med. 2008, 29, 67–71. [Google Scholar] [CrossRef]
- Khan-Merchant, N.; Penumetcha, M.; Meilhac, O.; Parthasarathy, S. Oxidized fatty acids promote atherosclerosis only in the presence of dietary cholesterol in low-density lipoprotein receptor knockout mice. J. Nutr. 2002, 132, 3256–3262. [Google Scholar] [CrossRef]
- Feig, J.E.; Feig, J.L.; Dangas, G.D. The role of HDL in plaque stabilization and regression: Basic mechanisms and clinical implications. Coron. Artery Dis. 2016, 27, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Pownall, H.J.; Ehnholm, C. The unique role of apolipoprotein A-I in HDL remodeling and metabolism. Curr. Opin. Lipidol. 2006, 17, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, C.G.; Anantharamaiah, G.M.; Engler, J.A.; Borhani, D.W. Structural models of human apolipoprotein A-I: A critical analysis and review. Biochim. Biophys. Acta 2001, 1531, 4–46. [Google Scholar] [CrossRef]
- Colvin, P.L.; Parks, J.S. Metabolism of high density lipoprotein subfractions. Curr. Opin. Lipidol. 1999, 10, 309–314. [Google Scholar] [CrossRef]
- Eggerman, T.L.; Hoeg, J.M.; Meng, M.S.; Tombragel, A.; Bojanovski, D.; Brewer, H.B., Jr. Differential tissue-specific expression of human apoA-I and apoA-II. J. Lipid Res. 1991, 32, 821–828. [Google Scholar] [CrossRef]
- Danielsen, E.M.; Hansen, G.H.; Rasmussen, K.; Niels-Christiansen, L.L.; Frenzel, F. Apolipoprotein A-1 (apoA-1) deposition in, and release from, the enterocyte brush border: A possible role in transintestinal cholesterol efflux (TICE)? Biochim. Biophys. Acta 2012, 1818, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J. High-density lipoproteins as an emerging therapeutic target for atherosclerosis. JAMA J. Am. Med. Assoc. 2003, 290, 2322–2324. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, G. Epidemiologic evidence for high-density lipoprotein cholesterol as a risk factor for coronary artery disease. Am. J. Cardiol. 2001, 88, 9N–13N. [Google Scholar] [CrossRef]
- Maron, D.J. The epidemiology of low levels of high-density lipoprotein cholesterol in patients with and without coronary artery disease. Am. J. Cardiol. 2000, 86, 11L–14L. [Google Scholar] [CrossRef]
- Libby, P.; Aikawa, M.; Schonbeck, U. Cholesterol and atherosclerosis. Biochim. Biophys. Acta 2000, 1529, 299–309. [Google Scholar] [CrossRef]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Fogelman, A.M. Mechanisms of disease: Proatherogenic HDL—An evolving field. Nat. Clin. Practice. Endocrinol. Metab. 2006, 2, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.; Rader, D.J. Emerging therapies targeting high-density lipoprotein metabolism and reverse cholesterol transport. Circulation 2006, 113, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K.; Valenta, D.T.; Hime, N.J.; Rye, K.A. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Trigatti, B.; Rigotti, A.; Krieger, M. The role of the high-density lipoprotein receptor SR-BI in cholesterol metabolism. Curr. Opin. Lipidol. 2000, 11, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Barnett, J.; Fong, L.G. High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim. Biophys. Acta 1990, 1044, 275–283. [Google Scholar] [CrossRef]
- Navab, M.; Ananthramaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Ansell, B.J.; Fonarow, G.C.; Vahabzadeh, K.; Hama, S.; Hough, G.; Kamranpour, N.; et al. The oxidation hypothesis of atherogenesis: The role of oxidized phospholipids and HDL. J. Lipid Res. 2004, 45, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.Y.; Cooke, C.J.; Anantharamaiah, G.M.; Chaddha, M.; Jin, L.; Subbanagounder, G.; Faull, K.F.; Reddy, S.T.; Miller, N.E.; et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: Step 1. J. Lipid Res. 2000, 41, 1481–1494. [Google Scholar] [CrossRef]
- Rong, R.; Ramachandran, S.; Penumetcha, M.; Khan, N.; Parthasarathy, S. Dietary oxidized fatty acids may enhance intestinal apolipoprotein A-I production. J. Lipid Res. 2002, 43, 557–564. [Google Scholar] [CrossRef]
- Cohn, J.S.; Marcoux, C.; Davignon, J. Detection, quantification, and characterization of potentially atherogenic triglyceride-rich remnant lipoproteins. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2474–2486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olofsson, S.O.; Borèn, J. Apolipoprotein B: A clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J. Intern. Med. 2005, 258, 395–410. [Google Scholar] [CrossRef]
- Staprãns, I.; Rapp, J.H.; Pan, X.M.; Kim, K.Y.; Feingold, K.R. Oxidized lipids in the diet are a source of oxidized lipid in chylomicrons of human serum. Arter. Thromb. 1994, 14, 1900–1905. [Google Scholar] [CrossRef]
- Penumetcha, M.; Khan, N.; Parthasarathy, S. Dietary oxidized fatty acids: An atherogenic risk? J. Lipid Res. 2000, 41, 1473–1480. [Google Scholar] [CrossRef]
- Porter, N.A. A perspective on free radical autoxidation: The physical organic chemistry of polyunsaturated fatty acid and sterol peroxidation. J. Org. Chem. 2013, 78, 3511–3524. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, N.; Ohkawa, H.; Miike, A.; Tatano, T.; Yagi, K. A new assay method for lipid peroxides using a methylene blue derivative. Biochem. Int. 1985, 10, 205–211. [Google Scholar]
- Kaneko, T.; Kaji, K.; Matsuo, M. Cytotoxicities of a linoleic acid hydroperoxide and its related aliphatic aldehydes toward cultured human umbilical vein endothelial cells. Chem. Biol. Interact. 1988, 67, 295–304. [Google Scholar] [CrossRef]
- Lopez, M.A.; Vicente, J.; Kulasekaran, S.; Vellosillo, T.; Martinez, M.; Irigoyen, M.L.; Cascon, T.; Bannenberg, G.; Hamberg, M.; Castresana, C. Antagonistic role of 9-lipoxygenase-derived oxylipins and ethylene in the control of oxidative stress, lipid peroxidation and plant defence. Plant J. Cell Mol. Biol. 2011, 67, 447–458. [Google Scholar] [CrossRef]
- Litvinov, D.; Selvarajan, K.; Garelnabi, M.; Brophy, L.; Parthasarathy, S. Anti-atherosclerotic actions of azelaic acid, an end product of linoleic acid peroxidation, in mice. Atherosclerosis 2010, 209, 449–454. [Google Scholar] [CrossRef]
- Muellner, M.K.; Schreier, S.M.; Laggner, H.; Hermann, M.; Esterbauer, H.; Exner, M.; Gmeiner, B.M.; Kapiotis, S. Hydrogen sulfide destroys lipid hydroperoxides in oxidized LDL. Biochem. J. 2009, 420, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Jaichander, P.; Selvarajan, K.; Garelnabi, M.; Parthasarathy, S. Induction of paraoxonase 1 and apolipoprotein A-I gene expression by aspirin. J. Lipid Res. 2008, 49, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Orsavova, J.; Misurcova, L.; Ambrozova, J.V.; Vicha, R.; Mlcek, J. Fatty Acids Composition of Vegetable Oils and Its Contribution to Dietary Energy Intake and Dependence of Cardiovascular Mortality on Dietary Intake of Fatty Acids. Int. J. Mol. Sci. 2015, 16, 12871–12890. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Furutera, A.; Seki, K.; Toyoda, Y.; Tanaka, K.; Sugimoto, Y. Malondialdehyde generated from peroxidized linolenic acid causes protein modification in heat-stressed plants. Plant Physiol. Biochem. 2008, 46, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Shokri, Y.; Variji, A.; Nosrati, M.; Khonakdar-Tarsi, A.; Kianmehr, A.; Kashi, Z.; Bahar, A.; Bagheri, A.; Mahrooz, A. Importance of paraoxonase 1 (PON1) as an antioxidant and antiatherogenic enzyme in the cardiovascular complications of type 2 diabetes: Genotypic and phenotypic evaluation. Diabetes Res. Clin. Pract. 2020, 161, 108067. [Google Scholar] [CrossRef] [PubMed]
- Staprans, I.; Hardman, D.A.; Pan, X.M.; Feingold, K.R. Effect of oxidized lipids in the diet on oxidized lipid levels in postprandial serum chylomicrons of diabetic patients. Diabetes Care 1999, 22, 300–306. [Google Scholar] [CrossRef]
- Staprans, I.; Pan, X.M.; Rapp, J.H.; Feingold, K.R. The role of dietary oxidized cholesterol and oxidized fatty acids in the development of atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1075–1082. [Google Scholar] [CrossRef]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef]
- Levy, E.; Mehran, M.; Seidman, E. Caco-2 cells as a model for intestinal lipoprotein synthesis and secretion. FASEB J. 1995, 9, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Hiebl, V.; Schachner, D.; Ladurner, A.; Heiss, E.H.; Stangl, H.; Dirsch, V.M. Caco-2 Cells for Measuring Intestinal Cholesterol Transport—Possibilities and Limitations. Biol. Proced. Online 2020, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Mehran, M.; Levy, E.; Bendayan, M.; Seidman, E. Lipid, apolipoprotein, and lipoprotein synthesis and secretion during cellular differentiation in Caco-2 cells. In Vitro Cell Dev. Biol. Anim. 1997, 33, 118–128. [Google Scholar] [CrossRef]
- Grefhorst, A.; Verkade, H.J.; Groen, A.K. The TICE Pathway: Mechanisms and Lipid-Lowering Therapies. Methodist Debakey Cardiovasc. J. 2019, 15, 70–76. [Google Scholar] [CrossRef]
- Aw, T.Y.; Williams, M.W. Intestinal absorption and lymphatic transport of peroxidized lipids in rats: Effect of exogenous GSH. Am. J. Physiol. 1992, 263, G665–G672. [Google Scholar] [CrossRef]
- Yuan, Z.X.; Rapoport, S.I.; Soldin, S.J.; Remaley, A.T.; Taha, A.Y.; Kellom, M.; Gu, J.; Sampson, M.; Ramsden, C.E. Identification and profiling of targeted oxidized linoleic acid metabolites in rat plasma by quadrupole time-of-flight mass spectrometry. Biomed. Chromatogr. BMC 2013, 27, 422–432. [Google Scholar] [CrossRef]
- Kanazawa, K.; Ashida, H. Dietary hydroperoxides of linoleic acid decompose to aldehydes in stomach before being absorbed into the body. Biochim. Biophys. Acta 1998, 1393, 349–361. [Google Scholar] [CrossRef]
- Zarei, M.; Uppin, V.; Acharya, P.; Talahalli, R. Ginger and turmeric lipid-solubles attenuate heated oil-induced oxidative stress in the brain via the upregulation of NRF2 and improve cognitive function in rats. Metab. Brain Dis. 2021, 36, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef]
- Grego, A.V.; Mingrone, G. Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: An update. Clin. Nutr. 1995, 14, 143–148. [Google Scholar] [CrossRef]
- Passi, S.; Picardo, M.; Mingrone, G.; Breathnach, A.S.; Nazzaro-Porro, M. Azelaic acid--biochemistry and metabolism. Acta Derm. Venereol. Suppl. 1989, 143, 8–13. [Google Scholar]
- Fitton, A.; Goa, K.L. Azelaic acid. A review of its pharmacological properties and therapeutic efficacy in acne and hyperpigmentary skin disorders. Drugs 1991, 41, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Sieber, M.A.; Hegel, J.K. Azelaic acid: Properties and mode of action. Ski. Pharmacol. Physiol. 2014, 27 (Suppl. S1), 9–17. [Google Scholar] [CrossRef] [PubMed]
- Schicho, R.; Marsche, G.; Storr, M. Cardiovascular complications in inflammatory bowel disease. Curr. Drug Targets 2015, 16, 181–188. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Deme, P.; Gupta, R.; Litvinov, D.; Burge, K.; Parthasarathy, S.; Narasimhulu, C.A. Intestinal and Hepatic Uptake of Dietary Peroxidized Lipids and Their Decomposition Products, and Their Subsequent Effects on Apolipoprotein A1 and Paraoxonase1. Antioxidants 2021, 10, 1258. https://doi.org/10.3390/antiox10081258
Jiang X, Deme P, Gupta R, Litvinov D, Burge K, Parthasarathy S, Narasimhulu CA. Intestinal and Hepatic Uptake of Dietary Peroxidized Lipids and Their Decomposition Products, and Their Subsequent Effects on Apolipoprotein A1 and Paraoxonase1. Antioxidants. 2021; 10(8):1258. https://doi.org/10.3390/antiox10081258
Chicago/Turabian StyleJiang, Xueting, Pragney Deme, Rajat Gupta, Dmitry Litvinov, Kathryn Burge, Sampath Parthasarathy, and Chandrakala Aluganti Narasimhulu. 2021. "Intestinal and Hepatic Uptake of Dietary Peroxidized Lipids and Their Decomposition Products, and Their Subsequent Effects on Apolipoprotein A1 and Paraoxonase1" Antioxidants 10, no. 8: 1258. https://doi.org/10.3390/antiox10081258
APA StyleJiang, X., Deme, P., Gupta, R., Litvinov, D., Burge, K., Parthasarathy, S., & Narasimhulu, C. A. (2021). Intestinal and Hepatic Uptake of Dietary Peroxidized Lipids and Their Decomposition Products, and Their Subsequent Effects on Apolipoprotein A1 and Paraoxonase1. Antioxidants, 10(8), 1258. https://doi.org/10.3390/antiox10081258