
Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle

 , and
, and {kind=link}
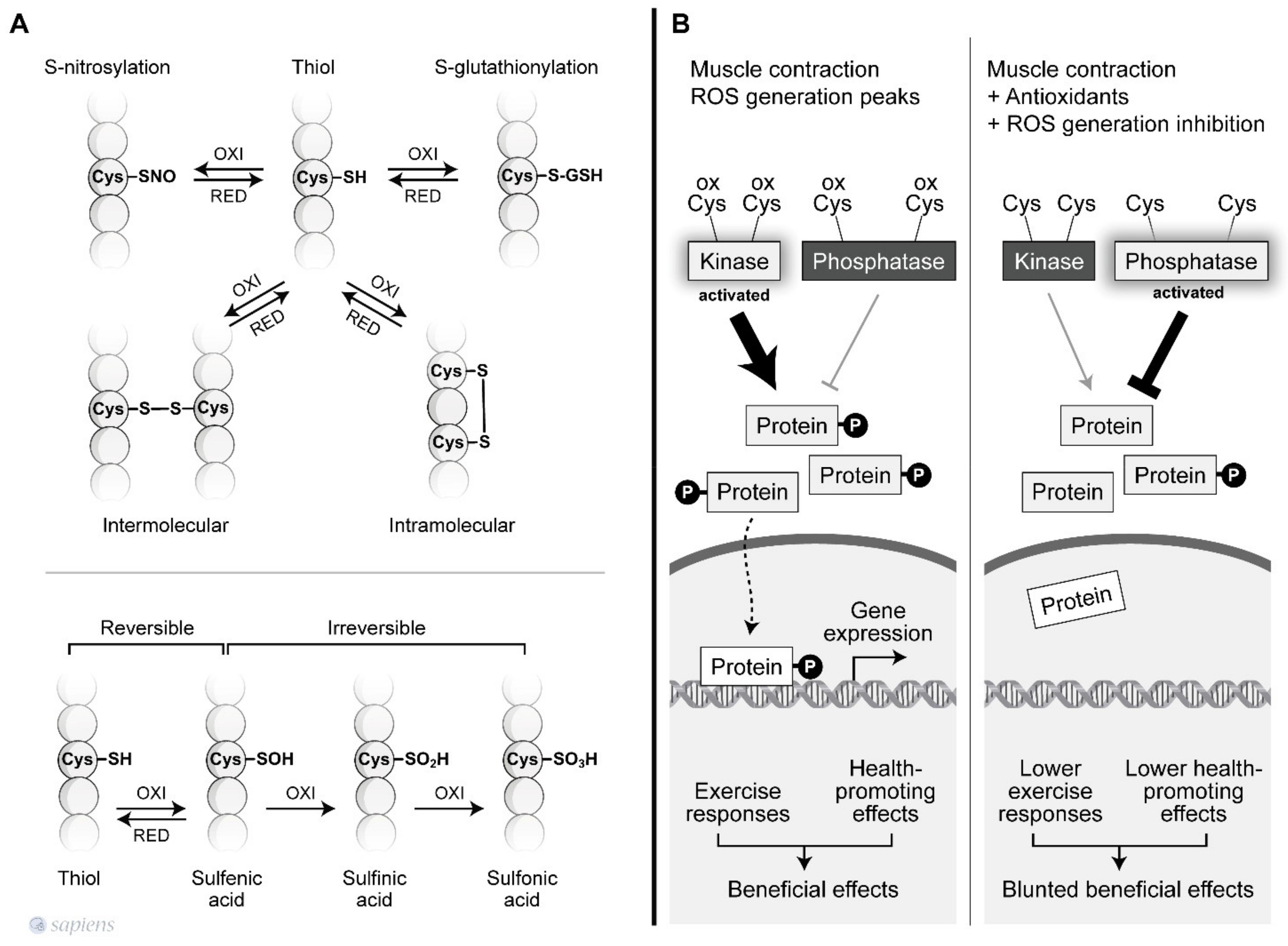{kind=link}
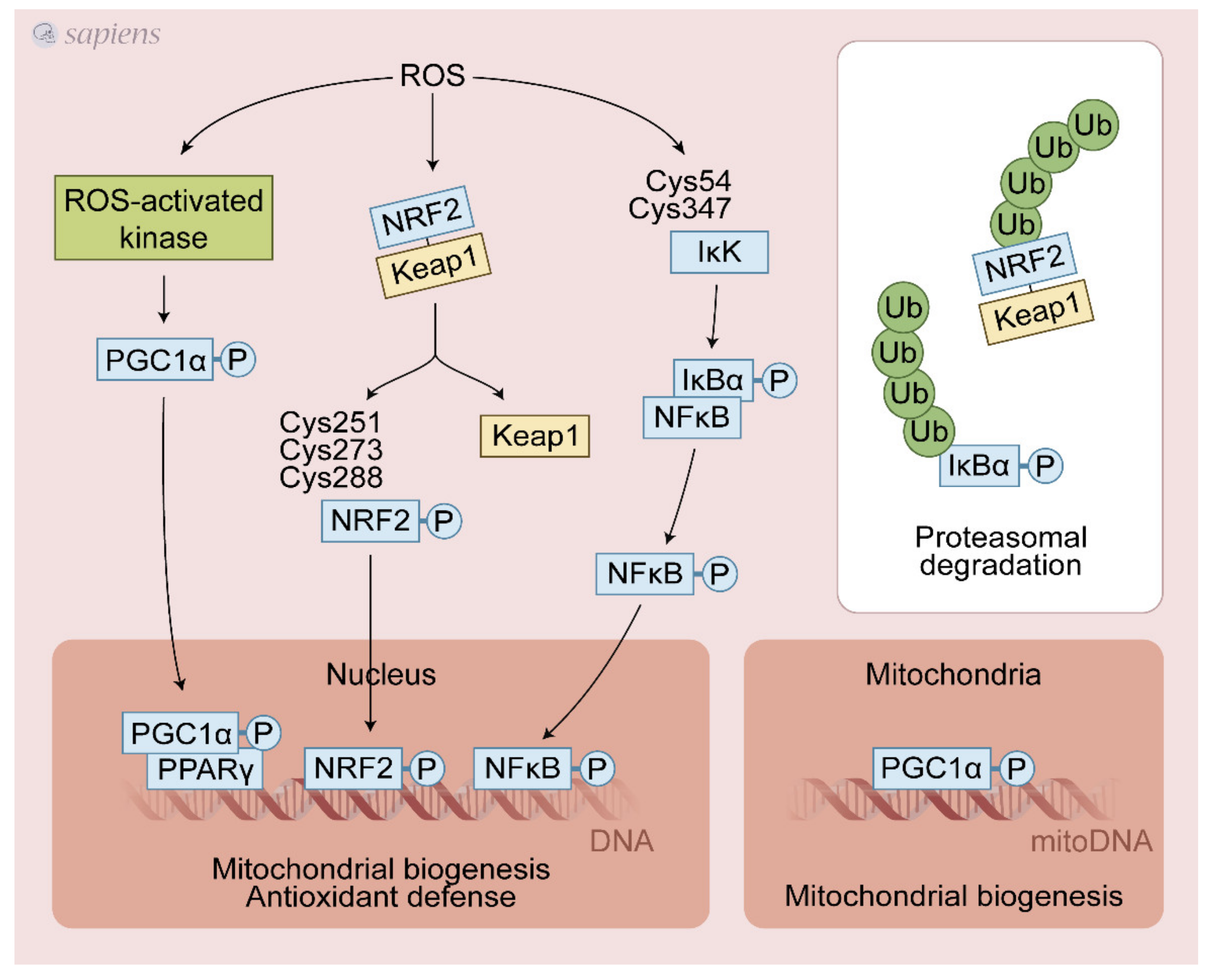{kind=link}
{kind=link}
Abstract
1. Introduction
2. Main Sources of ROS in Skeletal Muscle during Contraction
3. Intracellular Mechanisms Potentially Related to ROS Production Following Muscular Contraction
4. The Concept of ROS Modulating Phosphatases and Kinase Tone
5. Exercise-Generated ROS Activate Redox-Sensitive Transcription Factors
6. Exercise-Generated ROS Is Crucial to Muscle Glucose Uptake
7. The Role of ROS in Physical Exercise Adaptations
7.1. Mitochondrial Biogenesis and Antioxidant Defense
7.2. Hypertrophy and Mass Regulation
8. Fine-Tuned Redox-Mediated Signaling Promotes the Beneficial Effects of Exercise in Muscle
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hawley, J.A.; Hargreaves, M.; Joyner, M.J.; Zierath, J.R. Integrative Biology of Exercise. Cell 2014, 159, 738–749. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Saltin, B. Exercise as Medicine-Evidence for Prescribing Exercise as Therapy in 26 Different Chronic Diseases. Scand. J. Med. Sci. Sports 2015, 25, 1–72. [Google Scholar] [CrossRef]
- Louzada, R.A.; Bouviere, J.; Matta, L.P.; Werneck-de-Castro, J.P.; Dupuy, C.; Carvalho, D.P.; Fortunato, R.S. Redox Signaling in Widespread Health Benefits of Exercise. Antioxid. Redox Signal. 2020, 33, 745–760. [Google Scholar] [CrossRef]
- Jackson, M.J.; Vasilaki, A.; McArdle, A. Cellular Mechanisms Underlying Oxidative Stress in Human Exercise. Free Radic. Biol. Med. 2016, 98, 13–17. [Google Scholar] [CrossRef]
- Powers, S.K.; Radak, Z.; Ji, L.L. Exercise-Induced Oxidative Stress: Past, Present and Future. J. Physiol. 2016, 594, 5081–5092. [Google Scholar] [CrossRef]
- Liochev, S.I. Reactive Oxygen Species and the Free Radical Theory of Aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Commentary: Oxidative Stress Reconsidered. Genes Nutr. 2009, 4, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015; ISBN 978-0-19-180213-3. [Google Scholar]
- Hawley, J.A.; Lessard, S.J. Exercise Training-Induced Improvements in Insulin Action. Acta Physiol. 2008, 192, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants Prevent Health-Promoting Effects of Physical Exercise in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef]
- Clifford, T.; Jeffries, O.; Stevenson, E.J.; Davies, K.A.B. The Effects of Vitamin C and E on Exercise-Induced Physiological Adaptations: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Crit. Rev. Food Sci. Nutr. 2020, 60, 3669–3679. [Google Scholar] [CrossRef]
- Henriquez-Olguin, C.; Meneses-Valdes, R.; Jensen, T.E. Compartmentalized Muscle Redox Signals Controlling Exercise Metabolism-Current State, Future Challenges. Redox Biol. 2020, 35, 101473. [Google Scholar] [CrossRef] [PubMed]
- Margaritelis, N.V.; Paschalis, V.; Theodorou, A.A.; Kyparos, A.; Nikolaidis, M.G. Redox Basis of Exercise Physiology. Redox Biol. 2020, 35, 101499. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.S.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of Superoxide and Hydrogen Peroxide Production by Muscle Mitochondria Assessed Ex Vivo under Conditions Mimicking Rest and Exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of Mitochondrial and Nonmitochondrial Sources Implicate Nicotinamide Adenine Dinucleotide Phosphate Oxidase(s) in the Increased Skeletal Muscle Superoxide Generation That Occurs during Contractile Activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Close, G.L.; Kayani, A.; McArdle, A.; Viña, J.; Jackson, M.J. Effect of Xanthine Oxidase-Generated Extracellular Superoxide on Skeletal Muscle Force Generation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El-Hassani, R.; Schlumberger, M.; Dupuy, C. NADPH Oxidases: New Actors in Thyroid Cancer? Nat. Rev. Endocrinol. 2016, 12, 485–494. [Google Scholar] [CrossRef]
- Henríquez-Olguín, C.; Boronat, S.; Cabello-Verrugio, C.; Jaimovich, E.; Hidalgo, E.; Jensen, T.E. The Emerging Roles of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2 in Skeletal Muscle Redox Signaling and Metabolism. Antioxid. Redox Signal. 2019, 31, 1371–1410. [Google Scholar] [CrossRef]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.G.; Jackson, M.J. In Situ Detection and Measurement of Intracellular Reactive Oxygen Species in Single Isolated Mature Skeletal Muscle Fibers by Real Time Fluorescence Microscopy. Antioxid. Redox Signal. 2008, 10, 1463–1474. [Google Scholar] [CrossRef]
- Stofan, D.A.; Callahan, L.A.; DiMarco, A.F.; Nethery, D.E.; Supinski, G.S. Modulation of Release of Reactive Oxygen Species by the Contracting Diaphragm. Am. J. Respir. Crit. Care Med. 2000, 161, 891–898. [Google Scholar]
- Dikalov, S.I.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef]
- Maulucci, G.; Bačić, G.; Bridal, L.; Schmidt, H.H.; Tavitian, B.; Viel, T.; Utsumi, H.; Yalçın, A.S.; De Spirito, M. Imaging Reactive Oxygen Species-Induced Modifications in Living Systems. Antioxid. Redox Signal. 2016, 24, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Bačić, G.; Pavićević, A.; Peyrot, F. In Vivo Evaluation of Different Alterations of Redox Status by Studying Pharmacokinetics of Nitroxides Using Magnetic Resonance Techniques. Redox Biol. 2016, 8, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS Production by NADPH Oxidase 2 Regulates Muscle Glucose Uptake during Exercise. Nat. Commun. 2019, 10, 4623. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.N.; Zhao, H.; Chellappa, K.; Davis, J.G.; Nioka, S.; Baur, J.A.; Li, L.Z. Optical Redox Imaging of Fixed Unstained Muscle Slides Reveals Useful Biological Information. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2019, 21, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Herrero, A.; Barja, G. ADP-Regulation of Mitochondrial Free Radical Production Is Different with Complex I- or Complex II-Linked Substrates: Implications for the Exercise Paradox and Brain Hypermetabolism. J. Bioenerg. Biomembr. 1997, 29, 241–249. [Google Scholar] [CrossRef]
- Venditti, P.; Masullo, P.; Di Meo, S. Effect of Training on H2O2 Release by Mitochondria from Rat Skeletal Muscle. Arch. Biochem. Biophys. 1999, 372, 315–320. [Google Scholar] [CrossRef]
- Sun, Q.A.; Wang, B.; Miyagi, M.; Hess, D.T.; Stamler, J.S. Oxygen-Coupled Redox Regulation of the Skeletal Muscle Ryanodine Receptor/Ca2+ Release Channel (RyR1): Sites and Nature of Oxidative Modification. J. Biol. Chem. 2013, 288, 22961–22971. [Google Scholar] [CrossRef]
- Henríquez-Olguín, C.; Renani, L.B.; Arab-Ceschia, L.; Raun, S.H.; Bhatia, A.; Li, Z.; Knudsen, J.R.; Holmdahl, R.; Jensen, T.E. Adaptations to High-Intensity Interval Training in Skeletal Muscle Require NADPH Oxidase 2. Redox Biol. 2019, 24, 101188. [Google Scholar] [CrossRef]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol Ameliorates Muscular Pathology in the Dystrophic Mdx Mouse, a Model for Duchenne Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, A.C.C.; Rêgo-Monteiro, I.C.D.; Louzada, R.A.; Ortenzi, V.H.; Aguiar, A.P.D.; Abreu, E.S.D.; Cavalcanti-De-Albuquerque, J.P.A.; Hecht, F.; Oliveira, A.C.D.; Ceccatto, V.M.; et al. Differential Expression of NADPH Oxidases Depends on Skeletal Muscle Fiber Type in Rats. Oxid. Med. Cell. Longev. 2016, 2016, 6738701. [Google Scholar] [CrossRef]
- Louzada, R.A.; Corre, R.; Ameziane-El-Hassani, R.; Hecht, F.; Cazarin, J.; Buffet, C.; Carvalho, D.P.; Dupuy, C. Conformation of the N-Terminal Ectodomain Elicits Different Effects on DUOX Function: A Potential Impact on Congenital Hypothyroidism Caused by a H2O2 Production Defect. Thyroid Off. J. Am. Thyroid Assoc. 2018, 28, 1052–1062. [Google Scholar] [CrossRef]
- Carré, A.; Louzada, R.A.N.; Fortunato, R.S.; Ameziane-El-Hassani, R.; Morand, S.; Ogryzko, V.; de Carvalho, D.P.; Grasberger, H.; Leto, T.L.; Dupuy, C. When an Intramolecular Disulfide Bridge Governs the Interaction of DUOX2 with Its Partner DUOXA2. Antioxid. Redox Signal. 2015, 23, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.H.W.; Busse, R.; Brandes, R.P. Direct Interaction of the Novel Nox Proteins with P22phox Is Required for the Formation of a Functionally Active NADPH Oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 Functions as a Mitochondrial Energetic Sensor Coupling Cancer Metabolic Reprogramming to Drug Resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Michaelson, L.P.; Shi, G.; Ward, C.W.; Rodney, G.G. Mitochondrial Redox Potential during Contraction in Single Intact Muscle Fibers. Muscle Nerve 2010, 42, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Angélica Carrasco, M.; Jaimovich, E. Myotube Depolarization Generates Reactive Oxygen Species through NAD(P)H Oxidase; ROS-Elicited Ca2+ Stimulates ERK, CREB, Early Genes. J. Cell. Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sánchez, G.; Barrientos, G.; Aracena-Parks, P. A Transverse Tubule NADPH Oxidase Activity Stimulates Calcium Release from Isolated Triads via Ryanodine Receptor Type 1 S-Glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Vegas, A.; Campos, C.A.; Contreras-Ferrat, A.; Casas, M.; Buvinic, S.; Jaimovich, E.; Espinosa, A. ROS Production via P2Y1-PKC-NOX2 Is Triggered by Extracellular ATP after Electrical Stimulation of Skeletal Muscle Cells. PLoS ONE 2015, 10, e0129882. [Google Scholar] [CrossRef]
- Javeshghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N.A. Molecular Characterization of a Superoxide-Generating NAD(P)H Oxidase in the Ventilatory Muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418. [Google Scholar] [CrossRef]
- Ward, C.W.; Prosser, B.L.; Lederer, W.J. Mechanical Stretch-Induced Activation of ROS/RNS Signaling in Striated Muscle. Antioxid. Redox Signal. 2014, 20, 929–936. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH Oxidase-Dependent Redox Signalling in Cardiac Hypertrophy, Remodelling and Failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH Oxidases in Skeletal Muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- De Grey, A.D.N.J. A Hypothesis for the Minimal Overall Structure of the Mammalian Plasma Membrane Redox System. Protoplasma 2003, 221, 3–9. [Google Scholar] [CrossRef]
- Iverson, D.; DeChatelet, L.R.; Spitznagel, J.K.; Wang, P. Comparison of NADH and NADPH Oxidase Activities in Granules Isolated from Human Polymorphonuclear Leukocytes with a Fluorometric Assay. J. Clin. Investig. 1977, 59, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K. NADH in Human Skeletal Muscle during Short-Term Intense Exercise. Pflüg. Arch. Eur. J. Physiol. 1985, 403, 193–196. [Google Scholar] [CrossRef]
- Sahlin, K.; Katz, A.; Henriksson, J. Redox State and Lactate Accumulation in Human Skeletal Muscle during Dynamic Exercise. Biochem. J. 1987, 245, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Iatsenko, I.; Boquete, J.P.; Lemaitre, B. Microbiota-Derived Lactate Activates Production of Reactive Oxygen Species by the Intestinal NADPH Oxidase Nox and Shortens Drosophila Lifespan. Immunity 2018, 49, 929–942.e5. [Google Scholar] [CrossRef] [PubMed]
- Specht, K.S.; Kant, S.; Addington, A.K.; McMillan, R.; Hulver, M.W.; Learnard, H.; Campbell, M.; Donnelly, S.; Caliz, A.D.; Pei, Y.; et al. Nox4 Mediates Skeletal Muscle Metabolic Responses to Exercise. Mol. Metab. 2021, 45, 101160. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardo, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing Xanthine Oxidase-Mediated Oxidative Stress Prevents Useful Cellular Adaptations to Exercise in Rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef]
- Ryan, M.J.; Jackson, J.R.; Hao, Y.; Leonard, S.S.; Alway, S.E. Inhibition of Xanthine Oxidase Reduces Oxidative Stress and Improves Skeletal Muscle Function in Response to Electrically Stimulated Isometric Contractions in Aged Mice. Free Radic. Biol. Med. 2011, 51, 38–52. [Google Scholar] [CrossRef]
- Gómez-Cabrera, M.-C.; Pallardó, F.V.; Sastre, J.; Viña, J.; García-del-Moral, L. Allopurinol and Markers of Muscle Damage among Participants in the Tour de France. JAMA 2003, 289, 2503–2504. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.W.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-Based Regulation of Signal Transduction: Principles, Pitfalls, and Promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef]
- Wright, V.P.; Reiser, P.J.; Clanton, T.L. Redox Modulation of Global Phosphatase Activity and Protein Phosphorylation in Intact Skeletal Muscle. J. Physiol. 2009, 587, 5767–5781. [Google Scholar] [CrossRef]
- Barbieri, E.; Sestili, P. Reactive Oxygen Species in Skeletal Muscle Signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef]
- Shao, D.; Oka, S.I.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A Redox-Dependent Mechanism for Regulation of AMPK Activation by Thioredoxin1 during Energy Starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-Activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Doñate, F. Superoxide Dismutase 1 (SOD1) Is Essential for H2O 2-Mediated Oxidation and Inactivation of Phosphatases in Growth Factor Signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Gomez-Cabrera, M.-C.; Steinhafel, N.; Vina, J. Acute Exercise Activates Nuclear Factor (NF)-KappaB Signaling Pathway in Rat Skeletal Muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1499–1506. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Nuclear Factor Erythroid-Derived 2-like 2 (NFE2L2, Nrf2) Mediates Exercise-Induced Mitochondrial Biogenesis and the Anti-Oxidant Response in Mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Mansouri, A.; Van Remmen, H.; van der Meulen, J.H.; Larkin, L.; Richardson, A.G.; McArdle, A.; Faulkner, J.A.; Jackson, M.J. Free Radical Generation by Skeletal Muscle of Adult and Old Mice: Effect of Contractile Activity. Aging Cell 2006, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-Dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-Driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Lukosz, M.; Jakob, S.; Büchner, N.; Zschauer, T.C.; Altschmied, J.; Haendeler, J. Nuclear Redox Signaling. Antioxid. Redox Signal. 2010, 12, 713–742. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Wright, D.C.; Han, D.-H.; Garcia-Roves, P.M.; Geiger, P.C.; Jones, T.E.; Holloszy, J.O. Exercise-Induced Mitochondrial Biogenesis Begins before the Increase in Muscle PGC-1α Expression. J. Biol. Chem. 2007, 282, 194–199. [Google Scholar] [CrossRef]
- Katz, A. Modulation of Glucose Transport in Skeletal Muscle by Reactive Oxygen Species. J. Appl. Physiol. 2007, 102, 1671–1676. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.; Mei-ling, A.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Erickson, J.R.; Julie He, B.; Grumbach, I.M.; Anderson, M.E. CaMKII in the Cardiovascular System: Sensing Redox States. Physiol. Rev. 2011, 91, 889–915. [Google Scholar] [CrossRef] [PubMed]
- da Pinheiro, C.H.J.; Silveira, L.R.; Nachbar, R.T.; Vitzel, K.F.; Curi, R. Regulation of Glycolysis and Expression of Glucose Metabolism-Related Genes by Reactive Oxygen Species in Contracting Skeletal Muscle Cells. Free Radic. Biol. Med. 2010, 48, 953–960. [Google Scholar] [CrossRef]
- Silveira, L.R.; Pilegaard, H.; Kusuhara, K.; Curi, R.; Hellsten, Y. The Contraction Induced Increase in Gene Expression of Peroxisome Proliferator-Activated Receptor (PPAR)-γ Coactivator 1α (PGC-1α), Mitochondrial Uncoupling Protein 3 (UCP3) and Hexokinase II (HKII) in Primary Rat Skeletal Muscle Cells Is Dependent on Rea. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 969–976. [Google Scholar] [CrossRef]
- Higaki, Y.; Mikami, T.; Fujii, N.; Hirshman, M.F.; Koyama, K.; Seino, T.; Tanaka, K.; Goodyear, L.J. Oxidative Stress Stimulates Skeletal Muscle Glucose Uptake through a Phosphatidylinositol 3-Kinase-Dependent Pathway. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E889–E897. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.; Holman, G.D.; Schmitz, O.; Pedersen, O. Contraction Stimulates Translocation of Glucose Transporter GLUT4 in Skeletal Muscle through a Mechanism Distinct from That of Insulin. Proc. Natl. Acad. Sci. USA 1995, 92, 5817–5821. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.D.; Hansen, P.A.; Holloszy, J.O. Wortmannin Inhibits Insulin-Stimulated but Not Contraction-Stimulated Glucose Transport Activity in Skeletal Muscle. FEBS Lett. 1995, 361, 51–54. [Google Scholar] [CrossRef]
- Sakamoto, K.; Arnolds, D.E.; Fujii, N.; Kramer, H.F.; Hirshman, M.F.; Goodyear, L.J. Role of Akt2 in Contraction-Stimulated Cell Signaling and Glucose Uptake in Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1031–E1037. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Renga, M.; Hoppeler, H.; Baum, O. The Impact of Antioxidant Supplements and Endurance Exercise on Genes of the Carbohydrate and Lipid Metabolism in Skeletal Muscle of Mice. Cell Biochem. Funct. 2013, 31, 51–59. [Google Scholar] [CrossRef]
- Xie, Z.; Dong, Y.; Zhang, M.; Cui, M.Z.; Cohen, R.A.; Riek, U.; Neumann, D.; Schlattner, U.; Zou, M.H. Activation of Protein Kinase Cζ by Peroxynitrite Regulates LKB1-Dependent AMP-Activated Protein Kinase in Cultured Endothelial Cells. J. Biol. Chem. 2006, 281, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Kjøbsted, R.; Roll, J.L.W.; Jørgensen, N.O.; Birk, J.B.; Foretz, M.; Viollet, B.; Chadt, A.; Al-Hasani, H.; Wojtaszewski, J.F.P. AMPK and TBC1D1 Regulate Muscle Glucose Uptake After, but Not During, Exercise and Contraction. Diabetes 2019, 68, 1427–1440. [Google Scholar] [CrossRef]
- Hingst, J.R.; Kjøbsted, R.; Birk, J.B.; Jørgensen, N.O.; Larsen, M.R.; Kido, K.; Larsen, J.K.; Kjeldsen, S.A.S.; Fentz, J.; Frøsig, C.; et al. Inducible Deletion of Skeletal Muscle AMPKα Reveals That AMPK Is Required for Nucleotide Balance but Dispensable for Muscle Glucose Uptake and Fat Oxidation during Exercise. Mol. Metab. 2020, 40, 101028. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Atherton, P.J.; Babraj, J.; Smith, K.; Singh, J.; Rennie, M.J.; Wackerhage, H. Selective Activation of AMPK-PGC-1α or PKB-TSC2-MTOR Signaling Can Explain Specific Adaptive Responses to Endurance or Resistance Training-like Electrical Muscle Stimulation. FASEB J. 2005, 19, 786–788. [Google Scholar] [CrossRef]
- Olsen, R.H.; Krogh-Madsen, R.; Thomsen, C.; Booth, F.W.; Pedersen, B.K. Metabolic Responses to Reduced Daily Steps in Healthy Nonexercising Men. JAMA 2008, 299, 1261–1263. [Google Scholar] [CrossRef]
- Hawley, J.A. Adaptations of Skeletal Muscle to Prolonged, Intense Endurance Training. Clin. Exp. Pharmacol. Physiol. 2002, 29, 218–222. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B. The Role of Exercise and PGC1alpha in Inflammation and Chronic Disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. PGC-1 Coactivators and the Regulation of Skeletal Muscle Fiber-Type Determination. Cell Metab. 2011, 13, 351. [Google Scholar] [CrossRef]
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise Intensity-Dependent Regulation of Peroxisome Proliferator-Activated Receptor γ Coactivator-1α MRNA Abundance Is Associated with Differential Activation of Upstream Signalling Kinases in Human Skeletal Muscle. J. Physiol. 2010, 588, 1779–1790. [Google Scholar] [CrossRef]
- Little, J.P.; Safdar, A.; Cermak, N.; Tarnopolsky, M.A.; Gibala, M.J. Acute Endurance Exercise Increases the Nuclear Abundance of PGC-1α in Trained Human Skeletal Muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298. [Google Scholar] [CrossRef]
- Miura, S.; Kawanaka, K.; Kai, Y.; Tamura, M.; Goto, M.; Shiuchi, T.; Minokoshi, Y.; Ezaki, O. An Increase in Murine Skeletal Muscle Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α (PGC-1α) MRNA in Response to Exercise Is Mediated by β-Adrenergic Receptor Activation. Endocrinology 2007, 148, 3441–3448. [Google Scholar] [CrossRef]
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.P.; et al. Endurance Training in Humans Leads to Fiber Type-Specific Increases in Levels of Peroxisome Proliferator-Activated Receptor-γ Coactivator-1 and Peroxisome Proliferator-Activated Receptor-α in Skeletal Muscle. Diabetes 2003, 52, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARδ Agonists Are Exercise Mimetics. Cell 2008, 135, 189. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP Kinase Activity in the Regulation of PGC-1α Transcription in Skeletal Muscle Cells. Am. J. Physiol. Cell Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef]
- Jäer, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-Activated Protein Kinase (AMPK) Action in Skeletal Muscle via Direct Phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise Stimulates Pgc-1α Transcription in Skeletal Muscle through Activation of the P38 MAPK Pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of Mitochondrial Biogenesis in Skeletal Muscle by CaMK. Science 2002, 296, 349–352. [Google Scholar] [CrossRef]
- Lira, V.A.; Benton, C.R.; Yan, Z.; Bonen, A. PGC-1α Regulation by Exercise Training and Its Influences on Muscle Function and Insulin Sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E145–E161. [Google Scholar] [CrossRef]
- Yan, Z. Exercise, PGC-1α, and Metabolic Adaptation in Skeletal Muscle. Appl. Physiol. Nutr. Metab. 2009, 34, 424–427. [Google Scholar] [CrossRef]
- Bocco, B.M.L.C.; Louzada, R.A.N.; Silvestre, D.H.S.; Santos, M.C.S.; Anne-Palmer, E.; Rangel, I.F.; Abdalla, S.; Ferreira, A.C.; Ribeiro, M.O.; Gereben, B.; et al. Thyroid Hormone Activation by Type 2 Deiodinase Mediates Exercise-Induced Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Expression in Skeletal Muscle. J. Physiol. 2016, 594, 5255–5269. [Google Scholar] [CrossRef]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise Activation of Muscle Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Signaling Is Redox Sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef]
- Hashimoto, T.; Brooks, G.A. Mitochondrial Lactate Oxidation Complex and an Adaptive Role for Lactate Production. Med. Sci. Sports Exerc. 2008, 40, 486–494. [Google Scholar] [CrossRef]
- Nalbandian, M.; Radak, Z.; Takeda, M. N-Acetyl-L-Cysteine Prevents Lactate-Mediated PGC1-Alpha Expression in C2C12 Myotubes. Biology 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α Isoform Induced by Resistance Training Regulates Skeletal Muscle Hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Olguín, C.; Díaz-Vegas, A.; Utreras-Mendoza, Y.; Campos, C.; Arias-Calderón, M.; Llanos, P.; Contreras-Ferrat, A.; Espinosa, A.; Altamirano, F.; Jaimovich, E.; et al. NOX2 Inhibition Impairs Early Muscle Gene Expression Induced by a Single Exercise Bout. Front. Physiol. 2016, 7, 282. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.E.R.; Luecke, T.J.; Wendeln, H.K.; Toma, K.; Hagerman, F.C.; Murray, T.F.; Ragg, K.E.; Ratamess, N.A.; Kraemer, W.J.; Staron, R.S. Muscular Adaptations in Response to Three Different Resistance-Training Regimens: Specificity of Repetition Maximum Training Zones. Eur. J. Appl. Physiol. 2002, 88, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; De Rezende, F.F.; Rohrbach, S.; Zhang, M.; Schröder, K. Nox4 Is Dispensable for Exercise Induced Muscle Fibre Switch. PLoS ONE 2015, 10, e0130769. [Google Scholar] [CrossRef]
- Hancock, M.; Hafstad, A.D.; Nabeebaccus, A.A.; Catibog, N.; Logan, A.; Smyrnias, I.; Hansen, S.S.; Lanner, J.; Schröder, K.; Murphy, M.P.; et al. Myocardial NADPH Oxidase-4 Regulates the Physiological Response to Acute Exercise. eLife 2018, 7. [Google Scholar] [CrossRef]
- Hellsten-Westing, Y. Immunohistochemical Localization of Xanthine Oxidase in Human Cardiac and Skeletal Muscle. Histochemistry 1993, 100, 215–222. [Google Scholar] [CrossRef]
- Chen, H.-F.; Chuang, H.-C.; Tan, T.-H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.-I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote TNFalpha-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Wadley, G.D.; Nicolas, M.A.; Hiam, D.S.; McConell, G.K. Xanthine Oxidase Inhibition Attenuates Skeletal Muscle Signaling Following Acute Exercise but Does Not Impair Mitochondrial Adaptations to Endurance Training. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Musaró, A.; Giacinti, C.; Borsellino, G.; Dobrowolny, G.; Pelosi, L.; Cairns, L.; Ottolenghi, S.; Cossu, G.; Bernardi, G.; Battistini, L.; et al. Stem Cell-Mediated Muscle Regeneration Is Enhanced by Local Isoform of Insulin-like Growth Factor 1. Proc. Natl. Acad. Sci. USA 2004, 101, 1206–1210. [Google Scholar] [CrossRef]
- Handayaningsih, A.E.; Iguchi, G.; Fukuoka, H.; Nishizawa, H.; Takahashi, M.; Yamamoto, M.; Herningtyas, E.H.; Okimura, Y.; Kaji, H.; Chihara, K.; et al. Reactive Oxygen Species Play an Essential Role in IGF-I Signaling and IGF-I-Induced Myocyte Hypertrophy in C2C12 Myocytes. Endocrinology 2011, 152, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Spangenburg, E.E.; Le Roith, D.; Ward, C.W.; Bodine, S.C. A Functional Insulin-like Growth Factor Receptor Is Not Necessary for Load-Induced Skeletal Muscle Hypertrophy. J. Physiol. 2008, 586, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Arc-Chagnaud, C.; Salvador-Pascual, A.; Brioche, T.; Chopard, A.; Olaso-Gonzalez, G.; Viña, J. Redox Modulation of Muscle Mass and Function. Redox Biol. 2020, 35, 101531. [Google Scholar] [CrossRef]
- Parkington, J.D.; Siebert, A.P.; LeBrasseur, N.K.; Fielding, R.A. Differential Activation of MTOR Signaling by Contractile Activity in Skeletal Muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, 1086–1090. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/MTOR Pathway Is a Crucial Regulator of Skeletal Muscle Hypertrophy and Can Prevent Muscle Atrophy In Vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Crossland, H.; Kazi, A.A.; Lang, C.H.; Timmons, J.A.; Pierre, P.; Wilkinson, D.J.; Smith, K.; Szewczyk, N.J.; Atherton, P.J. Focal Adhesion Kinase Is Required for IGF-I-Mediated Growth of Skeletal Muscle Cells via a TSC2/MTOR/S6K1-Associated Pathway. Am. J. Physiol. Endocrinol. Metab. 2013, 305. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Abraham, A.G.; O’Neill, E. PI3K/Akt-Mediated Regulation of P53 in Cancer. Biochem. Soc. Trans. 2014, 42, 798–803. [Google Scholar] [CrossRef]
- Low, I.C.C.; Loh, T.; Huang, Y.; Virshup, D.M.; Pervaiz, S. Ser70 Phosphorylation of Bcl-2 by Selective Tyrosine Nitration of PP2A-B56δ Stabilizes Its Antiapoptotic Activity. Blood 2014, 124, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin Exerts an Antiapoptotic Effect by Regulating the Redox State of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef]
- Braakhuis, A.J.; Hopkins, W.G.; Lowe, T.E. Effects of Dietary Antioxidants on Training and Performance in Female Runners. Eur. J. Sport Sci. 2014, 14, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Viña, J. Oral Administration of Vitamin C Decreases Muscle Mitochondrial Biogenesis and Hampers Training-Induced Adaptations in Endurance Performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Bjørnsen, T.; Salvesen, S.; Berntsen, S.; Hetlelid, K.J.; Stea, T.H.; Lohne-Seiler, H.; Rohde, G.; Haraldstad, K.; Raastad, T.; Køpp, U.; et al. Vitamin C and E Supplementation Blunts Increases in Total Lean Body Mass in Elderly Men after Strength Training. Scand. J. Med. Sci. Sports 2016, 26, 755–763. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-Stimulated Glucose Uptake—Regulation and Implications for Glycaemic Control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef]
- Stephens, N.A.; Sparks, L.M. Resistance to the Beneficial Effects of Exercise in Type 2 Diabetes: Are Some Individuals Programmed to Fail? J. Clin. Endocrinol. Metab. 2015, 100, 43–52. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Selenoprotein P-Expression, Functions, and Roles in Mammals. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1441–1447. [Google Scholar] [CrossRef]
- Misu, H.; Takayama, H.; Saito, Y.; Mita, Y.; Kikuchi, A.; Ishii, K.A.; Chikamoto, K.; Kanamori, T.; Tajima, N.; Lan, F.; et al. Deficiency of the Hepatokine Selenoprotein P Increases Responsiveness to Exercise in Mice through Upregulation of Reactive Oxygen Species and AMP-Activated Protein Kinase in Muscle. Nat. Med. 2017, 23, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Takamura, T. Hepatokine Selenoprotein P-Mediated Reductive Stress Causes Resistance to Intracellular Signal Transduction. Antioxid. Redox Signal. 2020, 33, 517–524. [Google Scholar] [CrossRef]
- Morales-Alamo, D.; Ponce-González, J.G.; Guadalupe-Grau, A.; Rodríguez-García, L.; Santana, A.; Cusso, R.; Guerrero, M.; Dorado, C.; Guerra, B.; Calbet, J.A.L. Critical Role for Free Radicals on Sprint Exercise-Induced CaMKII and AMPKα Phosphorylation in Human Skeletal Muscle. J. Appl. Physiol. 2013, 114, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Barré, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-Activated Protein Kinase by Multisite Phosphorylation in Response to Agents That Elevate Cellular CAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef]
- Soltys, C.L.M.; Kovacic, S.; Dyck, J.R.B. Activation of Cardiac AMP-Activated Protein Kinase by LKB1 Expression or Chemical Hypoxia Is Blunted by Increased Akt Activity. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2472–H2479. [Google Scholar] [CrossRef]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute Exercise Stress Activates Nrf2/ARE Signaling and Promotes Antioxidant Mechanisms in the Myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Fathi, R.; Nasiri, K.; Akbari, A.; Ahmadi-KaniGolzar, F.; Farajtabar, Z. Exercise Protects against Ethanol-Induced Damage in Rat Heart and Liver through the Inhibition of Apoptosis and Activation of Nrf2/Keap-1/HO-1 Pathway. Life Sci. 2020, 256, 117958. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.-H.; Shih, C.-T.; Ching, C.-H.; Huang, J.-Y.; Jen, C.J.; Yu, L.; Kuo, Y.-M.; Wu, F.-S.; Chuang, J.-I. Treadmill Exercise Activates Nrf2 Antioxidant System to Protect the Nigrostriatal Dopaminergic Neurons from MPP+ Toxicity. Exp. Neurol. 2015, 263, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Matta, L.; Fonseca, T.S.; Faria, C.C.; Lima-Junior, N.C.; De Oliveira, D.F.; Maciel, L.; Boa, L.F.; Pierucci, A.P.T.R.; Ferreira, A.C.F.; Nascimento, J.H.M.; et al. The Effect of Acute Aerobic Exercise on Redox Homeostasis and Mitochondrial Function of Rat White Adipose Tissue. Available online: https://www.hindawi.com/journals/omcl/2021/4593496/ (accessed on 14 February 2021).
- Ji, L.L. Modulation of Skeletal Muscle Antioxidant Defense by Exercise: Role of Redox Signaling. Free Radic. Biol. Med. 2008, 44, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L. Redox Signaling in Skeletal Muscle: Role of Aging and Exercise. Adv. Physiol. Educ. 2015, 39, 352–359. [Google Scholar] [CrossRef]
- Hollander, J.; Fiebig, R.; Gore, M.; Bejma, J.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide Dismutase Gene Expression in Skeletal Muscle: Fiber-Specific Adaptation to Endurance Training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 277. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide Dismutase Derivative Reduces Oxidative Damage in Skeletal Muscle of Rats during Exhaustive Exercise. J. Appl. Physiol. 1995, 79, 129–135. [Google Scholar] [CrossRef]
- Kuwahara, H.; Horie, T.; Ishikawa, S.; Tsuda, C.; Kawakami, S.; Noda, Y.; Kaneko, T.; Tahara, S.; Tachibana, T.; Okabe, M.; et al. Oxidative Stress in Skeletal Muscle Causes Severe Disturbance of Exercise Activity without Muscle Atrophy. Free Radic. Biol. Med. 2010, 48, 1252–1262. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D.; Kang, C. Muscle Disuse Atrophy Caused by Discord of Intracellular Signaling. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Powers, S.K.; Ozdemir, M.; Hyatt, H. Redox Control of Proteolysis during Inactivity-Induced Skeletal Muscle Atrophy. Antioxid. Redox Signal. 2020, 33, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Ballarò, R.; Costelli, P. The Redox Balance: A Target for Interventions against Muscle Wasting in Cancer Cachexia? Antioxid. Redox Signal. 2020, 33, 542–558. [Google Scholar] [CrossRef]
- Viña, J.; Olaso-Gonzalez, G.; Arc-Chagnaud, C.; De la Rosa, A.; Gomez-Cabrera, M.C. Modulating Oxidant Levels to Promote Healthy Aging. Antioxid. Redox Signal. 2020, 33, 570–579. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-Induced Skeletal Muscle Atrophy Is Associated with Increased Mitochondrial ROS Production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293. [Google Scholar] [CrossRef]
- McArdle, A.; Pollock, N.; Staunton, C.A.; Jackson, M.J. Aberrant Redox Signalling and Stress Response in Age-Related Muscle Decline: Role in Inter- and Intra-Cellular Signalling. Free Radic. Biol. Med. 2019, 132, 50–57. [Google Scholar] [CrossRef]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-Induced Oxidative Stress: Friend or Foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Fortunato, R.S.; Louzada, R.A. Muscle Redox Signaling: Engaged in Sickness and in Health. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; García, A.; Härtel, S.; Hidalgo, C.; Jaimovich, E. NADPH Oxidase and Hydrogen Peroxide Mediate Insulin-Induced Calcium Increase in Skeletal Muscle Cells. J. Biol. Chem. 2009, 284, 2568–2575. [Google Scholar] [CrossRef]
- De Figueiredo, A.S.P.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 Mediates Skeletal Muscle Insulin Resistance Induced by a High Fat Diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef]
- Lambertucci, R.H.; Hirabara, S.M.; Silveira, L.D.R.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate Increases Superoxide Production through Mitochondrial Electron Transport Chain and NADPH Oxidase Activity in Skeletal Muscle Cells. J. Cell. Physiol. 2008, 216, 796–804. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-de-Castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants 2021, 10, 537. https://doi.org/10.3390/antiox10040537
Bouviere J, Fortunato RS, Dupuy C, Werneck-de-Castro JP, Carvalho DP, Louzada RA. Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants. 2021; 10(4):537. https://doi.org/10.3390/antiox10040537
Chicago/Turabian StyleBouviere, Jessica, Rodrigo S. Fortunato, Corinne Dupuy, Joao Pedro Werneck-de-Castro, Denise P. Carvalho, and Ruy A. Louzada. 2021. "Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle" Antioxidants 10, no. 4: 537. https://doi.org/10.3390/antiox10040537
APA StyleBouviere, J., Fortunato, R. S., Dupuy, C., Werneck-de-Castro, J. P., Carvalho, D. P., & Louzada, R. A. (2021). Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants, 10(4), 537. https://doi.org/10.3390/antiox10040537