
Metabolic Targets of Coenzyme Q10 in Mitochondria





Abstract
1. Introduction
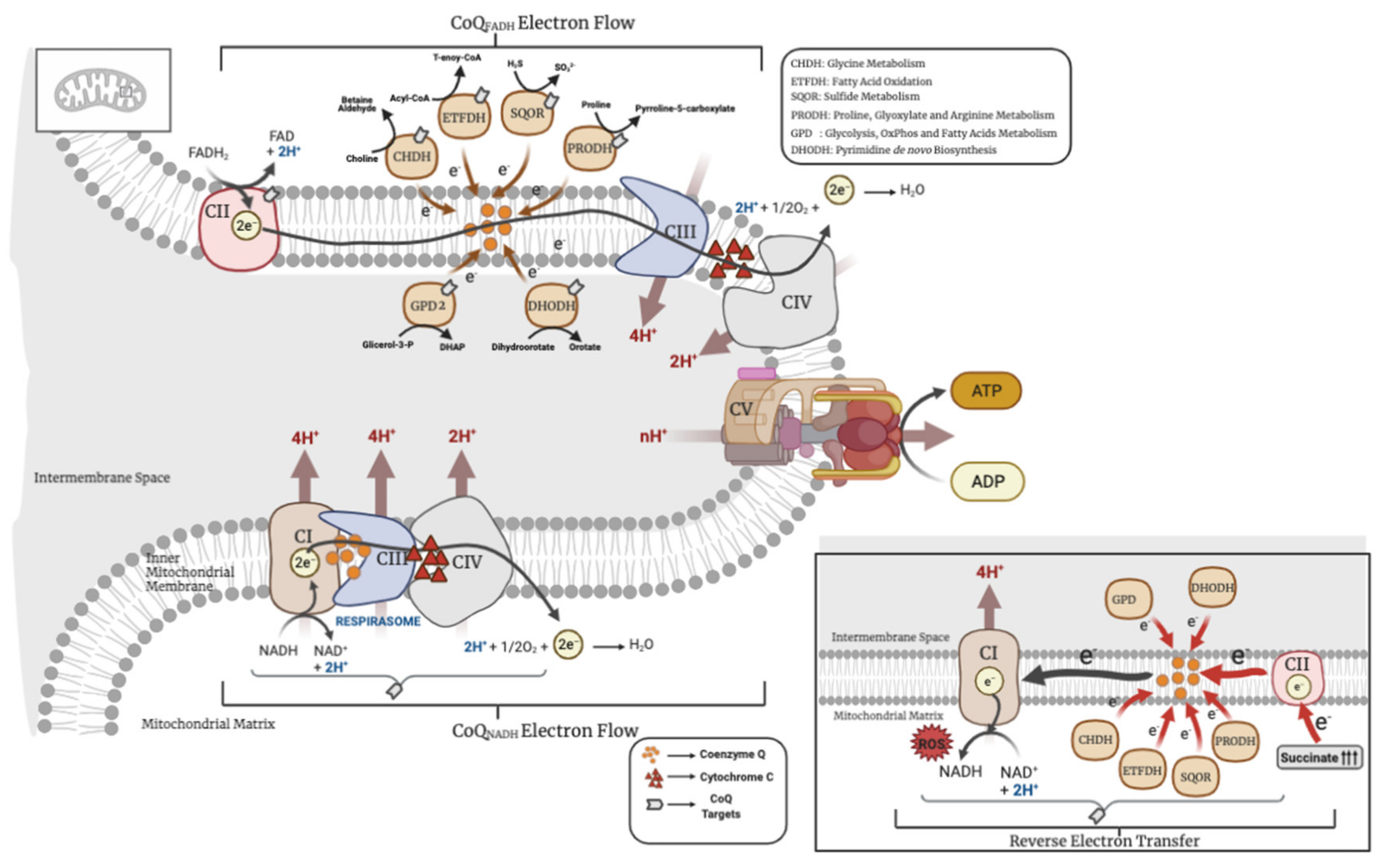
2. CoQ in the OxPhos System
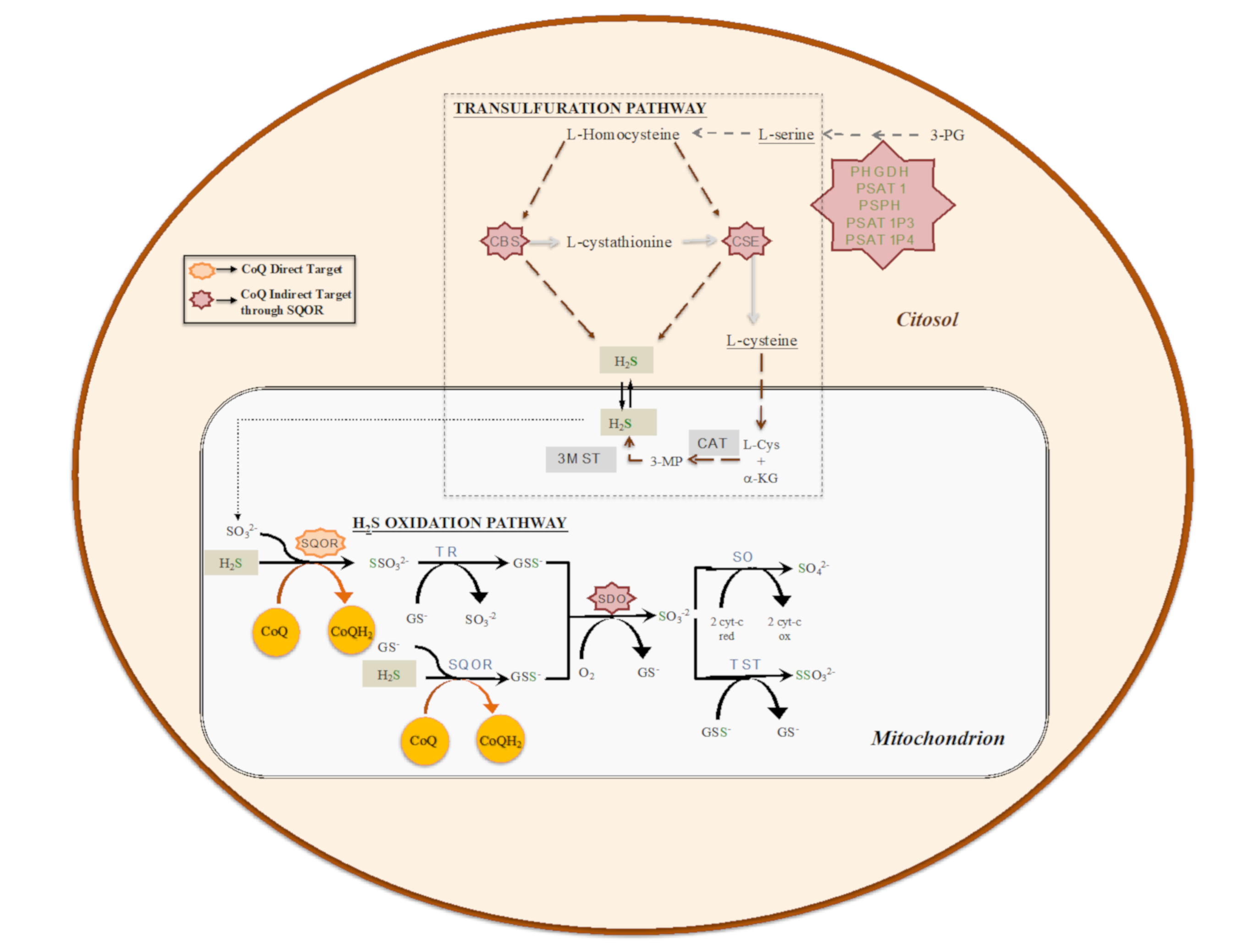
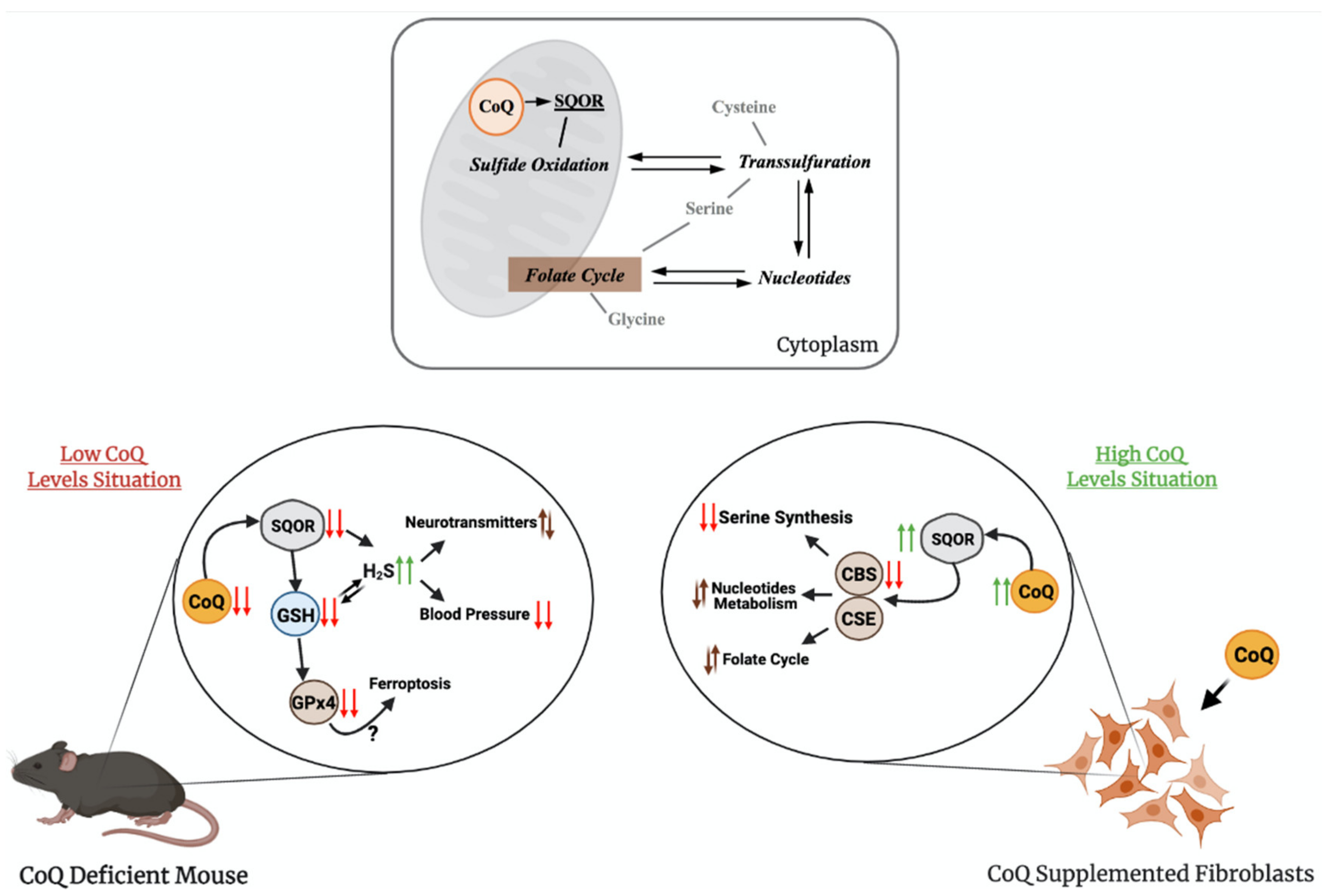
3. The Role of CoQ in the Regulation of Sulfide Metabolism and Others Linked Pathways
4. Other CoQ-Linked Reactions in Mitochondria
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of interest
References
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Garcia, J.A.; Guaras, A.; Acin-Perez, R.; Bullejos-Peregrin, J.; Lopez, A.; Escames, G.; Enriquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sanchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.P.; Medana, C.; Stainier, D.Y.; et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef]
- Kuhl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 2017, 6, e30952. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7. [Google Scholar] [CrossRef]
- Awad, A.M.; Bradley, M.C.; Fernandez-Del-Rio, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. The Complexity of Making Ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef]
- Diaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; Gonzalez-Garcia, P.; Battino, M.; Lopez, L.C.; Varela-Lopez, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Von-Moltke, J.; Naini, A.; Krishna, S.; Schuelke, M.; Salviati, L.; Navas, P.; DiMauro, S.; Hirano, M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J. 2008, 22, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Garone, C.; Emmanuele, V.; Tadesse, S.; Krishna, S.; Dorado, B.; Hirano, M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013, 27, 612–621. [Google Scholar] [CrossRef]
- Duberley, K.E.; Abramov, A.Y.; Chalasani, A.; Heales, S.J.; Rahman, S.; Hargreaves, I.P. Human neuronal coenzyme Q(10) deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2013, 36, 63–73. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef]
- Navas, P.; Villalba, J.M.; Lenaz, G. Coenzyme Q-dependent functions of plasma membrane in the aging process. Age 2005, 27, 139–146. [Google Scholar] [CrossRef]
- Barros, M.H.; Johnson, A.; Gin, P.; Marbois, B.N.; Clarke, C.F.; Tzagoloff, A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J. Biol. Chem. 2005, 280, 42627–42635. [Google Scholar] [CrossRef]
- Kemmerer, Z.A.; Robinson, K.P.; Schmitz, J.M.; Paulson, B.R.; Jochem, A.; Hutchins, P.D.; Coon, J.J.; Pagliarini, D.J. UbiB proteins regulate cellular CoQ distribution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Schagger, H. Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol. 2001, 65, 231–244. [Google Scholar]
- Sousa, P.M.; Silva, S.T.; Hood, B.L.; Charro, N.; Carita, J.N.; Vaz, F.; Penque, D.; Conrads, T.P.; Melo, A.M. Supramolecular organizations in the aerobic respiratory chain of Escherichia coli. Biochimie 2011, 93, 418–425. [Google Scholar] [CrossRef]
- Lenaz, G.; Genova, M.L. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: Random collisions vs. solid state electron channeling. Am. J. Physiol. Cell Physiol. 2007, 292, C1221–C1239. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Szibor, M.; Gainutdinov, T.; Fernandez-Vizarra, E.; Dufour, E.; Gizatullina, Z.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Viscomi, C.; Gellerich, F.; et al. Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148137. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef]
- Milliken, A.S.; Kulkarni, C.A.; Brookes, P.S. Acid enhancement of ROS generation by complex-I reverse electron transport is balanced by acid inhibition of complex-II: Relevance for tissue reperfusion injury. Redox Biol. 2020, 37, 101733. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Lopez-Martin, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sanchez-Alcazar, J.A.; et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef]
- Levavasseur, F.; Miyadera, H.; Sirois, J.; Tremblay, M.L.; Kita, K.; Shoubridge, E.; Hekimi, S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J. Biol. Chem. 2001, 276, 46160–46164. [Google Scholar] [CrossRef]
- Nakai, D.; Yuasa, S.; Takahashi, M.; Shimizu, T.; Asaumi, S.; Isono, K.; Takao, T.; Suzuki, Y.; Kuroyanagi, H.; Hirokawa, K.; et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem. Biophys. Res. Commun. 2001, 289, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, P.; Grunler, J.; Mattsson, J.; Sindelar, P.J.; Nordlund, P.; Berthold, D.A. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J. Biol. Chem. 2001, 276, 33297–33300. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Cueto-Urena, C.; Benabdellah, K.; Gutierrez-Guerrero, A.; Cobo, M.; Hidalgo-Gutierrez, A.; Rodriguez-Sevilla, J.J.; Martin, F.; Lopez, L.C. Gene Therapy Corrects Mitochondrial Dysfunction in Hematopoietic Progenitor Cells and Fibroblasts from Coq9R239X Mice. PLoS ONE 2016, 11, e0158344. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Vasta, V.; Hahn, S.; Gangoiti, J.A.; Opheim, E.; Sedensky, M.M.; Morgan, P.G. The role of DMQ(9) in the long-lived mutant clk-1. Mech. Ageing Dev. 2011, 132, 331–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Hidalgo-Gutierrez, A.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Lopez, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; DiMauro, S.; Hirano, M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: Time- and compound-dependent effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Renal Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuna-Castroviejo, D.; Lopez, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef]
- Hidalgo-Gutierrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Diaz-Casado, M.E.; Sanchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef]
- Di Meo, I.; Lamperti, C.; Tiranti, V. Mitochondrial diseases caused by toxic compound accumulation: From etiopathology to therapeutic approaches. EMBO Mol. Med. 2015, 7, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Banerjee, R. Sulfur as a signaling nutrient through hydrogen sulfide. Annu. Rev. Nutr. 2014, 34, 171–205. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Modis, K.; Coletta, C.; Erdelyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Kabil, H.; Kabil, O.; Banerjee, R.; Harshman, L.G.; Pletcher, S.D. Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. USA 2011, 108, 16831–16836. [Google Scholar] [CrossRef]
- Hine, C.; Mitchell, J.R. Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp. Gerontol. 2015, 68, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.J.; Duan, C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Trevino-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, P.; Hidalgo-Gutierrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sanchez-Hernandez, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.W.; Elias, A.F.; Kuster, A.; Laugwitz, L.; Larson, A.A.; Landry, A.P.; Ellwood-Digel, L.; Mirsky, D.M.; Dimmock, D.; Haven, J.; et al. Pathogenic variants in SQOR encoding sulfide:quinone oxidoreductase are a potentially treatable cause of Leigh disease. J. Inherit. Metab. Dis. 2020, 43, 1024–1036. [Google Scholar] [CrossRef]
- Mottawea, W.; Chiang, C.K.; Muhlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine-beta-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsstrom, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamaki, J.; Roivainen, A.; Marjamaki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef]
- Forsstrom, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054.e1047. [Google Scholar] [CrossRef]
- Krug, A.K.; Gutbier, S.; Zhao, L.; Poltl, D.; Kullmann, C.; Ivanova, V.; Forster, S.; Jagtap, S.; Meiser, J.; Leparc, G.; et al. Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP+. Cell Death Dis. 2014, 5, e1222. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkila, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjosund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef]
- Bao, X.R.; Ong, S.E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016, 5. [Google Scholar] [CrossRef]
- Ziosi, M.; Di Meo, I.; Kleiner, G.; Gao, X.H.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.J.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2017, 9, 96–111. [Google Scholar] [CrossRef]
- Luna-Sanchez, M.; Hidalgo-Gutierrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, A.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2017, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef]
- Rosenfeldt, F.L. Metabolic supplementation with orotic acid and magnesium orotate. Cardiovasc. Drugs Ther. 1998, 12 (Suppl. s2), 147–152. [Google Scholar] [CrossRef]
- Montero, R.; Sanchez-Alcazar, J.A.; Briones, P.; Navarro-Sastre, A.; Gallardo, E.; Bornstein, B.; Herrero-Martin, D.; Rivera, H.; Martin, M.A.; Marti, R.; et al. Coenzyme Q10 deficiency associated with a mitochondrial DNA depletion syndrome: A case report. Clin. Biochem. 2009, 42, 742–745. [Google Scholar] [CrossRef]
- Montero, R.; Grazina, M.; Lopez-Gallardo, E.; Montoya, J.; Briones, P.; Navarro-Sastre, A.; Land, J.M.; Hargreaves, I.P.; Artuch, R.; Coenzyme, Q.D.S.G. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 2013, 13, 337–341. [Google Scholar] [CrossRef]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416.e310. [Google Scholar] [CrossRef] [PubMed]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta 2010, 1797, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Ohkuma, A.; Hayashi, Y.K.; Lopez, L.C.; Hirano, M.; Nonaka, I.; Noguchi, S.; Chen, L.H.; Jong, Y.J.; Nishino, I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009, 19, 212–216. [Google Scholar] [CrossRef]
- Summitt, C.B.; Johnson, L.C.; Jonsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef]
- Moxley, M.A.; Tanner, J.J.; Becker, D.F. Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch. Biochem. Biophys. 2011, 516, 113–120. [Google Scholar] [CrossRef]
- Rauchova, H.; Battino, M.; Fato, R.; Lenaz, G.; Drahota, Z. Coenzyme Q-pool function in glycerol-3-phosphate oxidation in hamster brown adipose tissue mitochondria. J. Bioenerg. Biomembr. 1992, 24, 235–241. [Google Scholar] [CrossRef]
- Barrett, M.C.; Dawson, A.P. The reaction of choline dehydrogenase with some electron acceptors. Biochem. J. 1975, 151, 677–683. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Frischmuth, K.; Klingenberg, M. Uncoupling proteins 2 and 3 are highly active H+ transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. USA 2001, 98, 1416–1421. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef]
- Jaburek, M.; Garlid, K.D. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef]
- Esteves, T.C.; Echtay, K.S.; Jonassen, T.; Clarke, C.F.; Brand, M.D. Ubiquinone is not required for proton conductance by uncoupling protein 1 in yeast mitochondria. Biochem. J. 2004, 379, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Sluse, F.E.; Jarmuszkiewicz, W.; Navet, R.; Douette, P.; Mathy, G.; Sluse-Goffart, C.M. Mitochondrial UCPs: New insights into regulation and impact. Biochim. Biophys. Acta 2006, 1757, 480–485. [Google Scholar] [CrossRef]
- Walter, L.; Miyoshi, H.; Leverve, X.; Bernard, P.; Fontaine, E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic. Res. 2002, 36, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E.; Bernardi, P. Progress on the mitochondrial permeability transition pore: Regulation by complex I and ubiquinone analogs. J. Bioenerg. Biomembr. 1999, 31, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E.; Ichas, F.; Bernardi, P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef]
- Belliere, J.; Devun, F.; Cottet-Rousselle, C.; Batandier, C.; Leverve, X.; Fontaine, E. Prerequisites for ubiquinone analogs to prevent mitochondrial permeability transition-induced cell death. J. Bioenerg. Biomembr. 2012, 44, 207–212. [Google Scholar] [CrossRef]
- Devun, F.; Walter, L.; Belliere, J.; Cottet-Rousselle, C.; Leverve, X.; Fontaine, E. Ubiquinone analogs: A mitochondrial permeability transition pore-dependent pathway to selective cell death. PLoS ONE 2010, 5, e11792. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tissue | ||||
|---|---|---|---|---|
| Protein | Kidneys | Heart | Cerebellum | |
| SQOR | −1.2755 | −1.1292 | ||
| GPD2 | −0.0407 | 0.2659 | 0.0454 | |
| ETFDH | 0.1131 | −0.1072 | 0.0665 | |
| CHDH | 0.1572 | |||
| DHODH | 0.2237 | |||
| PRODH | 0.4076 | 0.1372 | 0.5226 | |
| PRODH2 | 0.6400 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hidalgo-Gutiérrez, A.; González-García, P.; Díaz-Casado, M.E.; Barriocanal-Casado, E.; López-Herrador, S.; Quinzii, C.M.; López, L.C. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants 2021, 10, 520. https://doi.org/10.3390/antiox10040520
Hidalgo-Gutiérrez A, González-García P, Díaz-Casado ME, Barriocanal-Casado E, López-Herrador S, Quinzii CM, López LC. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants. 2021; 10(4):520. https://doi.org/10.3390/antiox10040520
Chicago/Turabian StyleHidalgo-Gutiérrez, Agustín, Pilar González-García, María Elena Díaz-Casado, Eliana Barriocanal-Casado, Sergio López-Herrador, Catarina M. Quinzii, and Luis C. López. 2021. "Metabolic Targets of Coenzyme Q10 in Mitochondria" Antioxidants 10, no. 4: 520. https://doi.org/10.3390/antiox10040520
APA StyleHidalgo-Gutiérrez, A., González-García, P., Díaz-Casado, M. E., Barriocanal-Casado, E., López-Herrador, S., Quinzii, C. M., & López, L. C. (2021). Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants, 10(4), 520. https://doi.org/10.3390/antiox10040520