Abstract
Neurotransmitter depletion and mitochondrial dysfunction are among the multiple pathological events that lead to neurodegeneration. Following our previous studies related with the development of multitarget mitochondriotropic antioxidants, this study aims to evaluate whether the π-system extension on the chemical scaffolds of AntiOXCIN2 and AntiOXCIN3 affects their bioactivity and safety profiles. After the synthesis of four triphenylphosphonium (TPP+) conjugates (compounds 2–5), we evaluated their antioxidant properties and their effect on neurotransmitter-metabolizing enzymes. All compounds were potent equine butyrylcholinesterase (eqBChE) and moderate electric eel acetylcholinesterase (eeAChE) inhibitors, with catechols 4 and 5 presenting lower IC50 values than AntiOXCIN2 and AntiOXCIN3, respectively. However, differences in the inhibition potency and selectivity of compounds 2–5 towards non-human and human cholinesterases (ChEs) were observed. Co-crystallization studies with compounds 2–5 in complex with human ChEs (hChEs) showed that these compounds exhibit different binging modes to hAChE and hBChE. Unlike AntiOXCINs, compounds 2–5 displayed moderate human monoamine oxidase (hMAO) inhibitory activity. Moreover, compounds 4 and 5 presented higher ORAC-FL indexes and lower oxidation potential values than the corresponding AntiOXCINs. Catechols 4 and 5 exhibited broader safety windows in differentiated neuroblastoma cells than benzodioxole derivatives 2 and 3. Compound 4 is highlighted as a safe mitochondria-targeted antioxidant with dual ChE/MAO inhibitory activity. Overall, this work is a contribution for the development of dual therapeutic agents addressing both mitochondrial oxidative stress and neurotransmitter depletion.
1. Introduction
Neurodegeneration is a complex process that results from multiple mechanisms acting concurrently [1]. The main neuropathological hallmarks of Alzheimer’s disease (AD) and Parkinson’s Disease (PD) are the neuronal loss with consequent decrease of neurotransmitter levels, and the formation of protein aggregates [2,3]. Based on these observations, enzymes involved in neurotransmitters breakdown (e.g., cholinesterases (ChEs); monoamine oxidases (MAOs)) are among the main biological targets for the development of new therapeutics [1].
Mitochondria are also central players involved in the pathogenesis of AD and PD [4], since they are both one of the primary sources and one of the critical targets of reactive species (RS) [5]. Mitochondria play essential roles in the ATP synthesis, homeostasis of intracellular second messengers (calcium; RS), and apoptosis [6,7]. The high energy demand required for neuronal survival and excitability in the central nervous system (CNS) is mainly dependent on mitochondrial ATP generation [8]. Improper function of mitochondria may increase the neurons’ susceptibility to oxidative stress [9] and compromise neuronal survival [6]. Indeed, mitochondrial dysfunction is associated with increased RS production, intracellular calcium dyshomeostasis, and decreased ATP generation [4,10].
Considering the pivotal role of mitochondria in fundamental cellular processes [11], molecules that act on or accumulate in mitochondria may have great therapeutic potential [12]. To be able to act on mitochondrial targets, compounds usually need to be specifically directed towards these organelles [13]. One of the most commonly used strategies to deliver bioactive molecules to mitochondria is their conjugation with lipophilic cations such as triphenylphosphonium (TPP+) [13,14]. The selective accumulation of these lipophilic cations into the mitochondrial matrix occurs against the concentration gradient [15] and is driven by the plasma and mitochondrial membrane potentials [16].
Our research group recently developed mitochondria-targeted antioxidants, in which lipophilic TPP+ cations were conjugated with hydroxycinnamic (AntiOXCINs) and hydroxybenzoic (AntiOXBENs) acids [17,18,19,20,21,22]. We showed that these compounds accumulated within the mitochondrial matrix of rat liver mitochondria [18,19] and exhibited remarkable antioxidant properties [17,18,19,20,21]. Recently, we showed that they also displayed moderate to potent ChE inhibitory activities [17,22]. To understand the bioactivity and safety profiles of AntiOXCINs, we evaluated the effect of the modification of some of their substructures through structure-activity-toxicity studies. Accordingly, we evaluated the influence of the type of spacer between the phenolic ring and the carboxamide group (none, vinyl, methylene, ethylene), the length of the alkyl linker between the carboxamide group and the TPP+ moiety (six- or ten-carbon), and the substitution pattern of the phenolic ring (catechol or pyrogallol) [21,22].
Within this framework, we aim to evaluate whether the elongation of the α,β-unsaturated chain of AntiOXCINs while maintaining the compounds’ overall lipophilicity affects their bioactivity and safety profiles. Interestingly, this type of substructure (5-phenyl-2,4-pentadienyl moiety) can be found in the chemical structure of piperine (1-[5-(1,3-benzodioxol-5-yl)-1-oxo-2,4-pentadienyl]piperidine), compound 1, Figure 1), the main alkaloid found in numerous piper species [23]. Recent pharmacological studies in AD and PD animal models showed that piperine improves cognitive function [24,25], attenuates rotenone-induced motor function and mitigates neuronal loss in substantia nigra [26] and in the hippocampus [25]. Piperine also protected neurons against rotenone-induced mitochondrial damage [26] and oxidative stress [25,27], and presented MAO inhibition properties [28,29].
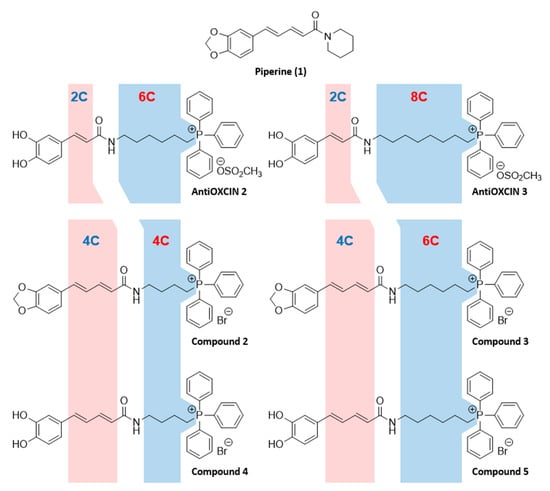
Figure 1.
Rational design of mitochondria-targeted compounds 2–5 based on AntiOXCIN2, AntiOXCIN3, and piperine (compound 1).
Taken together these plausible assumptions, herein we report the synthesis of four new TPP+ cations (compounds 2–5, Figure 1) inspired on the chemical structures of AntiOXCIN2, AntiOXCIN3 and piperine. In general, the rational design consisted of introducing an additional ethylenic group, with a concomitant shortening of the alkyl linker between the carboxamide group and the TPP+ moiety (Figure 1). This strategy was used to obtain new catechol antioxidants (compounds 4 and 5) with lipophilicity similar to AntiOXCIN2 and AntiOXCIN3. Due to the relevant biological activities of piperine, benzodioxole derivatives 2 and 3 were also included in this study. We then evaluated the compounds’ inhibition profile and binding mode towards cholinesterases (acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8)) and MAOs (MAO-A and MAO-B, EC 1.4.3.4), as well as their antioxidant and redox properties. Finally, we studied their cytotoxicity in differentiated neuroblastoma (SH-SY5Y) cells and predicted their ability to cross the blood-brain barrier.
2. Materials and Methods
2.1. Chemistry
The reagents, general methods and apparatus are described in supplementary information.
2.1.1. Synthesis of AntiOXCIN2 and AntiOXCIN3
Synthesis and structural analysis were previously reported by Teixeira et al. [19].
2.1.2. Synthesis of Mitochondria-Targeted Agents Inspired on Piperine
General Procedure for the Obtention of Phtalimidylalkylltriphenylphosphonium Salts
A mixture containing the appropriate N-(bromoalkyl)phthalimide (1 mmol) and triphenylphosphine (1.2 mmol) was mixed under argon atmosphere and heated at 130 °C, protected from the light. The solid obtained was recrystallized from dichloromethane/diethyl ether. The procedure was adapted from Cheng et al. 2016 [30] with some modifications.
(4-(1,3-Dioxoisoindolin-2-yl)butyl)triphenylphosphonium bromide (8). Compound 8 was obtained in the following conditions: compound 6 (1.00 g, 3.56 mmol), triphenylphosphine (1.06 g, 4.03 mmol). The mixture was heated at 130 °C for 10 h. η = 97 %. 1H NMR (CDCl3-d1): δ (ppm) = 1.62 (m, 2H, N(CH2)2CH2CH2), 2.15 (m, 2H, NCH2CH2(CH2)2), 3.77 (t, J = 6.3 Hz, 2H, N(CH2)3CH2), 4.01 (m, 2H, NCH2(CH2)3), 7.71 (m, 13 H, PPh3, H5, H6, H7, H8), 7.85 (m, 6H, PPh3). 13C NMR (CDCl3-d1): δ (ppm) = 19.5 (d, J = 3.9 Hz, NCH2CH2(CH2)2), 21.7 (d, J = 50.4 Hz, N(CH2)3CH2), 28.6 (d, J = 16.8 Hz, N(CH2)2CH2CH2), 36.4 (NCH2(CH2)3), 118.3 (d, J = 86.0 Hz, C1’), 123.2 (C5, C8), 130.4 (d, J = 12.5 Hz, C3’, C5’), 131.9 (C4, C9), 133.8 (d, J = 9.9 Hz, C2’, C6’), 134.1 (C6, C7), 135.0 (d, J = 2.9 Hz, C4’), 168.4 (C1, C3). ESI/MS m/z (%): 464 (M+-Br, 100).
(6-(1,3-Dioxoisoindolin-2-yl)hexyl)triphenylphosphonium bromide (9). Compound 9 was obtained in the following conditions: compound 7 (0.50 g, 1.61 mmol), triphenylphosphine (0.63 g, 2.42 mmol). The mixture was heated at 130 °C for 1.5 h. η = 87 %. 1H NMR (CDCl3-d1): δ (ppm) = 1.32 (m, 2H, NCH2CH2(CH2)4), 1.61 (m, 4H, N(CH2)2CH2(CH2)3, N(CH2)3CH2(CH2)2), 1.72 (m, 2H, N(CH2)4CH2CH2)), 3.62 (t, J = 7.0 Hz, 2H, NCH2(CH2)5), 3.87 (m, 2H, N(CH2)5CH2), 7.77 (m, 19H, PPh3, H5, H6, H7, H8). 13C NMR (CDCl3-d1): δ (ppm) = 22.1 (d, J = 4.9 Hz, N(CH2)3CH2(CH2)2), 22.8 (d, J = 49.1 Hz, N(CH2)5CH2), 26.5 (N(CH2)2CH2(CH2)3), 28.2 (NCH2CH2(CH2)4), 29.8 (d, J = 15.7 Hz, N(CH2)4CH2CH2), 37.8 (NHCH2(CH2)5), 118.5 (d, J = 85.8 Hz, C1’), 123.2 (C5, C8), 130.5 (d, J = 12.5 Hz, C3’, C5’), 132.2 (C4, C9), 133.8 (d, J = 10.1 Hz, C2’, C6’), 133.9 (C6, C7), 134.9 (d, J = 2.9 Hz, C4’), 168.4 (C1, C3). ESI/MS m/z (%): 492 (M+-Br, 100).
General Procedure for the Obtention of Aminoalkylltriphenylphosphonium Salts
In a round bottom flask with compounds 8 or 9 (1 mmol) dissolved in ethanol (5 mL), n-butylamine (10–24 mmol) was added. The mixture was refluxed until the reaction was complete. Then, the solvent was partially concentrated, and water was added. The solid obtained was filtered off and the filtrate was extracted with dichloromethane. Then, the aqueous phase was concentrated. The material was used without further purification in the next step. The procedure was adapted from Cheng et al. [30] 2016 with some modifications.
(4-Aminobutyl)triphenylphosphonium bromide (10). Compound 10 was obtained in the following conditions: compound 8 (0.93 g, 1.71 mmol), ethanol (10 mL), n-butylamine (2.2 mL, 40.6 mmol). The mixture was refluxed for 10 h. η = 86 %. 1H NMR (MeOD-d4): δ (ppm) = 1.74 (m, 4H, NH2CH2CH2(CH2)2, NH2(CH2)2CH2CH2), 2.73 (t, J = 6.8 Hz, 2H, NH2CH2(CH2)3), 3.44 (m, 2H, NH2(CH2)3CH2), 7.77 (m, 15H, PPh3). 13C NMR (MeOD-d4): δ (ppm) = 19.7 (d, J = 4.2 Hz, NH2CH2CH2(CH2)2), 21.3 (d, J = 51.5 Hz, NH2(CH2)3CH2), 31.8 (d, J = 16.8 Hz, NH2(CH2)2CH2CH2), 39.8 (NH2CH2(CH2)3), 118.5 (d, J = 86.6 Hz, C1’), 130.2 (d, J = 12.6 Hz, C3’, C5’), 133.5 (d, J = 10.1 Hz, C2’, C6’), 134.9 (d, J = 3.1 Hz, C4’). ESI/MS m/z (%): 334 (M+-Br, 100).
(6-Aminohexyl)triphenylphosphonium bromide (11). Compound 11 was obtained in the following conditions: compound 9 (2.00 g, 3.49 mmol), ethanol (24 mL), butylamine (3.5 mL, 34.9 mmol). The mixture was refluxed for 1.5 h. η = 80 %. 1H NMR (MeOD-d4): δ (ppm) = 1.44 (m, 2H, NHCH2CH2(CH2)4), 1.68 (m, 6H, NH2(CH2)2CH2(CH2)3, NH2(CH2)3CH2(CH2)2, NH2(CH2)4CH2CH2)), 2.92 (t, J = 6.8 Hz, 2H, NH2CH2(CH2)5), 3.42 (m, 2H, NH2(CH2)5CH2), 7.84 (m, 15H, PPh3). 13C NMR (MeOD-d4): δ (ppm) = 21.3 (d, J = 51.4 Hz, NH2(CH2)5CH2), 22.0 (d, J = 4.3 Hz, NH2(CH2)3CH2(CH2)2), 25.3 (NH2(CH2)2CH2(CH2)3), 26.9 (NH2CH2CH2(CH2)4), 29.6 (d, J = 16.6 Hz, NH(CH2)4CH2CH2), 39.1 (NHCH2(CH2)5), 118.5 (d, J = 86.4 Hz, C1’), 130.2 (d, J = 12.6 Hz, C3’, C5’), 133.4 (d, J = 9.9 Hz, C2’, C6’), 134.9 (d, J = 3.0 Hz, C4’). ESI/MS m/z (%): 362 (M+-Br, 100).
Synthesis of (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)Penta-2,4-Dienoic Acid (12)
Synthesis and structural analysis described in literature [29].
Piperic Acid Amidation
Piperic acid (compound 2, 1 mmol) was dissolved in dichloromethane (2.5 mL) and Et3N (2 mmol). To the stirred solution kept in an ice bath, ethyl chloroformate (2 mmol) was added dropwise. After stirring for 2 h at room temperature, the mixture was cooled again in an ice bath and the appropriate amine (compounds 10 or 11, 2 mmol) was slowly added. The reaction was stirred overnight at room temperature. Upon completion, dichloromethane (30 mL) was added. The mixture was extracted with HBr 1 M (3 × 10 mL) and washed with water. The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated. The crude product was purified by flash column chromatography (silica, dichloromethane/methanol (9:1)) and recrystallized from methanol/water. The procedure was adapted from Teixeira et al. [20] with some modifications.
(4-((2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)penta-2,4-dienamido)butyl)triphenylphosphonium bromide (2). Compound 2 was obtained in the following conditions: compound 12 (0.42 g, 1.93 mmol), dichloromethane (8 mL), Et3N (700 µL, 3.65 mmol), ethyl chloroformate (362 µL, 4.18 mmol), amine 10 (1.09 g, 2.46 mmol). η = 44 %. 1H NMR (CDCl3-d1): δ (ppm) = 1.76 (m, 2H, NHCH2CH2(CH2)2), 1.98 (m, 2H, NH(CH2)2CH2CH2), 3.47 (dd, J = 11.7 Hz, 5.8 Hz, 2H, NHCH2(CH2)3), 3.65 (m, 2H, NH(CH2)3CH2), 5.98 (s, 2H, OCH2O), 6.43 (d, J = 15.0 Hz, 1H, Hα), 6.77 (m, 3H, H5, Hγ, Hδ), 6.90 (dd, J = 1.6 Hz, 8.2 Hz, 1H, H6), 6.99 (d, J = 1.6 Hz, 1H, H2), 7.31 (dd, J = 10.4 Hz, 15.0 Hz, 1H, Hβ), 7.74 (m, 15 H, PPh3), 8.55 (t, J = 5.6 Hz, 1H, CONH). 13C NMR (CDCl3-d1): δ (ppm) = 19.5 (d, J = 4.0 Hz, NHCH2CH2(CH2)2), 22.4 (d, J = 50.4 Hz, NH(CH2)3CH2), 28.7 (d, J = 16.6 Hz, NH(CH2)2CH2CH2), 37.4 (NHCH2(CH2)3), 101.2 (OCH2O), 105.9 (C2), 108.4 (C5), 118.2 (d, J = 86.0 Hz, C1’), 122.2 (Cα), 125.0 (C6), 125.7 (Cγ), 130.5 (d, J = 12.5 Hz, C3’, C5’), 131.3 (C1), 133.7 (d, J = 10.0 Hz, C2’, C6’), 135.1 (d, J = 2.9 Hz, C4’), 137.5 (Cδ), 139.5 (Cβ), 147.9 (C3), 148.2 (C4), 167.3 (CONH). ESI/MS m/z (%): 534 (M+-Br, 100).
(6-((2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)penta-2,4-dienamido)hexyl)triphenylphosphonium bromide (3). Compound 3 was obtained in the following conditions: compound 12 (0.76 g, 3.48 mmol), dichloromethane (20 mL), Et3N (1 mL, 7.12 mmol), ethyl chloroformate (665 µL, 6.96 mmol), amine 11 (2.00 g, 4.52 mmol). η = 46 %. 1H NMR (CDCl3-d1): δ (ppm) = 1.60 (m, 8H, NHCH2CH2(CH2)4, NH(CH2)2CH2(CH2)3, NH(CH2)3CH2(CH2)2, NH(CH2)4CH2CH2), 3.37 (m, 2H, NHCH2(CH2)5), 3.68 (m, 2H, NH(CH2)5CH2), 5.97 (s, 2H, OCH2O), 6.61 (d, J = 15.0 Hz, 1H, Hα), 6.67 (d, J = 15.6 Hz, 1H, Hδ), 6.76 (m, 2H, H5, Hγ), 6.86 (dd, J = 1.6 Hz, 8.2 Hz, 1H, H6), 6.95 (d, J = 1.6 Hz, 1H, H2), 7.34 (dd, J = 11.6 Hz, 15.0 Hz, 1H, Hβ), 7.71 (m, 6H, PPh3), 7.82 (m, 9H, PPh3), 8.24 (t, J = 4.9 Hz, 1H, CONH). 13C NMR (CDCl3-d1): δ (ppm) = 22.1 (d, J = 4.4 Hz, NH(CH2)3CH2(CH2)2), 22.3 (d, J = 50.2 Hz, NH(CH2)5CH2), 25.5 (NH(CH2)2CH2(CH2)3), 28.4 (NHCH2CH2(CH2)4), 29.2 (d, J = 16.2 Hz, NH(CH2)4CH2CH2), 38.7 (NHCH2(CH2)5), 101.2 (OCH2O), 105.8 (C2), 108.4 (C5), 118.3 (d, J = 85.8 Hz, C1’), 122.2 (Cα), 125.2 (C6), 126.0 (Cγ), 130.5 (d, J = 12.5 Hz, C3’, C5’), 131.4 (C1), 133.6 (d, J = 10.0 Hz, C2’, C6’), 135.1 (d, J = 2.9 Hz, C4’), 137.3 (Cδ), 139.6 (Cβ), 147.8 (C3), 148.1 (C4), 167.1 (CONH). ESI/MS m/z (%): 562 (M+-Br, 100).
Demethylenation Reaction
In a round bottom flask, compounds 2 or 3 (1 mmol) were maintained under argon atmosphere. Then, the compounds were dissolved in anhydrous dichloromethane and BBr3.S(CH3)2 (4 mmol) was added. The mixture was protected from the light and refluxed for 6 h. Upon completion, water was added to destroy BBr3.S(CH3)2. The solid formed was isolated by filtration and purified by flash column chromatography (silica, dichloromethane/methanol (95:5)). The procedure was adapted from Williard et al. [31] with some modifications.
(4-((2E,4E)-5-(3,4-Dihydroxyphenyl)penta-2,4-dienamido)butyl)triphenylphosphonium bromide (4). Compound 4 was obtained in the following conditions: compound 2 (1.28 g, 2.10 mmol), dichloromethane (35 mL), BBr3.S(CH3)2 (2.85 g, 9.1 mmol). η = 29 %. 1H NMR (DMSO-d6): 1.55 (m, 2H, NH(CH2)2CH2CH2), 1.64 (m, 2H, NHCH2CH2(CH2)2), 3.77 (dd, J = 3.7 Hz, 7.8 Hz, 2H, NH(CH2)3CH2), 4.01 (m, 2H, NHCH2(CH2)3), 5.98 (d, J = 15.0 Hz, 1H, Hα), 6.74 (m, 3H, Hδ, H5, Hγ), 6.85 (dd, J = 2.0 Hz, 8.2 Hz, 1H, H6), 6.95 (d, J = 1.9 Hz, 1H, H2), 7.12 (dd, J = 10.2 Hz, 15.0 Hz, 1H, Hβ), 7.78 (m, 12H, PPh3), 7.89 (m, 3H, PPh3), 8.00 (t, J = 5.4 Hz, 1H, CONH), 9.11 (s, 2H, 2×OH). 13C NMR (DMSO-d6): δ (ppm) = 19.1 (d, J = 3.9 Hz, NHCH2CH2(CH2)2), 19.8 (d, J = 50.1 Hz, NH(CH2)3CH2), 29.8 (d, J = 16.9 Hz, NH(CH2)2CH2CH2), 37.1 (NCH2(CH2)3), 113.7 (C2), 115.6 (C5), 118.4 (d, J = 85.7 Hz, C1’), 119.3 (Cα), 123.2 (C6), 123.4 (Cγ), 127.8 (C1), 130.2 (d, J = 12.4 Hz, C3’, C5’), 133.5 (d, J = 10.1 Hz, C2’, C6’), 134.8 (d, J = 2.5 Hz, C4’), 138.7 (Cδ), 139.6 (Cβ), 145.4 (C3), 146.5 (C4), 165.4 (CONH). ESI/MS m/z (%): 522 (M+-Br, 100).
(6-((2E,4E)-5-(3,4-Dihydroxyphenyl)penta-2,4-dienamido)hexyl)triphenylphosphonium bromide (5). Compound 5 was obtained in the following conditions: compound 3 (1.11 g, 1.72 mmol), dichloromethane (30 mL), BBr3.S(CH3)2 (2.35 g, 7.52 mmol). η = 44 %. 1H NMR (DMSO-d6): δ (ppm) = 1.28 (m, 2H, NH(CH2)2CH2(CH2)3), 1.38 (m, 2H, NH(CH2)3CH2(CH2)2), 1.49 (m, 4H, NHCH2CH2(CH2)4, NH(CH2)4CH2CH2), 3.11 (m, 2H, NHCH2(CH2)5), 3.58 (m, 2H, NH(CH2)5CH2), 6.03 (d, J = 15.0 Hz, 1H, Hα), 6.71 (m, 3H, Hδ, H5, Hγ), 6.83 (dd, J = 2.0 Hz, 8.3 Hz, 1H, H6), 6.94 (d, J = 2.0 Hz, 1H, H2), 7.11 (dd, J = 9.8 Hz, 15.1 Hz, 1H, Hβ), 7.79 (m, 12H, PPh3), 7.90 (m, 3H, PPh3), 7.94 (t, J = 5.6 Hz, 1H, CONH), 8.98 (s, 1H, OH), 9.27 (s, 1H, OH). 13C NMR (DMSO-d6): δ (ppm) = 20.1 (d, J = 49.9 Hz, NH(CH2)5CH2), 21.6 (d, J = 4.3 Hz, NH(CH2)3CH2(CH2)2), 25.5 (NH(CH2)2CH2(CH2)3), 28.8 (NHCH2CH2(CH2)4), 29.4 (d, J = 16.7 Hz, NH(CH2)4CH2CH2), 38.3 (NHCH2(CH2)5), 113.6 (C2), 115.6 (C5), 118.5 (d, J = 85.6 Hz, C1’), 119.2 (Cα), 123.5 (C6), 123.5 (Cγ), 127.8 (C1), 130.1 (d, J = 12.4 Hz, C3’, C5’), 133.5 (d, J = 10.1 Hz, C2’, C6’), 134.8 (d, J = 2.8 Hz, C4’), 138.4 (Cδ), 139.3 (Cβ), 145.4 (C3), 146.4 (C4), 165.1 (CONH). ESI/MS m/z (%): 550 (M+-Br, 100).
2.2. Enzymatic Assays
2.2.1. Acetylcholinesterase and Butyrylcholinesterase
Evaluation of Eel Acetylcholinesterase and Equine Butyrylcholinesterase Inhibitory Activity
The eeAChE and eqBChE inhibitory activities of the compounds under study were determined following the Ellman’s method [32,33] (see supplementary information).
Evaluation of Human Acetylcholinesterase and Human Butyrylcholinesterase Inhibitory Activities
Recombinant hAChE and hBChE were produced and purified as previously described [34,35]. Test compounds were dissolved in methanol 100 % to prepare stock solutions at 40 mM and subsequently diluted in water to reach the desired concentrations. Recombinant hAChE and hBChE activities were measured in presence of appropriate compound concentrations by spectrophotometry at 412 nm and 25 °C in a 1 mL cuvette containing Ellman’s buffer (0.5 mM DTNB, 0.1% BSA, 0.1 M phosphate, pH 7.4). Measurements were performed at least in duplicate for each tested concentration and final methanol concentrations were kept below 5%. The compound concentration producing 50 % inhibition was determined by nonlinear fitting with ProFit (Quantumsoft) using Equation (1).
Crystallization, X-ray Data Collection and Processing
Recombinant hAChE and hBChE crystals were grown by hanging drop vapor diffusion at 20 °C. For hAChE, a crystallization buffer containing lithium sulfate 1.6 M, HEPES 100 mM pH 7 and magnesium sulfate 60 mM was used. For hBChE, the crystallization buffer was MES 0.1 M pH 6.5 and ammonium sulfate 2.15 M. Crystals of hAChE and hBChE were soaked overnight in presence of compounds 2–5 at 1 mM. Crystals were then washed with a cryoprotectant solution composed of the crystallization buffer with glycerol 20% and flash-cooled in liquid nitrogen.
Diffraction data were collected at the Proxima 2 beam line, synchrotron SOLEIL (Gif-sur-Yvette), at 100 K on an EIGER X 9M area detector (λ = 0.9801 Å). Data were processed with XDS [36] and scaled with XSCALE. The structures were solved by molecular replacement with PHASER [37] using PDB 4EY4 as a starting model for hAChE and PDB 1P0I for hBChE. The models were built by iterative cycles of model building using Coot [38] and refinement using Phenix [39]. Geometry restrains of compound 3 were generated using Phenix.eLBOW and the AM1 semi-empirical quantum mechanical method. Table 1 shows the crystallographic data collection and refinement statistics of the hAChE (PDB: 6ZWE) and the hBChE (PDB: 6ZWI) in complex with compound 3.

Table 1.
Data collection and refinement statistics. Calculated using Phenix [39]. R-work=Σ |fo − |fc||/Σ |fo|, fo and fc are observed and calculated structure factors, R-free set uses about thousands randomly chosen reflections. Statistics for the highest-resolution shell are shown in parentheses.
2.2.2. Evaluation of Human Monoamine Oxidase Inhibitory Activity
The inhibitory activity of lipophilic TPP+ cations on hMAO-A and hMAO-B was studied using an experimental protocol described elsewhere [29,40] (see supplementary information).
2.3. Oxygen Radical Absorbance Capacity (ORAC-FL) Assay
The ORAC-FL assay was performed using an experimental protocol adapted from the literature [41,42] and described in Supplementary Information.
2.4. Electrochemical Measurements
DPV experiments were performed as described in literature [43] (see supplementary information).
2.5. In Vitro Toxicology
2.5.1. Materials
All reagents used were of analytical grade or of the highest grade available. Neutral red (NR) solution, resazurin, Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L glucose, retinoic acid and 12-O-tetradecanoylphorbol-13-acetate (TPA) were obtained from Sigma Aldrich. Reagents used in cell culture such as heat-inactivated fetal bovine serum (FBS), antibiotic (10,000 U/mL penicillin, 10,000 μg/mL streptomycin), MEM Non-Essential Amino Acids solution (100×) (MEM NEAA), 0.25% trypsin/1 mM ethylenediamine tetraacetic acid (EDTA) and Hanks’ balanced salt solution (HBSS) were purchased from Gibco Laboratories (Lenexa, KS, USA). Dimethylsulfoxide (DMSO), absolute ethanol, and acetic acid were obtained from Merck (Darmstadt, Germany).
2.5.2. Cell Lines and Culture Conditions
Human SH-SY5Y neuroblastoma cells and human epithelial colorectal adenocarcinoma (Caco-2) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cell culture and cell differentiation were performed as previously described by Fernandes et al. [44] and in supplementary information.
2.5.3. Cytotoxicity
Stock solutions of the test compounds (100 mM) were freshly prepared in DMSO. Final concentrations of the test compounds were obtained by diluting them into cell culture medium immediately before use, giving a final maximum concentration of 0.1% DMSO.
For cytotoxicity studies, differentiated SH-SY5Y cells were incubated with increasing concentrations of the test compounds (0.20–100 µM) for 24 h. Controls were treated with culture media containing 0.1% DMSO. Cell viability was estimated using two different methods: resazurin reduction assay and NR uptake assay.
2.5.4. Statistical Analysis
The data obtained are expressed as mean ± standard error mean (SEM) of at least three independent experiments (n = 3). All statistical analyses were performed using GraphPad PRISM version 6 for Windows. The normality of the data distribution was evaluated using three normality tests: KS normality test, D’Agostino and Pearson omnibus normality test, and Shapiro−Wilk normality test. Statistical comparisons between groups were estimated using the parametric method two-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test. In all cases, p values lower than 0.05 were considered significant.
2.6. Evaluation of the Chromatographic Hydrophobicity Index
Chromatographic hydrophobicity indexes (CHIs) at pH 2.3 were determined using an experimental protocol described elsewhere [45,46] and included in the supplementary information.
2.7. Estimation of Drug-Like Properties
The calculation of molecular weight (MW), topological polar surface area (TPSA), number of hydrogen bond donors (HBD) and acceptors (HBA), and number of rotatable bonds (RB) was performed using SwissADME (http://swissadme.ch/index.php (accessed 16 February 2021)).
3. Results and Discussion
3.1. Chemistry
The synthetic route used to obtain TPP+ conjugates 2–5 is depicted in Scheme 1. Aminoalkyltriphenylphosphonium salts 10 and 11 were prepared in 2 steps. The appropriate bromoalkylphthalymides (compounds 6 and 7) were heated with triphenylphosphine (TPP) to obtain compounds 8 and 9 (Scheme 1, step a), which in turn were refluxed with n-butylamine in ethanol to induce the phthalymidyl ring cleavage, yielding compounds 10 and 11 (Scheme 1, step b). Piperic acid (compound 12) was obtained by alkaline hydrolysis of piperine (compound 1) (Scheme 1, step c). Piperic acid was then acylated with ethyl chloroformate in alkaline media and reacted with amines 10 and 11 to afford compounds 2 and 3, respectively (Scheme 1, step d). Catechol derivatives 4 and 5 were prepared by the demethylenation of compounds 2 and 3, respectively, using boron tribromide dimethyl sulfide complex (BBr3.S(CH3)2). (Scheme 1, step e). The synthesis of AntiOXCIN2 and AntiOXCIN3 was performed as reported by Teixeira et al. [19].
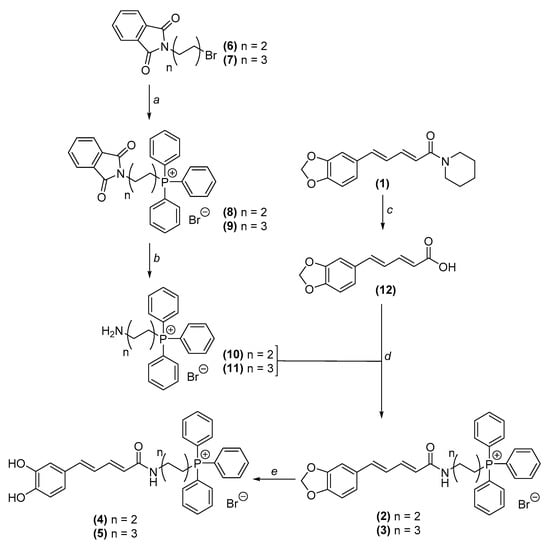
Scheme 1.
General procedure pursued to obtain the mitochondria-targeted compounds 2–5. (a) TPP, argon atmosphere, 130 °C, 1.5–4 h; (b) BuNH2, ethanol, reflux, 1.5–10 h; (c) Methanolic solution of NaOH 2 M, reflux, 24 h; (d) 1. dichloromethane, Et3N, ethyl chloroformate, rt, 2 h; 2. Compounds 10 and 11, rt, overnight; (e) BBr3.S(CH3)2, dichloromethane, argon atmosphere, reflux, 6 h.
3.2. Cholinesterase Inhibition Studies
3.2.1. Evaluation of Electric Eel Acetylcholinesterase and Equine Butyrylcholinesterase Inhibitory Activities
The eeAChE and eqBChE inhibitory activities of piperine and derivatives thereof were evaluated using the Ellman’s method [32,33]. Donepezil, a reversible and selective AChE inhibitor commonly prescribed for AD [47,48], was used as a reference. The results are expressed as IC50 values and are displayed in Table 2.
Table 2.
Evaluation of ChE (eeAChE, eqBChE, hAChE, hBChE) inhibitory activity of piperine (compound 1), compounds 2–5, AntiOXCIN2 and AntiOXCIN3 and reference inhibitor.
While piperine (1) did not display significant eeAChE and eqBChE inhibitory activities at 10 µM, compounds 2–5, AntiOXCIN2 and AntiOXCIN3 inhibited both eeAChE and eqBChE in our experimental conditions. Concerning eeAChE inhibition, the IC50 values were within the micromolar range. Compounds 4 and 5 displayed lower eeAChE IC50 values than AntiOXCIN2 and AntiOXCIN3, respectively, suggesting that the increased rigidification of the aliphatic chain improves eeAChE inhibition. Catechol derivatives 4 and 5 showed lower IC50 values than the counterparts containing the benzodioxole ring (compounds 2 and 3, respectively). The spacer length (four or six carbon) did not significantly influence eeAChE inhibition.
Remarkably, compounds 2–5, AntiOXCIN2 and AntiOXCIN3 displayed higher potency towards eqBChE, presenting IC50 values within the nanomolar range. As observed in eeAChE inhibition, catechol derivatives with conjugated double bonds (compounds 4 and 5) exhibited lower eqBChE than the related cinnamoyl counterparts (AntiOXCIN2 and AntiOXCIN3, respectively). Moreover, the derivatives bearing a six-carbon chain (compounds 3 and 5) were better eqBChE inhibitors than the four-carbon chain analogues (compounds 2 and 4). In contrast to the results obtained with eeAChE, benzodioxole derivatives 2 and 3 showed lower IC50 values than catechols 4 and 5, respectively.
The eqBChE selectivity index, determined from the ratio of the IC50 values of eeAChE and eqBChE, showed that benzodioxole derivatives 2 and 3 were more selective towards eqBChE (226-fold and 314-fold, respectively) than catechols 4 and 5 (36-fold and 62-fold, respectively). In addition, compounds 4, 5, AntiOXCIN2 and AntiOXCIN3 presented eqBChE selectivity index within the same range, indicating that the selectivity towards eqBChE was maintained with the increased rigidification of the aliphatic chain.
3.2.2. Evaluation of Human Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activities
Based on the promising results obtained in eeAChE and eqBChE inhibition studies, compounds 2–5 were selected to further investigate their inhibitory activities towards human ChEs (hChEs). The data obtained are presented in Table 2.
Compared with the results obtained with eeAChE and eqBChE, differences in the inhibition potency and selectivity of compounds 2–5 towards hChEs were observed. In general, TPP+ conjugates 2–5 were less active towards hChEs, with the differences being more pronounced between the data obtained with hBChE and eqBChE. The lower potency of several compounds towards hChEs was also observed in other studies [49,50]. Although eeAChE and eqBChE share > 88 % of amino acid identity with the human ChEs [50,51], they may present distinct structural and dynamic features. For instance, eqBChE and hBChE present different volume of the active site and exhibit three specific residue variations in the gorge site (Val305, Asp311 and Leu313 in eqBChE are replaced with Ala277, Gly283 and Pro285 in hBChE, respectively) [50]. These differences may invariably influence the binding of compounds to the enzymes and reflect in the ChE inhibitory activities.
All TPP+ conjugates presented IC50 values towards hAChE and hBChE within the low micromolar or high nanomolar range. While compounds containing a four-carbon chain were more selective towards hBChE (compound 2: SI = 55; compound 4: SI = 7.3), compounds bearing a six-carbon chain exhibited higher selectivity for hAChE (compound 3: SI = 0.67; compound 5: SI = 0.39). Interestingly, compounds containing six-carbon alkyl chains (compounds 3 and 5) displayed lower hAChE IC50 values and higher hBChE IC50 values than the four-carbon chain counterparts (compounds 2 and 4, respectively). Indeed, compound 3 was the most potent hAChE inhibitor of the series (compound 3: hAChE = 2 µM), while compound 2 presented the lowest IC50 towards hBChE (compound 2: hAChE = 200 nM). Moreover, benzodioxole derivatives 2 and 3 were more potent hAChE and hBChE inhibitors than catechol derivatives 4 and 5, respectively. Overall, these results suggest different binding modality and interaction of TPP+ conjugates towards hAChE and hBChE.
3.2.3. Crystallographic Studies with Human Cholinesterases
To gain insight into the binding mode of TPP+ conjugates to hAChE and hBChE, attempts to solve the crystallographic structures of the complexes formed between compounds 2–5 and recombinant hChEs were conducted. Structures of the complexes formed between compounds 2–5 and hAChE were solved (data not shown). Structures of hBChE in complex with compounds 3 and 5 were also obtained (data not shown). Compounds 2–5 bind similarly to hAChE and compounds 3 and 5 bind similarly to hBChE, although distinct binding modes of compounds to hAChE and hBChE were observed. Figure 2 shows an overview of the binding of compound 3 to hAChE and hBChE (Top) and a corresponding closer view of the molecule inside the active site of each enzyme (Bottom).
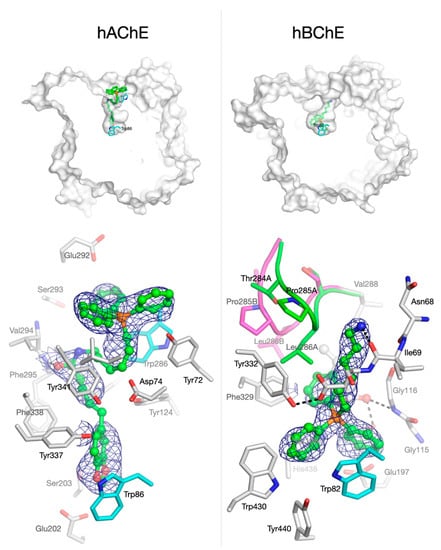
Figure 2.
Views of compound 3 in complex with hAChE (PDB 6ZWE) and hBChE (PDB 6ZWI). Compound 3 is shown in green ball and sticks. Top: Overview of compound 3 location inside the gorge of hChEs defined by the solvent accessible surface, showing the TPP+ moiety outside the gorge of hAChE (left) and inside and at the bottom of the gorge of hBChE (right). Bottom: Closer view of compound 3 inside the gorge of hAChE and hBChE. A 1-σ feature-enhanced map is represented as a blue mesh [52]. Residues lining the gorge are represented in sticks with carbon in white. Key peripheral (Trp286) and active site tryptophan residues (Trp86 and Trp82) are represented in sticks with carbon in cyan. The two alternate conformations of the acyl-loop of hBChE are represented in stick and cartoon, with carbons in green (conformation A) and magenta (conformation B).
For hAChE, the electron density map corresponding to compound 3 can be observed in the active site of the enzyme, identifying clearly the location of TPP+ moiety at the entrance of the active-site gorge of the enzyme (Figure 2, bottom left). Strong stacking interaction between aromatic amino acids located at the rim of the gorge of hAChE and the TPP+ moiety was not detected. The electron density map locates the benzodioxole moiety of compound 3 at the bottom of the gorge in a pseudo T-staking interaction with Trp86. Parts of the electron density of the linker between the TPP+ moiety and the benzodioxole ring of compound 3 are missing. Nevertheless, electron density is clearly present around the oxygen atom of the amide function interacting with Phe295.
Compound 3 appears to bind hBChE in an opposite way than hAChE: the electron density map of compound 3 clearly indicates the location of the TPP+ moiety at the bottom of hBChE active site (Figure 1, bottom right). The wider opening and overall volume of hBChE active site (500 Å3), compared to hAChE (300 Å3) [53], allows the TPP+ moiety to enter the gorge and bind in the vicinity of the catalytic site. A π-stacking interaction between Trp82 and two phenyl groups of the TPP+ moiety may stabilize the molecule inside the active site. It is important to note that, in previous studies of our research group, both the fitting orientation of TPP+ compounds within the active site gorge of hBChE and their interactions with Trp82 were predicted in docking simulations [17,22]. Missing electron density of compound 3 indicates that the piperoyl moiety is not visible. This moiety may be freely spanning outside the active site of hBChE. The binding of compound 3 to hBChE induces a position shift of Phe329 compared to 1P0I, but the most striking result is the coexistence of alternate conformations of the acyl-binding loop (Thr284-Pro285-Leu 286; Figure 2, bottom right). Such an extensive rearrangement of the acyl-loop has never been observed before for hBChE. In addition, as usually seen in the X-ray structures of this enzyme, an unidentified 5-carbon long carboxylic acid is bound in the vicinity of the catalytic serine extending its alkyl chain in the acyl-binding pocket.
Thus, the data obtained from the resolved crystal complex of compound 3 with hBChE is highly relevant for two main reasons. First, this is the first time the binding mode of TPP+ conjugates with hBChE is experimentally demonstrated; second, the data constitutes per se a validation of our molecular docking data obtained in previous studies [17,22].
3.3. Monoamine Oxidase Inhibition Studies
We evaluated the hMAO inhibition properties of piperine (compound 1) and TPP+ conjugates 2–5, using kynuramine as substrate and recombinant hMAO-A and -B isoforms [29,40]. The hMAO-A and hMAO-B inhibitory potency (IC50) and selectivity (SI) data of the compounds under study and reference inhibitors (clorgyline for hMAO-A and (R)-(−)-deprenyl, rasagiline, safinamide for hMAO-B) are reported in Table 3.
Table 3.
Evaluation of hMAOs (hMAO-A and hMAO-B) inhibitory activity of piperine (compound 1), compounds 2–5, AntiOXCIN2, AntiOXCIN3 and reference inhibitors.
In our previous reports, piperine showed moderate and selective hMAO-B inhibitory activity (compound 1, IC50 = 1.05 µM, SI = 10) [29]. AntiOXCIN2 and AntiOXCIN3 did not significantly inhibit hMAOs at 10 µM (% inhibition < 50 %). In contrast, compounds 2–5 inhibited both hMAO isoforms, presenting IC50 values within the micromolar or high nanomolar range. The differences in the compounds’ rigidity and/or in the position of the carbonyl group may be underlying the different hMAO inhibition profiles of compounds 2–5 and AntiOXCINs. Compounds 2 and 3 displayed similar or lower IC50 values towards hMAOs than compounds 4 and 5, respectively. Therefore, the benzodioxole ring opening had a negative effect on the ability to inhibit both hMAO isoforms. Concerning the spacer length, compounds containing a six-carbon chain (compounds 3 and 5) were more effective hMAO-B inhibitors than compounds with a four-carbon chain (compounds 2 and 4). However, no correlation was found between hMAO-A inhibition and spacer length.
3.4. Antioxidant Activity
To evaluate the radical scavenging activity of piperine (compound 1), compounds 2–5, AntiOXCIN2 and AntiOXCIN3, we used the oxygen radical absorbance capacity (ORAC) assay. This method measures the antioxidant activity against peroxyl radical (ROO•)-induced oxidation of fluorescein, a fluorescent probe, into a non-fluorescent product [54]. While piperine (compound 1) and compounds 2 and 3 were unable to scavenge ROO• radicals, catechols 4 and 5, AntiOXCIN2 and AntiOXCIN3 effectively protected fluorescein from oxidation (Table 4). Compounds 4 and 5 displayed lower ORAC-FL indexes than AntiOXCIN2 and AntiOXCIN3, respectively. Thus, the π-system extension in the chemical structure of AntiOXCINs enhanced the compounds’ ability to act as chain-breaking antioxidants against ROO• radicals.
Table 4.
ORAC-FL indexes and redox potentials (Ep) of piperine (compound 1), compounds 2–5, AntiOXCIN2 and AntiOXCIN3.
3.5. Electrochemical Studies
The redox behaviour of compounds 2–5 and AntiOXCINs was studied by differential pulse voltammetry (DPV) at physiological pH 7.4, using a glassy carbon-working electrode. Benzodioxole derivatives 2 and 3 presented two overlapped anodic peaks at physiological pH. The high values of redox potential (Ep) values obtained for these compounds are consistent with molecules that exhibit poor antioxidant activity. Indeed, compounds 2 and 3 do not present aromatic hydroxyl groups that can be oxidized, being the conjugated dienone system the most probable site for oxidation [55]. In contrast, catechols 4 and 5 displayed well-defined anodic peaks associated with the oxidation of the catechol groups (Figure 3). The redox potential (Ep) values obtained for compounds 4 and 5 were 125 mV and 144 mV (Table 4), respectively. The Ep values of compounds 4 and 5 were lower than those obtained with AntiOXCIN2 (Ep = 166 mV) and AntiOXCIN3 (Ep = 164 mV) [19]. Therefore, the higher electron delocalization between the aromatic ring and the carbonyl group shifted the peak potentials towards less positive values, thus enhancing the reducing power of catechols.
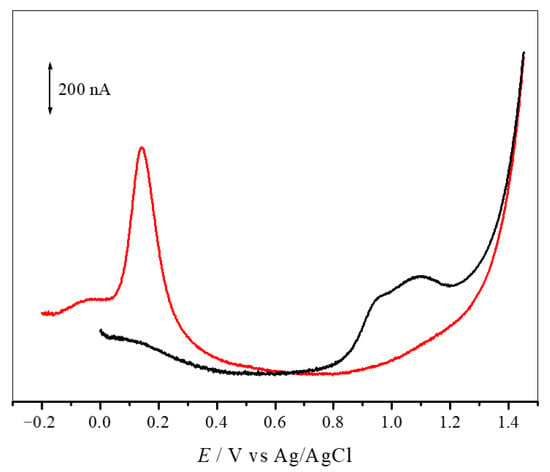
Figure 3.
Differential pulse voltammogram for 0.1 mM solutions of (▬) compound 3 and (▬) compound 5 in physiological pH 7.4 supporting electrolyte. Scan rate: 5 mV·s−1.
3.6. Evaluation of Cytotoxicity Profile
To evaluate the cytotoxicity of TPP+ conjugates 2–5, differentiated SH-SY5Y cells were incubated with increasing concentrations of the test compounds (0.20–100 µM) for 24 h. Cellular cytotoxicity was assessed using the resazurin reduction assay, which estimates metabolic activity of viable cells [56], and the neutral red (NR) uptake assay, which relies on the lysosomal incorporation of the dye NR in living cells [57]. The results, presented as mean resorufin fluorescence (% of control) ± SEM and NR uptake (% of control) ± SEM (n = 3), are depicted in Figure 4.
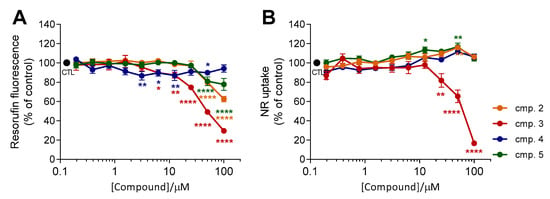
Figure 4.
Cellular viability of differentiated SH-SY5Y neuroblastoma cells after a 24 h treatment with increasing concentrations of compounds 2–5 (0.20–100 µM). Cellular viability was evaluated using the resazurin reduction assay (A) and the NR uptake assay (B). Results are expressed as the mean % of untreated controls ± SEM (n = 3). Statistical comparisons were made using two-way ANOVA followed by Dunnet’s multiple comparison test. In all cases, p values lower than 0.05 were considered significant (* p < 0.05, ** p < 0.01, **** p < 0.0001 vs untreated cells).
Data obtained in our previous reports showed that piperine (compound 1) did not exhibit cytotoxic effects at concentrations up to 50 µM [58]. Similarly, AntiOXCIN2 and AntiOXCIN3 did not markedly affect both resazurin reduction and NR uptake at concentrations up to 100 µM (Figure S1). However, a significant dose-dependent decrease in resorufin fluorescence was observed with compounds 2, 3, and 5 (Figure 4A). Compound 3 presented the most significant decreases in resazurin reduction (resorufin fluorescence < 85%) at concentrations above 12.5 µM (Figure 4A). This tendency was also observed with compounds 2 and 5 at concentrations above 25 µM (Figure 4A). On the other hand, incubation of neuroblastoma cells with increasing concentrations of compound 4 up to 100 µM did not markedly affect resazurin reduction (Figure 4A).
Resazurin reduction was influenced by the substitution pattern of the aromatic ring and by the length of the lipophilic spacer. The effects of the structural components of TPP+ cations 2–5 were particularly noticeable at the highest tested concentration (100 µM). In fact, decreases of resazurin reduction were more prominent by treatment with benzodioxoles (compounds 2 and 3) than by catechols (compounds 4 and 5) (Figure 4A). Moreover, compounds bearing a four-carbon alkyl chain (compounds 2 and 4) led to a lower decrease in resazurin reduction than the six-carbon alkyl chain analogues (compounds 3 and 5, respectively) (Figure 4A).
Interestingly, the same conclusions were not similar when using the NR uptake assay (Figure 4B). Neutral red (NR) uptake remained constant with increasing concentrations of compounds 2, 4 and 5 (Figure 4B), even at concentrations in which the resazurin reduction was significantly decreased (Figure 4A, compounds 2 and 5, ≥ 50 µM). Only compound 3 displayed the same tendency observed with the resazurin reduction assay, showing significant lysosomal toxicity at concentrations above 12.5 µM.
In summary, these results indicate that catechol TPP+ conjugates are less prone to decrease cellular metabolism at high concentrations than the corresponding benzodioxole derivatives. Moreover, catechols 4 and 5 share the same cytotoxicity profile of the parent AntiOXCINs.
3.7. Evaluation of Drug-Like Properties
To study the drug-likeness of TPP+ conjugates 2–5, AntiOXCIN2 and AntiOXCIN3, we first determined their chromatographic hydrophobic index (CHI) LogPoct (CHI LogPoct). The CHI parameter is derived from the retention times obtained in a fast gradient reversed-phase HPLC method [59]. Chromatographic hydrophobicity index (CHI) values of neutral molecules are directly correlated with the compounds’ lipophilicity [59]. In our case, the determination of CHI was carried out under acidic conditions (pH 2.3) to prevent the ionization of the catechol moiety. The values of CHI were then used to calculate CHI LogPoct. The results are presented in Table 5.
Table 5.
Calculated CHI and CHI Log Poct and predicted drug-like properties of piperine (compound 1) and TPP+ conjugates 2–5, AntiOXCIN2 and AntiOXCIN3.
Chromatographic hydrophobicity index LogPoct (CHI LogPoct) values of compounds 2–5 were considerably lower than the obtained with piperine (Table 5). Therefore, despite the high lipophilicity of the TPP+ moiety, the positive charge of TPP+ conjugates may decrease the affinity towards the hydrophobic stationary phase. Moreover, CHI LogPoct values increased with the presence of a benzodioxole ring (compounds 2 and 3 vs compounds 4 and 5, respectively) and longer alkyl linkers (compounds 2 and 4 vs. compounds 3 and 5, respectively) (Table 4). Finally, CHI LogPoct values of compounds 4 and 5 were similar to those of AntiOXCIN2 and AntiOXCIN3, respectively, suggesting that the π-system extension maintained the compounds’ hydrophobicity.
We also calculated several physicochemical properties to predict the ability of compounds 2–5, AntiOXCIN2 and AntiOXCIN3 to attain the CNS. These include molecular weight (MW), topological polar surface area (TPSA in Å), number of hydrogen acceptors (HBA), number of hydrogen donors (HBD) and number of rotatable bonds (RB) (Table 5). Among the estimated parameters, MW and RB values of TPP+ conjugates exceeded the limits suggested for CNS-active drugs (MW < 500 g·mol−1; RB < 8). Nonetheless, despite their high volume, several lipophilic TPP+ cations were taken up by mitochondria within several tissues (brain, heart, liver) following long-term oral administration [63]. In addition, our previous studies showed that catechol and pyrogallol lipophilic TPP+ conjugates can cross human cerebral microvascular endothelial (hCMEC/D3) cells, an in vitro cellular model of human BBB, in a dose-dependent manner [21]. As expected, compounds 4 and 5 presented a lower number of RB than AntiOXCIN2 and AntiOXCIN3, respectively. The other predicted physicochemical properties (TPSA, HBA, HBD) fell within the proposed limits.
4. Conclusions
In this work, we compared the bioactivity and toxicity profiles of two AntiOXCINs with bioisosteres with extended electron delocalization and similar lipophilicity. The increased rigidity of lipophilic TPP+ conjugates enhanced eeAChE and eqBChE inhibition while preserving the selectivity index towards eqBChE. Co-crystallization studies with hChEs demonstrated that the TPP+ derivatives bind differently to the active sites of hAChE and hBChE. While the TPP+ moiety was present at the entrance of the active-site gorge of hAChE, in hBChE it was located within the gorge in the vicinity of the active site. These data validated our results from docking simulations performed for similar derivatives that predicted the binding mode of lipophilic TPP+ conjugates to the active site of hBChE [17,22]. Unlike AntiOXCINs, compounds 2–5 are endowed with moderate hMAO inhibition properties. In addition, catechols 4 and 5 scavenged ROO• radicals more efficiently and presented lower Ep values than AntiOXCIN2 and AntiOXCIN3, respectively. Cytotoxicity studies showed that catechols compounds 4 and 5 are less cytotoxic than the related benzodioxole-containing compounds (compounds 2 and 3). Moreover, the safety profile of compounds 4 and 5 at concentrations up to 100 µM was similar to that of AntiOXCIN2 and AntiOXCIN3, respectively.
Compound 4 stands out as the best mitochondria-targeted agent of the series. In addition to the dual ChE/MAO inhibition profile, compound 4 is endowed with antioxidant activity. Compound 4 also presents a safe cytotoxicity profile in differentiated neuroblastoma cells (<100 µM) and favourable drug-like properties.
Overall, this work represents an advance towards the development of mitochondria-targeted antioxidants with multitarget activity. These results can potentially propel the discovery of new compounds able to tackle simultaneously neurotransmitter depletion and mitochondria-associated oxidative stress in NDs.
Supplementary Materials
The following are available online: https://www.mdpi.com/2076-3921/10/2/329/s1. Experimental details, Tables, Figures. Table S1. Retention times (tR) of the standard mixture obtained by LC/UV at pH 2.3. Figure S1. Cellular viability of differentiated SH-SY5Y neuroblastoma cells after a 24 h treatment with AntiOXCIN2 and AntiOXCIN3 at eight different concentrations (0.20–100 µM). Figure S2. Linear correlation obtained by plotting the retention times (tR) of each of the individual standard mixture compounds against the CHI values at pH 2.3 (CHI0 pH 2.3). Figure S3. 1H and 13C NMR spectra of compound 2 (NMR spectra obtained in CDCl3-d1). Figure S4. 1H and 13C NMR spectra of compound 3 (NMR spectra obtained in CDCl3-d1). Figure S5. 1H and 13C NMR spectra of compound 4 (NMR spectra obtained in DMSO-d6). Figure S6. 1H and 13C NMR spectra of compound 5 (NMR spectra obtained in DMSO-d6).
Author Contributions
D.C., P.J.O. and F.B. designed research; D.C., S.B. (Sofia Benfeito), F.C., S.B. (Sandra Barreiro), O.D.S., X.B., F.N., P.S. and J.G. performed research; D.C., S.B. (Sofia Benfeito), S.B. (Sandra Barreiro), O.D.S. and F.N. analyzed data; D.C., O.D.S. and F.N. wrote the paper; F.B., P.J.O., F.R. and J.D. reviewed and edited the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Foundation for Science and Technology (FCT) and FEDER/COMPETE (Grants UIDB/00081/2020, PTDC/MED-QUI/29164/2017, POCI-01-0145-FEDER-29164, PTDC/BIA-MOL/28607/2017, POCI-01-0145-FEDER-028607). D. Chavarria, S. Benfeito and F. Cagide grants are supported by FCT and FEDER/COMPETE. The authors also gratefully acknowledge the Direction Générale de l’Armement (DGA) and Service de Santé des Armées (SSA) of the French Ministry of Armed Forces for sustained financial support to O.D.S, J.D, X.B and F.N (grant NBC-5-C-4210).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within the article and its supplementary material. Other data that support the findings of this study are available upon request from the corresponding authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ramsay, R.R.; Majekova, M.; Medina, M.; Valoti, M. Key Targets for Multi-Target Ligands Designed to Combat Neurodegeneration. Front. Neurosci. 2016, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef] [PubMed]
- Elmabruk, A.; Das, B.; Yedlapudi, D.; Xu, L.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Design, Synthesis, and Pharmacological Characterization of Carbazole Based Dopamine Agonists as Potential Symptomatic and Neuroprotective Therapeutic Agents for Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 396–411. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef]
- Fetisova, E.K.; Avetisyan, A.V.; Izyumov, D.S.; Korotetskaya, M.V.; Chernyak, B.V.; Skulachev, V.P. Mitochondria-targeted antioxidant SkQR1 selectively protects MDR (Pgp 170)-negative cells against oxidative stress. FEBS Lett. 2010, 584, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Facecchia, K.; Fochesato, L.A.; Ray, S.D.; Stohs, S.J.; Pandey, S. Oxidative toxicity in neurodegenerative diseases: Role of mitochondrial dysfunction and therapeutic strategies. J. Toxicol. 2011, 2011, 683728. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Biasutto, L.; Mattarei, A.; La Spina, M.; Azzolini, M.; Parrasia, S.; Szabò, I.; Zoratti, M. Strategies to target bioactive molecules to subcellular compartments. Focus on natural compounds. Eur. J. Med. Chem. 2019, 181, 11557. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control. Release 2015, 207, 40–58. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; Weissig, V. Mitochondria-Targeted Agents: Mitochondriotropics, Mitochondriotoxics, and Mitocans. Handb. Exp. Pharmacol. 2017, 240, 423–438. [Google Scholar] [CrossRef]
- He, H.; Li, D.W.; Yang, L.Y.; Fu, L.; Zhu, X.J.; Wong, W.K.; Jiang, F.L.; Liu, Y. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci. Rep. 2015, 5, 13543. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Porteous, C.M.; Logan, A.; Evans, C.; Ledgerwood, E.C.; Menon, D.K.; Aigbirhio, F.; Smith, R.A.; Murphy, M.P. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: Implications for mitochondria-specific therapies and probes. Biochim. Biophys. Acta 2010, 1800, 1009–1017. [Google Scholar] [CrossRef]
- Oliveira, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Sequeira, L.; Mesiti, F.; Silva, T.; Garrido, J.; Remiao, F.; Vilar, S.; et al. Hydroxybenzoic Acid Derivatives as Dual-Target Ligands: Mitochondriotropic Antioxidants and Cholinesterase Inhibitors. Front. Chem. 2018, 6, 126. [Google Scholar] [CrossRef]
- Teixeira, J.; Oliveira, C.; Amorim, R.; Cagide, F.; Garrido, J.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; Oliveira, P.J.; et al. Development of hydroxybenzoic-based platforms as a solution to deliver dietary antioxidants to mitochondria. Sci. Rep. 2017, 7, 6842. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Cagide, F.; Benfeito, S.; Soares, P.; Garrido, J.; Baldeiras, I.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; et al. Development of a Mitochondriotropic Antioxidant Based on Caffeic Acid: Proof of Concept on Cellular and Mitochondrial Oxidative Stress Models. J. Med. Chem. 2017, 60, 7084–7098. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Soares, P.; Benfeito, S.; Gaspar, A.; Garrido, J.; Murphy, M.P.; Borges, F. Rational discovery and development of a mitochondria-targeted antioxidant based on cinnamic acid scaffold. Free Radic. Res. 2012, 46, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Benfeito, S.; Oliveira, C.; Fernandes, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Garrido, J.; Martins, C.; Sarmento, B.; Silva, R.; et al. Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur. J. Med. Chem. 2019, 167, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Benfeito, S.; Fernandes, C.; Vilar, S.; Remião, F.; Uriarte, E.; Borges, F. Exploring the multi-target performance of mitochondriotropic antioxidants against the pivotal Alzheimer’s disease pathophysiological hallmarks. Molecules 2020, 25, 276. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, Y.H.; Liu, H.; Qu, H.D. Neuroprotective effects of piperine on the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease mouse model. Int. J. Mol. Med. 2015, 36, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Wattanathorn, J.; Chonpathompikunlert, P.; Muchimapura, S.; Priprem, A.; Tankamnerdthai, O. Piperine, the potential functional food for mood and cognitive disorders. Food Chem. Toxicol. 2008, 46, 3106–3110. [Google Scholar] [CrossRef]
- Chonpathompikunlert, P.; Wattanathorn, J.; Muchimapura, S. Piperine, the main alkaloid of Thai black pepper, protects against neurodegeneration and cognitive impairment in animal model of cognitive deficit like condition of Alzheimer’s disease. Food Chem. Toxicol. 2010, 48, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.Q.; Huang, Z.; Ip, S.P.; Xian, Y.F.; Che, C.T. Protective effects of piperine against corticosterone-induced neurotoxicity in PC12 cells. Cell. Mol. Neurobiol. 2012, 32, 531–537. [Google Scholar] [CrossRef]
- Lee, S.A.; Hong, S.S.; Han, X.H.; Hwang, J.S.; Oh, G.J.; Lee, K.S.; Lee, M.K.; Hwang, B.Y.; Ro, J.S. Piperine from the fruits of Piper longum with inhibitory effect on monoamine oxidase and antidepressant-like activity. Chem. Pharm. Bull. 2005, 53, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, D.; Cagide, F.; Pinto, M.; Gomes, L.R.; Low, J.N.; Borges, F. Development of piperic acid-based monoamine oxidase inhibitors: Synthesis, structural characterization and biological evaluation. J. Mol. Struct. 2019, 1182, 298–307. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [PubMed]
- Williard, P.G.; Fryhle, C.B. Boron trihalide-methyl sulfide complexes as convenient reagents for dealkylation of aryl ethers. Tetrahedron Lett. 1980, 21, 3731–3734. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Borloz, A.; Urbain, A.; Marston, A.; Hostettmann, K.; Carrupt, P.A.; Reist, M. In vitro screening assays to identify natural or synthetic acetylcholinesterase inhibitors: Thin layer chromatography versus microplate methods. Eur. J. Pharm. Sci. 2008, 33, 109–119. [Google Scholar] [CrossRef]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef]
- Brazzolotto, X.; Wandhammer, M.; Ronco, C.; Trovaslet, M.; Jean, L.; Lockridge, O.; Renard, P.Y.; Nachon, F. Human butyrylcholinesterase produced in insect cells: Huprine-based affinity purification and crystal structure. FEBS J. 2012, 279, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallog. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Hagenow, S.; Stasiak, A.; Ramsay, R.R.; Stark, H. Ciproxifan, a histamine H3 receptor antagonist, reversibly inhibits monoamine oxidase A and B. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Mura, F.; Silva, T.; Castro, C.; Borges, F.; Zuniga, M.C.; Morales, J.; Olea-Azar, C. New insights into the antioxidant activity of hydroxycinnamic and hydroxybenzoic systems: Spectroscopic, electrochemistry, and cellular studies. Free Radic. Res. 2014, 48, 1473–1484. [Google Scholar] [CrossRef]
- Garrido, J.; Gaspar, A.; Garrido, E.M.; Miri, R.; Tavakkoli, M.; Pourali, S.; Saso, L.; Borges, F.; Firuzi, O. Alkyl esters of hydroxycinnamic acids with improved antioxidant activity and lipophilicity protect PC12 cells against oxidative stress. Biochimie 2012, 94, 961–967. [Google Scholar] [CrossRef]
- Fernandes, C.; Pinto, M.; Martins, C.; Gomes, M.J.; Sarmento, B.; Oliveira, P.J.; Remião, F.; Borges, F. Development of a PEGylated-Based Platform for Efficient Delivery of Dietary Antioxidants Across the Blood-Brain Barrier. Bioconjug. Chem. 2018, 29, 1677–1689. [Google Scholar] [CrossRef]
- Camurri, G.; Zaramella, A. High-throughput liquid chromatography/mass spectrometry method for the determination of the chromatographic hydrophobicity index. Anal. Chem. 2001, 73, 3716–3722. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reynolds, D.P. Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, B. Donepezil: A review. Expert Opin. Drug Metab. Toxicol. 2005, 1, 527–536. [Google Scholar] [CrossRef]
- Kumar, A.; Pintus, F.; Di Petrillo, A.; Medda, R.; Caria, P.; Matos, M.J.; Vina, D.; Pieroni, E.; Delogu, F.; Era, B.; et al. Novel 2-pheynlbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018, 8, 4424. [Google Scholar] [CrossRef]
- Karlsson, D.; Fallarero, A.; Brunhofer, G.; Mayer, C.; Prakash, O.; Mohan, C.G.; Vuorela, P.; Erker, T. The exploration of thienothiazines as selective butyrylcholinesterase inhibitors. Eur. J. Pharm. Sci. 2012, 47, 190–205. [Google Scholar] [CrossRef]
- Moorad, D.R.; Luo, C.; Saxena, A.; Doctor, B.P.; Garcia, G.E. Purification and determination of the amino acid sequence of equine serum butyrylcholinesterase. Toxicol. Methods 1999, 9, 219–227. [Google Scholar] [CrossRef]
- Afonine, P.V.; Moriarty, N.W.; Mustyakimov, M.; Sobolev, O.V.; Terwilliger, T.C.; Turk, D.; Urzhumtsev, A.; Adams, P.D. FEM: Feature-enhanced map. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 646–666. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Deng, Z.; Wu, T.; Liu, R.; Loewen, S.; Tsao, R. Microwave-assisted extraction of phenolics with maximal antioxidant activities in tomatoes. Food Chem. 2012, 130, 928–936. [Google Scholar] [CrossRef]
- Carp, O.E.; Moraru, A.; Pinteala, M.; Arvinte, A. Electrochemical behaviour of piperine. Comparison with control antioxidants. Food Chem. 2021, 339. [Google Scholar] [CrossRef]
- Silva, F.S.; Starostina, I.G.; Ivanova, V.V.; Rizvanov, A.A.; Oliveira, P.J.; Pereira, S.P. Determination of Metabolic Viability and Cell Mass Using a Tandem Resazurin/Sulforhodamine B Assay. Curr. Protoc. Toxicol. 2016, 68, 2–24. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Chavarria, D.; Fernandes, C.; Silva, V.; Silva, C.; Gil-Martins, E.; Soares, P.; Silva, T.; Silva, R.; Remiao, F.; Oliveira, P.J.; et al. Design of novel monoamine oxidase-B inhibitors based on piperine scaffold: Structure-activity-toxicity, drug-likeness and efflux transport studies. Eur. J. Med. Chem. 2020, 185, 111770. [Google Scholar] [CrossRef]
- Valko, K.; Bevan, C.; Reynolds, D. Chromatographic Hydrophobicity Index by Fast-Gradient RP-HPLC: A High-Throughput Alternative to log P/log D. Anal. Chem. 1997, 69, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A.; Pennington, L.D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.; Du, C.M.; Bevan, C.; Reynolds, D.P.; Abraham, M.H. Rapid method for the estimation of octanol/water partition coefficient (Log Poct) from gradient RP-HPLC retention and a hydrogen bond acidity term (∑α2H). Curr. Med. Chem. 2001, 8, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).