
Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy

 ,
,  and
and 
Abstract
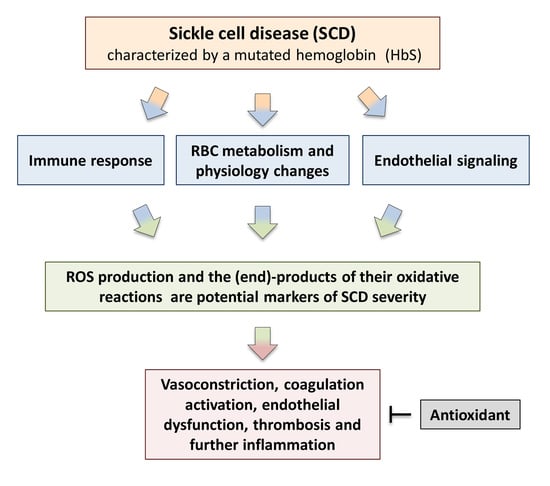
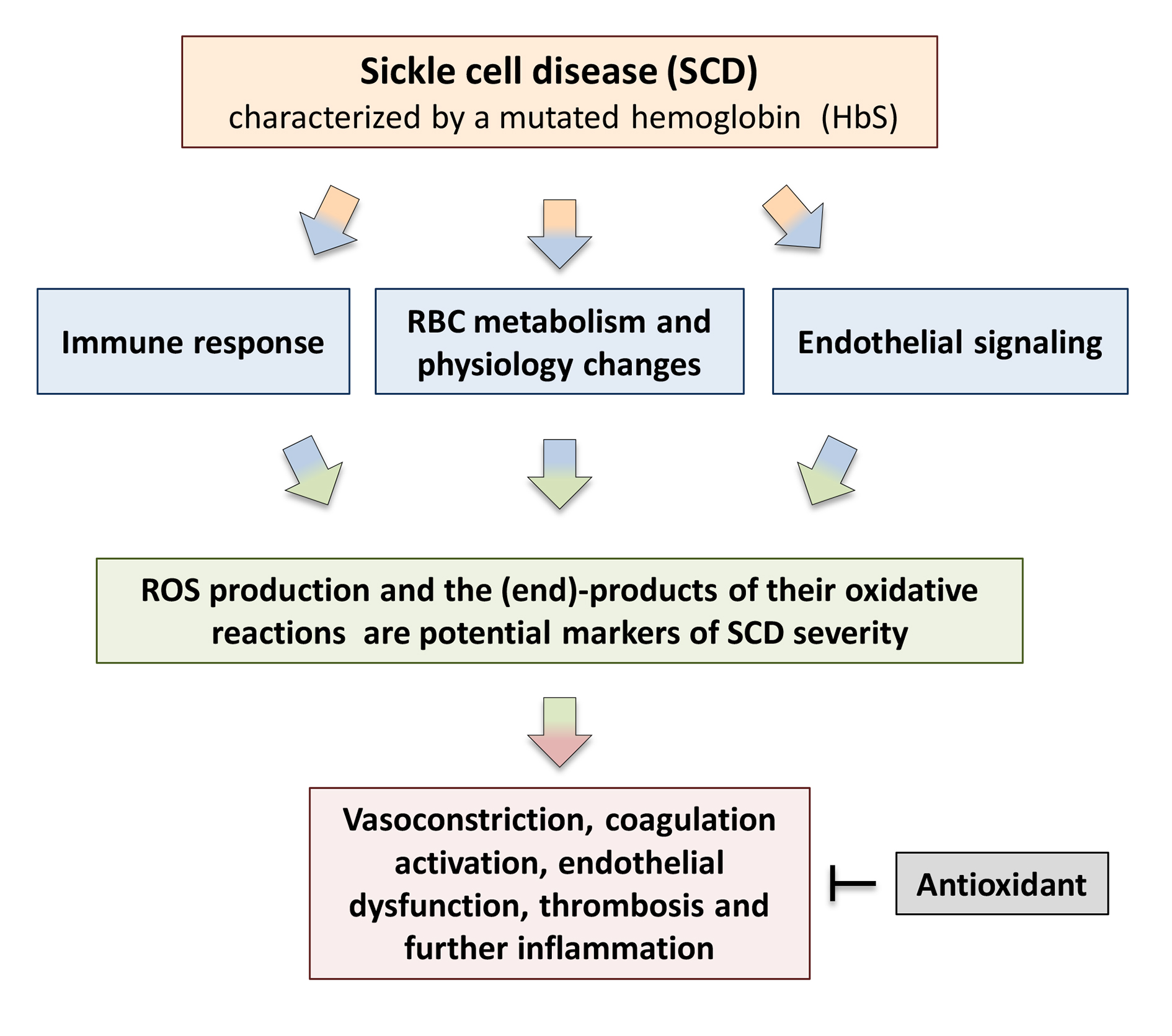
1. Introduction
2. Source of ROS in SCD
2.1. Increased Activity of Several Oxidases
2.2. HbS Autoxidation
2.3. Heme and Iron Release
2.4. Decreased •NO Bioavailability
2.5. Uncoupling of NOS Activity
2.6. Increased Asymmetric Dimethylarginine
3. Oxidative Damage to Intracellular Components in SCD RBCs
3.1. Oxidative Damage to SCD RBCs Cytoskeletal Proteins
3.2. Oxidative Damage to SCD RBCs Membrane Lipids
4. RBC Hemolysis
5. Cell Adhesion and Vessel Occlusion
6. Advanced Glycoxidation and Lipoxidation End-Products
AGEs and ALEs in SCD

{kind=link}
{kind=link}
| Compound | Specificity in SCD | Tissue | References |
|---|---|---|---|
| Advanced lipoxidation end-products (ALEs) (adducts with oxylipin carbonyl compounds) | |||
| F2-isoprostanes adducts | Biomarker of lipid peroxidation | Plasma | [119,120,121] |
| Malondialdehyde (MDA) adducts | Biomarker of lipid peroxidation | Plasma | [122,124,125] |
| Mixed advanced lipoxidation end-products (ALEs)/advanced glycoxidation end-products (AGEs) | |||
| Nε-carboxymethyllysine (CML) | Biomarker of hemolysis-related organ complications and vascular pathology | Red blood cells, plasma | [110,111] |
| Nε-carboxyethyllysine (CEL) | Biomarker of hemolysis-related organ complications and vascular pathology | Red blood cells, plasma | [110,111] |
| Adducts generated by the Maillard reaction | |||
| Pentosidine * | Biomarker of hemolysis-related organ complications and vascular pathology | Red blood cells, plasma | [111] |
7. Antioxidant Defenses in SCD
8. Antioxidant Therapy for SCD
8.1. L-Glutamine
| Antioxidants | Mechanisms | Effects in SCD Patients | Comments |
|---|---|---|---|
| L- Glutamine | Acts through the formation of reduced NADH. | Improves cellular redox potential and adhesion of sickle RBCs to the endothelium; facilitates protein and glutathione synthesis. | FDA approved; [155,156] |
| N-Acetylcysteine | Substrate for GSH generation. | Reduces the number of RBCs expressing phosphatidylserine, marker of peroxidative damage to inner membrane of RBCs. | NCT01800526 phase 2 trial; NCT01849016 phase 3 trial |
| Zinc supplementation | Zinc deficiency is associated with high incidence of infections, vaso-occlusion events and the chronic oxidative stress. | In young SCD patients improves linear growth and weight gain. Has beneficial effects on immunity, inflammatory state and oxidative stress. | NS; [159,160] |
| Nitric oxide | Reduced •NO concentration can be associated to increased levels of free O2•−. | Inhaled •NO improves tissue oxygenation and reduces pain in SCD patients with pulmonary hypertension. | NCT00094887 phase 2 trial |
| L-arginine | Induces GSH synthesis. | Improves •NO bioavailability. | NCT02447874 phase 2 trial |
| Alfa-lipoic acid | Induces GSH synthesis. | Increases glutathione level. | NCT01054768 phase 2 trial |
| L-acetyl-L-carnitine | Improves mitochondrial metabolism, facilitating entry of long-chain fatty acids into mitochondria and decreasing lipid peroxidation in tissue. | Decreases lipid peroxidation. | NCT01054768 phase 2 trial |
| Gum Arabic | Acts as immuno-modulatory. | Increases total antioxidant capacity and decreased MDA and H2O2 levels. | NCT04191213 phase 2 trial |
| Omega-3 fatty acids | O3FA deficiency correlates with an increase in plasma levels of the inflammatory biomarker | Have beneficial effects on vascular activation, inflammation and antioxidant systems. | [156,157,158,159,160,161,162,163,164,165,166,167,168,169,170] |
| Curcumin | Can modulate the activity of enzymes active in the neutralization of free radicals and it can inhibit ROS-generating enzymes. | Mitigates the effects of iron induced oxidative stress on lipid peroxidation and •NO levels. | [169,170] |
| Vitamins A, C and E | Their deficiency increases susceptibility to infection and hemolysis. | Conflicting results about the effectiveness of their supplementation on oxidative stress. | NCT03903133 phase 4 trial |
| Iron chelators | Avoids excessive iron overload and the consequent ROS generation. | Have a central role in the treatment of transfusion-dependent hemoglobinopathy. | NS; [158] |
8.2. N-Acetylcysteine (NAC)
8.3. Zinc Supplementation
8.4. Nitric Oxide and L-Arginine
8.5. α-Lipoic Acid and Acetyl-L-Carnitine
8.6. Other Antioxidant Agents
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chaturvedi, S.; DeBaun, M.R. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am. J. Hematol. 2016, 91, 5–14. [Google Scholar] [CrossRef]
- Thein, M.S.; Igbineweka, N.E.; Thein, S.L. Sickle cell disease in the older adult. Pathology 2017, 49, 1–9. [Google Scholar] [CrossRef]
- Aygun, B.; Odame, I. A global perspective on sickle cell disease. Pediatr. Blood Cancer 2012, 59, 386–390. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 377, 305. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Ashley-Koch, A.; Yang, Q.; Olney, R.S. Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review. Am. J. Epidemiol. 2000, 151, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360. [Google Scholar] [CrossRef]
- Tarasev, M.; Muchnik, M.; Light, L.; Alfano, K.; Chakraborty, S. Individual variability in response to a single sickling event for normal, sickle cell, and sickle trait erythrocytes. Transl. Res. 2017, 181, 96–107. [Google Scholar] [CrossRef]
- Chirico, E.N.; Pialoux, V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life 2012, 64, 72–80. [Google Scholar] [CrossRef]
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 2019, 3, 2679–2687. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Deshmukh, P.; Goswami, K.; Garg, N. Antioxidant vitamin levels in sickle cell disorders. Natl. Med. J. India 2007, 20, 11–13. [Google Scholar]
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematology 2019, 24, 1–9. [Google Scholar] [CrossRef]
- Al-Naama, L.M.; Hassan, M.K.; Mehdi, J.K. Association of erythrocytes antioxidant enzymes and their cofactors with markers of oxidative stress in patients with sickle cell anemia. Qatar Med. J. 2015, 2015, 14. [Google Scholar] [CrossRef]
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872. [Google Scholar] [CrossRef]
- Wood, K.C.; Hebbel, R.P.; Granger, D.N. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005, 19, 989–991. [Google Scholar] [CrossRef]
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006, 40, 1443–1453. [Google Scholar] [CrossRef]
- Wood, K.C.; Granger, D.N. Sickle cell disease: Role of reactive oxygen and nitrogen metabolites. Clin. Exp. Pharmacol. Physiol. 2007, 34, 926–932. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef]
- Aslan, M.; Ryan, T.M.; Adler, B.; Townes, T.M.; Parks, D.A.; Thompson, J.A.; Tousson, A.; Gladwin, M.T.; Patel, R.P.; Tarpey, M.M.; et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc. Natl. Acad. Sci. USA 2001, 98, 15215–15220. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Rifkind, J.M.; Mohanty, J.G.; Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2014, 5, 500. [Google Scholar] [CrossRef]
- Gizi, A.; Papassotiriou, I.; Apostolakou, F.; Lazaropoulou, C.; Papastamataki, M.; Kanavaki, I.; Kalotychou, V.; Goussetis, E.; Kattamis, A.; Rombos, I.; et al. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status. Blood Cells Mol. Dis. 2011, 46, 220–225. [Google Scholar] [CrossRef]
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle Cell Hemoglobin in the Ferryl State Promotes betaCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J. Biol. Chem. 2015, 290, 27939–27958. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Azarov, I.; He, X.; Jeffers, A.; Basu, S.; Ucer, B.; Hantgan, R.R.; Levy, A.; Kim-Shapiro, D.B. Rate of nitric oxide scavenging by hemoglobin bound to haptoglobin. Nitric Oxide 2008, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Moller, H.J.; Moestrup, S.K. Hemoglobin and heme scavenger receptors. Antioxid. Redox Signal. 2010, 12, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248. [Google Scholar] [CrossRef]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef]
- Mano, J. Reactive carbonyl species: Their production from lipid peroxides, action in environmental stress, and the detoxification mechanism. Plant. Physiol. Biochem. 2012, 59, 90–97. [Google Scholar] [CrossRef]
- Aslan, M.; Freeman, B.A. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease—Mechanisms and consequences. Cell Mol. Biol. 2004, 50, 95–105. [Google Scholar]
- Jeffers, A.; Gladwin, M.T.; Kim-Shapiro, D.B. Computation of plasma hemoglobin nitric oxide scavenging in hemolytic anemias. Free Radic. Biol. Med. 2006, 41, 1557–1565. [Google Scholar] [CrossRef]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Shiva, S.; Wang, X.; Ringwood, L.A.; Xu, X.; Yuditskaya, S.; Annavajjhala, V.; Miyajima, H.; Hogg, N.; Harris, Z.L.; Gladwin, M.T. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat. Chem. Biol. 2006, 2, 486–493. [Google Scholar] [CrossRef]
- Grubina, R.; Basu, S.; Tiso, M.; Kim-Shapiro, D.B.; Gladwin, M.T. Nitrite reductase activity of hemoglobin S (sickle) provides insight into contributions of heme redox potential versus ligand affinity. J. Biol. Chem. 2008, 283, 3628–3638. [Google Scholar] [CrossRef]
- Rees, D.C.; Gibson, J.S. Biomarkers in sickle cell disease. Br. J. Haematol. 2012, 156, 433–445. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Aboagye, G.; Campbell, A.D. Correlation of lipid peroxidation and nitric oxide metabolites, trace elements, and antioxidant enzymes in patients with sickle cell disease. J. Clin. Lab. Anal. 2020, 34, e23294. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.D.; Gladwin, M.T. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr. Opin. Hematol. 2003, 10, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Dror, Y.; Wallis, K. Arginase activity in erythrocytes of healthy and ill children. Clin. Chim Acta 1970, 28, 391–396. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef]
- Elias, D.B.; Barbosa, M.C.; Rocha, L.B.; Dutra, L.L.; Silva, H.F.; Martins, A.M.; Goncalves, R.P. L-arginine as an adjuvant drug in the treatment of sickle cell anaemia. Br. J. Haematol. 2013, 160, 410–412. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef]
- Baldus, S.; Eiserich, J.P.; Brennan, M.L.; Jackson, R.M.; Alexander, C.B.; Freeman, B.A. Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases. Free Radic. Biol. Med. 2002, 33, 1010. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Akinsheye, I.; Klings, E.S. Sickle cell anemia and vascular dysfunction: The nitric oxide connection. J. Cell Physiol. 2010, 224, 620–625. [Google Scholar] [CrossRef]
- Sharma, S.; Smith, A.; Kumar, S.; Aggarwal, S.; Rehmani, I.; Snead, C.; Harmon, C.; Fineman, J.; Fulton, D.; Catravas, J.D.; et al. Mechanisms of nitric oxide synthase uncoupling in endotoxin-induced acute lung injury: Role of asymmetric dimethylarginine. Vascul. Pharmacol. 2010, 52, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, X.; Shang, R.; Chen, Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide 2018, 78, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Landburg, P.P.; Teerlink, T.; van Beers, E.J.; Muskiet, F.A.; Kappers-Klunne, M.C.; van Esser, J.W.; Mac Gillavry, M.R.; Biemond, B.J.; Brandjes, D.P.; Duits, A.J.; et al. Association of asymmetric dimethylarginine with sickle cell disease-related pulmonary hypertension. Haematologica 2008, 93, 1410–1412. [Google Scholar] [CrossRef]
- Kato, G.J.; Wang, Z.; Machado, R.F.; Blackwelder, W.C.; Taylor, J.G.t.; Hazen, S.L. Endogenous nitric oxide synthase inhibitors in sickle cell disease: Abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br. J. Haematol. 2009, 145, 506–513. [Google Scholar] [CrossRef] [PubMed]
- El-Shanshory, M.; Badraia, I.; Donia, A.; Abd El-Hameed, F.; Mabrouk, M. Asymmetric dimethylarginine levels in children with sickle cell disease and its correlation to tricuspid regurgitant jet velocity. Eur. J. Haematol. 2013, 91, 55–61. [Google Scholar] [CrossRef]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef]
- Barodka, V.M.; Nagababu, E.; Mohanty, J.G.; Nyhan, D.; Berkowitz, D.E.; Rifkind, J.M.; Strouse, J.J. New insights provided by a comparison of impaired deformability with erythrocyte oxidative stress for sickle cell disease. Blood Cells Mol. Dis. 2014, 52, 230–235. [Google Scholar] [CrossRef]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Arena, A.; Giorgi, A.; di Francesco, L.; Schinina, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; et al. Polyubiquitinylation Profile in Down Syndrome Brain Before and After the Development of Alzheimer Neuropathology. Antioxid. Redox Signal. 2017, 26, 280–298. [Google Scholar] [CrossRef]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Strader, M.B.; Jana, S.; Meng, F.; Heaven, M.R.; Shet, A.S.; Thein, S.L.; Alayash, A.I. Post-translational modification as a response to cellular stress induced by hemoglobin oxidation in sickle cell disease. Sci. Rep. 2020, 10, 14218. [Google Scholar] [CrossRef] [PubMed]
- Noomuna, P.; Risinger, M.; Zhou, S.; Seu, K.; Man, Y.; An, R.; Sheik, D.A.; Wan, J.; Little, J.A.; Gurkan, U.A.; et al. Inhibition of Band 3 tyrosine phosphorylation: A new mechanism for treatment of sickle cell disease. Br. J. Haematol. 2020, 190, 599–609. [Google Scholar] [CrossRef]
- Ferru, E.; Pantaleo, A.; Carta, F.; Mannu, F.; Khadjavi, A.; Gallo, V.; Ronzoni, L.; Graziadei, G.; Cappellini, M.D.; Turrini, F. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica 2014, 99, 570–578. [Google Scholar] [CrossRef]
- Baliga, S.; Chaudhary, M.; Bhat, S.; Bhansali, P.; Agrawal, A.; Gundawar, S. Estimation of malondialdehyde levels in serum and saliva of children affected with sickle cell anemia. J. Indian Soc. Pedod. Prev. Dent. 2018, 36, 43–47. [Google Scholar] [CrossRef]
- Shaklai, N.; Sharma, V.S.; Ranney, H.M. Interaction of sickle cell hemoglobin with erythrocyte membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Fabry, M.E.; Nagel, R.L.; Rifkind, J.M. Heme degradation and oxidative stress in murine models for hemoglobinopathies: Thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Mol. Dis. 2008, 41, 60–66. [Google Scholar] [CrossRef]
- Olatunya, O.S.; Lanaro, C.; Longhini, A.L.; Penteado, C.F.F.; Fertrin, K.Y.; Adekile, A.; Saad, S.T.O.; Costa, F.F. Red blood cells microparticles are associated with hemolysis markers and may contribute to clinical events among sickle cell disease patients. Ann. Hematol. 2019, 98, 2507–2521. [Google Scholar] [CrossRef]
- Villaescusa, R.; Arce, A.A.; Lalanne-Mistrih, M.L.; Lamarre, Y.; Hierso, R.; Hernandez, C.; Hardy-Dessources, M.D.; CAREST Study Group. Natural antiband 3 antibodies in patients with sickle cell disease. Comptes Rendus Biologies 2013, 336, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef]
- Vercellotti, G.M. Special delivery: Microparticles convey heme. Blood 2015, 125, 3677–3678. [Google Scholar] [CrossRef][Green Version]
- Awojoodu, A.O.; Keegan, P.M.; Lane, A.R.; Zhang, Y.; Lynch, K.R.; Platt, M.O.; Botchwey, E.A. Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood 2014, 124, 1941–1950. [Google Scholar] [CrossRef]
- Niki, E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef]
- Kuypers, F.A. Hemoglobin s polymerization and red cell membrane changes. Hematol. Oncol. Clin. N. Am. 2014, 28, 155–179. [Google Scholar] [CrossRef]
- Hannemann, A.; Rees, D.C.; Brewin, J.N.; Noe, A.; Low, B.; Gibson, J.S. Oxidative stress and phosphatidylserine exposure in red cells from patients with sickle cell anaemia. Br. J. Haematol. 2018, 182, 567–578. [Google Scholar] [CrossRef]
- Setty, B.N.; Kulkarni, S.; Stuart, M.J. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 2002, 99, 1564–1571. [Google Scholar] [CrossRef]
- Kuypers, F.A.; Styles, L.A. The role of secretory phospholipase A2 in acute chest syndrome. Cell Mol. Biol. 2004, 50, 87–94. [Google Scholar]
- Neidlinger, N.A.; Larkin, S.K.; Bhagat, A.; Victorino, G.P.; Kuypers, F.A. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J. Biol. Chem. 2006, 281, 775–781. [Google Scholar] [CrossRef]
- Butikofer, P.; Yee, M.C.; Schott, M.A.; Lubin, B.H.; Kuypers, F.A. Generation of phosphatidic acid during calcium-loading of human erythrocytes. Evidence for a phosphatidylcholine-hydrolyzing phospholipase D. Eur. J. Biochem. 1993, 213, 367–375. [Google Scholar] [CrossRef]
- Hsu, L.L.; Champion, H.C.; Campbell-Lee, S.A.; Bivalacqua, T.J.; Manci, E.A.; Diwan, B.A.; Schimel, D.M.; Cochard, A.E.; Wang, X.; Schechter, A.N.; et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 2007, 109, 3088–3098. [Google Scholar] [CrossRef]
- Taylor, J.G.t.; Nolan, V.G.; Mendelsohn, L.; Kato, G.J.; Gladwin, M.T.; Steinberg, M.H. Chronic hyper-hemolysis in sickle cell anemia: Association of vascular complications and mortality with less frequent vasoocclusive pain. PLoS ONE 2008, 3, e2095. [Google Scholar] [CrossRef]
- Pearson, H.A.; Gallagher, D.; Chilcote, R.; Sullivan, E.; Wilimas, J.; Espeland, M.; Ritchey, A.K. Developmental pattern of splenic dysfunction in sickle cell disorders. Pediatrics 1985, 76, 392–397. [Google Scholar]
- Potoka, K.P.; Gladwin, M.T. Vasculopathy and pulmonary hypertension in sickle cell disease. Am. J. Physiol. Lung Cell. Mol. Physiol 2015, 308, L314–L324. [Google Scholar] [CrossRef] [PubMed]
- van Beers, E.J.; Yang, Y.; Raghavachari, N.; Tian, X.; Allen, D.T.; Nichols, J.S.; Mendelsohn, L.; Nekhai, S.; Gordeuk, V.R.; Taylor, J.G.t.; et al. Iron, inflammation, and early death in adults with sickle cell disease. Circ. Res. 2015, 116, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Mahaseth, H.; Welch, T.E.; Otterbein, L.E.; Hebbel, R.P.; Vercellotti, G.M. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J. Clin. Investig. 2006, 116, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef]
- Klings, E.S.; Farber, H.W. Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respir. Res. 2001, 2, 280–285. [Google Scholar] [CrossRef]
- Alam, M.Z.; Devalaraja, S.; Haldar, M. The Heme Connection: Linking Erythrocytes and Macrophage Biology. Front. Immunol. 2017, 8, 33. [Google Scholar] [CrossRef]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef]
- Chaar, V.; Picot, J.; Renaud, O.; Bartolucci, P.; Nzouakou, R.; Bachir, D.; Galacteros, F.; Colin, Y.; Le Van Kim, C.; El Nemer, W. Aggregation of mononuclear and red blood cells through an {alpha}4{beta}1-Lu/basal cell adhesion molecule interaction in sickle cell disease. Haematologica 2010, 95, 1841–1848. [Google Scholar] [CrossRef]
- Okpala, I. The intriguing contribution of white blood cells to sickle cell disease—A red cell disorder. Blood Rev. 2004, 18, 65–73. [Google Scholar] [CrossRef]
- Baynes, J.W. Chemical modification of proteins by lipids in diabetes. Clin. Chem. Lab. Med. 2003, 41, 1159–1165. [Google Scholar] [CrossRef]
- Sugiyama, S.; Miyata, T.; Inagi, R.; Kurokawa, K. Implication of the glycoxidation and lipoxidation reactions in the pathogenesis of dialysis-related amyloidosis (Review). Int. J. Mol. Med. 1998, 2, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bekker, P.; Tsimikas, S. Advanced glycation end products and diabetic cardiovascular disease. Cardiol. Rev. 2012, 20, 177–183. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef]
- Pamplona, R. Mitochondrial DNA damage and animal longevity: Insights from comparative studies. J. Aging Res. 2011, 2011, 807108. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. S1), 3–27. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef]
- Hammes, H.P.; Brownlee, M.; Lin, J.; Schleicher, E.; Bretzel, R.G. Diabetic retinopathy risk correlates with intracellular concentrations of the glycoxidation product Nepsilon-(carboxymethyl) lysine independently of glycohaemoglobin concentrations. Diabetologia 1999, 42, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W. The role of AGEs in aging: Causation or correlation. Exp. Gerontol. 2001, 36, 1527–1537. [Google Scholar] [CrossRef]
- Brownlee, M. Negative consequences of glycation. Metabolism 2000, 49, 9–13. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J.; et al. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef]
- Spickett, C.M.; Pitt, A.R. Modification of proteins by reactive lipid oxidation products and biochemical effects of lipoxidation. Essays Biochem. 2020, 64, 19–31. [Google Scholar] [CrossRef]
- Wei, X.; Yin, H. Covalent modification of DNA by alpha, beta-unsaturated aldehydes derived from lipid peroxidation: Recent progress and challenges. Free Radic. Res. 2015, 49, 905–917. [Google Scholar] [CrossRef]
- Somjee, S.S.; Warrier, R.P.; Thomson, J.L.; Ory-Ascani, J.; Hempe, J.M. Advanced glycation end-products in sickle cell anaemia. Br. J. Haematol. 2005, 128, 112–118. [Google Scholar] [CrossRef]
- Nur, E.; Brandjes, D.P.; Schnog, J.J.; Otten, H.M.; Fijnvandraat, K.; Schalkwijk, C.G.; Biemond, B.J.; CURAMA Study Group. Plasma levels of advanced glycation end products are associated with haemolysis-related organ complications in sickle cell patients. Br. J. Haematol. 2010, 151, 62–69. [Google Scholar] [CrossRef]
- Safwat, N.A.; Kenny, M.A. Soluble receptor for advanced glycation end products as a vasculopathy biomarker in sickle cell disease. Pediatr. Res. 2018, 84, 869–874. [Google Scholar] [CrossRef]
- Zhang, F.; Banker, G.; Liu, X.; Suwanabol, P.A.; Lengfeld, J.; Yamanouchi, D.; Kent, K.C.; Liu, B. The novel function of advanced glycation end products in regulation of MMP-9 production. J. Surg. Res. 2011, 171, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef]
- Schmidt, A.M. Soluble RAGEs—Prospects for treating & tracking metabolic and inflammatory disease. Vascul. Pharmacol. 2015, 72, 1–8. [Google Scholar] [CrossRef]
- Essien, E.U. Increased susceptibility of erythrocyte membrane lipids to peroxidation in sickle cell disease. Cent. Afr. J. Med. 1994, 40, 217–220. [Google Scholar] [PubMed]
- Belcher, J.D.; Marker, P.H.; Geiger, P.; Girotti, A.W.; Steinberg, M.H.; Hebbel, R.P.; Vercellotti, G.M. Low-density lipoprotein susceptibility to oxidation and cytotoxicity to endothelium in sickle cell anemia. J. Lab. Clin. Med. 1999, 133, 605–612. [Google Scholar] [CrossRef]
- Klings, E.S.; Christman, B.W.; McClung, J.; Stucchi, A.F.; McMahon, L.; Brauer, M.; Farber, H.W. Increased F2 isoprostanes in the acute chest syndrome of sickle cell disease as a marker of oxidative stress. Am. J. Respir. Crit. Care Med. 2001, 164, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Detterich, J.A.; Liu, H.; Suriany, S.; Kato, R.M.; Chalacheva, P.; Tedla, B.; Shah, P.M.; Khoo, M.C.; Wood, J.C.; Coates, T.D.; et al. Erythrocyte and plasma oxidative stress appears to be compensated in patients with sickle cell disease during a period of relative health, despite the presence of known oxidative agents. Free Radic. Biol. Med. 2019, 141, 408–415. [Google Scholar] [CrossRef]
- Akohoue, S.A.; Shankar, S.; Milne, G.L.; Morrow, J.; Chen, K.Y.; Ajayi, W.U.; Buchowski, M.S. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr. Res. 2007, 61, 233–238. [Google Scholar] [CrossRef]
- Mockesch, B.; Connes, P.; Charlot, K.; Skinner, S.; Hardy-Dessources, M.D.; Romana, M.; Jumet, S.; Petras, M.; Divialle-Doumdo, L.; Martin, C.; et al. Association between oxidative stress and vascular reactivity in children with sickle cell anaemia and sickle haemoglobin C disease. Br. J. Haematol. 2017, 178, 468–475. [Google Scholar] [CrossRef]
- Yalamanoglu, A.; Deuel, J.W.; Hunt, R.C.; Baek, J.H.; Hassell, K.; Redinius, K.; Irwin, D.C.; Schaer, D.J.; Buehler, P.W. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L765–L774. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med. Sci. 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Reno, C.O.; Barbosa, A.R.; de Carvalho, S.S.; Pinheiro, M.B.; Rios, D.R.; Cortes, V.F.; Barbosa, L.A.; Santos, H.L. Oxidative stress assessment in sickle cell anemia patients treated with hydroxyurea. Ann. Hematol. 2020, 99, 937–945. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [CrossRef]
- Rusanova, I.; Escames, G.; Cossio, G.; de Borace, R.G.; Moreno, B.; Chahboune, M.; Lopez, L.C.; Diez, T.; Acuna-Castroviejo, D. Oxidative stress status, clinical outcome, and beta-globin gene cluster haplotypes in pediatric patients with sickle cell disease. Eur. J. Haematol. 2010, 85, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Chaves, N.A.; Alegria, T.G.P.; Dantas, L.S.; Netto, L.E.S.; Miyamoto, S.; Bonini Domingos, C.R.; da Silva, D.G.H. Impaired antioxidant capacity causes a disruption of metabolic homeostasis in sickle erythrocytes. Free Radic. Biol. Med. 2019, 141, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Manfredini, V.; Lazzaretti, L.L.; Griebeler, I.H.; Santin, A.P.; Brandao, V.D.; Wagner, S.; Castro, S.M.; Peralba Mdo, C.; Benfato, M.S. Blood antioxidant parameters in sickle cell anemia patients in steady state. J. Natl. Med. Assoc. 2008, 100, 897–902. [Google Scholar] [CrossRef]
- Cho, C.S.; Kato, G.J.; Yang, S.H.; Bae, S.W.; Lee, J.S.; Gladwin, M.T.; Rhee, S.G. Hydroxyurea-induced expression of glutathione peroxidase 1 in red blood cells of individuals with sickle cell anemia. Antioxid. Redox Signal. 2010, 13, 1–11. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Bindoli, A.; Rigobello, M.P. Principles in redox signaling: From chemistry to functional significance. Antioxid. Redox Signal. 2013, 18, 1557–1593. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid. Redox Signal. 2008, 10, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Zerez, C.R.; Lachant, N.A.; Lee, S.J.; Tanaka, K.R. Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease. Blood 1988, 71, 512–515. [Google Scholar] [CrossRef]
- Nur, E.; Brandjes, D.P.; Teerlink, T.; Otten, H.M.; Oude Elferink, R.P.; Muskiet, F.; Evers, L.M.; Ten Cate, H.; Biemond, B.J.; Duits, A.J.; et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann. Hematol. 2012, 91, 1097–1105. [Google Scholar] [CrossRef]
- Morris, C.R.; Suh, J.H.; Hagar, W.; Larkin, S.; Bland, D.A.; Steinberg, M.H.; Vichinsky, E.P.; Shigenaga, M.; Ames, B.; Kuypers, F.A.; et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 2008, 111, 402–410. [Google Scholar] [CrossRef]
- Darghouth, D.; Koehl, B.; Madalinski, G.; Heilier, J.F.; Bovee, P.; Xu, Y.; Olivier, M.F.; Bartolucci, P.; Benkerrou, M.; Pissard, S.; et al. Pathophysiology of sickle cell disease is mirrored by the red blood cell metabolome. Blood 2011, 117, e57–e66. [Google Scholar] [CrossRef]
- Rogers, S.C.; Ross, J.G.; d’Avignon, A.; Gibbons, L.B.; Gazit, V.; Hassan, M.N.; McLaughlin, D.; Griffin, S.; Neumayr, T.; Debaun, M.; et al. Sickle hemoglobin disturbs normal coupling among erythrocyte O2 content, glycolysis, and antioxidant capacity. Blood 2013, 121, 1651–1662. [Google Scholar] [CrossRef]
- Reid, M.; Badaloo, A.; Forrester, T.; Jahoor, F. In vivo rates of erythrocyte glutathione synthesis in adults with sickle cell disease. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E73–E79. [Google Scholar] [CrossRef]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef]
- Garel, M.C.; Domenget, C.; Caburi-Martin, J.; Prehu, C.; Galacteros, F.; Beuzard, Y. Covalent binding of glutathione to hemoglobin. I. Inhibition of hemoglobin S polymerization. J. Biol. Chem. 1986, 261, 14704–14709. [Google Scholar] [CrossRef]
- Moore, R.B.; Shriver, S.K.; Jenkins, L.D.; Mankad, V.N.; Shah, A.K.; Plishker, G.A. Calpromotin, a cytoplasmic protein, is associated with the formation of dense cells in sickle cell anemia. Am. J. Hematol. 1997, 56, 100–106. [Google Scholar] [CrossRef]
- Basu, A.; Saha, S.; Karmakar, S.; Chakravarty, S.; Banerjee, D.; Dash, B.P.; Chakrabarti, A. 2D DIGE based proteomics study of erythrocyte cytosol in sickle cell disease: Altered proteostasis and oxidative stress. Proteomics 2013, 13, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M.; Hampton, M.B.; Peskin, A.V.; Winterbourn, C.C. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 2007, 109, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; Low, P.S.; Turrini, F.; Bertoldi, M.; Campanella, M.E.; Spano, D.; Pantaleo, A.; Siciliano, A.; De Franceschi, L. Peroxiredoxin-2 expression is increased in beta-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free Radic. Biol. Med. 2010, 49, 457–466. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.U.; Kwon, T.H.; Lee, D.S.; Ha, H.L.; Park, D.S.; Woo, E.J.; Lee, S.H.; Kim, J.M.; Chae, H.B.; et al. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem. Biophys. Res. Commun. 2012, 426, 427–432. [Google Scholar] [CrossRef]
- Wicher, K.B.; Fries, E. Evolutionary aspects of hemoglobin scavengers. Antioxid. Redox Signal. 2010, 12, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Alayash, A.I. Haptoglobin: Old protein with new functions. Clin. Chim. Acta 2011, 412, 493–498. [Google Scholar] [CrossRef]
- Noyes, W.D.; Garby, L. Rate of haptoglobin in synthesis in normal man. Determinations by the return to normal levels following hemoglobin infusion. Scand. J. Clin. Lab. Investig. 1967, 20, 33–38. [Google Scholar] [CrossRef]
- Thomas, A.M.; Gerogianni, A.; McAdam, M.B.; Floisand, Y.; Lau, C.; Espevik, T.; Nilsson, P.H.; Mollnes, T.E.; Barratt-Due, A. Complement Component C5 and TLR Molecule CD14 Mediate Heme-Induced Thromboinflammation in Human Blood. J. Immunol. 2019, 203, 1571–1578. [Google Scholar] [CrossRef]
- Santiago, R.P.; Guarda, C.C.; Figueiredo, C.V.B.; Fiuza, L.M.; Aleluia, M.M.; Adanho, C.S.A.; Carvalho, M.O.S.; Pitanga, T.N.; Zanette, D.L.; Lyra, I.M.; et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol. Dis. 2018, 72, 34–36. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Chadebech, P.; Bodivit, G.; Vieira-Martins, P.; Grunenwald, A.; Boudhabhay, I.; Poillerat, V.; Pakdaman, S.; Kiger, L.; Jouard, A.; et al. Complement activation in sickle cell disease: Dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am. J. Hematol. 2020, 95, 456–464. [Google Scholar] [CrossRef]
- Ji, X.; Feng, Y.; Tian, H.; Meng, W.; Wang, W.; Liu, N.; Zhang, J.; Wang, L.; Wang, J.; Gao, H. The Mechanism of Proinflammatory HDL Generation in Sickle Cell Disease Is Linked to Cell-Free Hemoglobin via Haptoglobin. PLoS ONE 2016, 11, e0164264. [Google Scholar] [CrossRef]
- Vendrame, F.; Olops, L.; Saad, S.T.O.; Costa, F.F.; Fertrin, K.Y. Differences in heme and hemopexin content in lipoproteins from patients with sickle cell disease. J. Clin. Lipidol. 2018, 12, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef]
- Niihara, Y.; Macan, H.; Eckman, J.R.; Koh, H.; Cooper, M.L.; Ziegler, T.R.; Razon, R.; Tanaka, K.R.; Stark, C.W.; Johnson, C.S. L-Glutamine Therapy Reduces Hospitalization for Sickle Cell Anemia and Sickle β°-Thalassemia Patients at Six Months—A Phase II Randomized Trial. Clin. Pharmacol. Biopharm. 2014, 3, 116. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. A New Step in the Treatment of Sickle Cell DiseasePublished as part of the Biochemistry series “Biochemistry to Bedside”. Biochemistry 2018, 57, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.G.H.; Belini Junior, E.; de Almeida, E.A.; Bonini-Domingos, C.R. Oxidative stress in sickle cell disease: An overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic. Biol. Med. 2013, 65, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Zemel, B.S.; Kawchak, D.A.; Fung, E.B.; Ohene-Frempong, K.; Stallings, V.A. Effect of zinc supplementation on growth and body composition in children with sickle cell disease. Am. J. Clin. Nutr. 2002, 75, 300–307. [Google Scholar] [CrossRef]
- Bao, B.; Prasad, A.S.; Beck, F.W.; Snell, D.; Suneja, A.; Sarkar, F.H.; Doshi, N.; Fitzgerald, J.T.; Swerdlow, P. Zinc supplementation decreases oxidative stress, incidence of infection, and generation of inflammatory cytokines in sickle cell disease patients. Transl. Res. 2008, 152, 67–80. [Google Scholar] [CrossRef]
- Aboursheid, T.; Albaroudi, O.; Alahdab, F. Inhaled nitric oxide for treating pain crises in people with sickle cell disease. Cochrane Database Syst. Rev. 2019, 10, CD011808. [Google Scholar] [CrossRef]
- Dasgupta, T.; Hebbel, R.P.; Kaul, D.K. Protective effect of arginine on oxidative stress in transgenic sickle mouse models. Free Radic. Biol. Med. 2006, 41, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.K.; Zhang, X.; Dasgupta, T.; Fabry, M.E. Arginine therapy of transgenic-knockout sickle mice improves microvascular function by reducing non-nitric oxide vasodilators, hemolysis, and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H39–H47. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Vivar, J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108–1119. [Google Scholar] [CrossRef]
- Lal, A.; Suh, J.H.; Atamna, W.; Canty, B.; Hagar, W.; Vichinsky, E.F.; Ames, B. Anti-oxidant treatment with alipoic acid and acetyl L-carnitine in hemoglobinopathies. Blood 2007, 11, 3799. [Google Scholar] [CrossRef]
- Lal, A.; Atamna, W.; Killilea, D.W.; Suh, J.H.; Ames, B.N. Lipoic acid and acetyl-carnitine reverse iron-induced oxidative stress in human fibroblasts. Redox Rep. 2008, 13, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kaddam, L.; Fadl-Elmula, I.; Eisawi, O.A.; Abdelrazig, H.A.; Salih, M.A.; Lang, F.; Saeed, A.M. Gum Arabic as novel anti-oxidant agent in sickle cell anemia, phase II trial. BMC Hematol. 2017, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Kalish, B.T.; Matte, A.; Andolfo, I.; Iolascon, A.; Weinberg, O.; Ghigo, A.; Cimino, J.; Siciliano, A.; Hirsch, E.; Federti, E.; et al. Dietary omega-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica 2015, 100, 870–880. [Google Scholar] [CrossRef]
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Curcumin Attenuates Iron Accumulation and Oxidative Stress in the Liver and Spleen of Chronic Iron-Overloaded Rats. PLoS ONE 2015, 10, e0134156. [Google Scholar] [CrossRef] [PubMed]
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Correction: Curcumin attenuates iron accumulation and oxidative stress in the liver and spleen of chronic iron-overloaded rats. PLoS ONE 2020, 15, e0243398. [Google Scholar] [CrossRef] [PubMed]
- Belini Junior, E.; da Silva, D.G.; Torres Lde, S.; de Almeida, E.A.; Cancado, R.D.; Chiattone, C.; Bonini-Domingos, C.R. Oxidative stress and antioxidant capacity in sickle cell anaemia patients receiving different treatments and medications for different periods of time. Ann. Hematol. 2012, 91, 479–489. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. https://doi.org/10.3390/antiox10020296
Vona R, Sposi NM, Mattia L, Gambardella L, Straface E, Pietraforte D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants. 2021; 10(2):296. https://doi.org/10.3390/antiox10020296
Chicago/Turabian StyleVona, Rosa, Nadia Maria Sposi, Lorenza Mattia, Lucrezia Gambardella, Elisabetta Straface, and Donatella Pietraforte. 2021. "Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy" Antioxidants 10, no. 2: 296. https://doi.org/10.3390/antiox10020296
APA StyleVona, R., Sposi, N. M., Mattia, L., Gambardella, L., Straface, E., & Pietraforte, D. (2021). Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants, 10(2), 296. https://doi.org/10.3390/antiox10020296