
Genistein Prevents Nitric Oxide Deficiency-Induced Cardiac Dysfunction and Remodeling in Rats

 , and
, and
Abstract
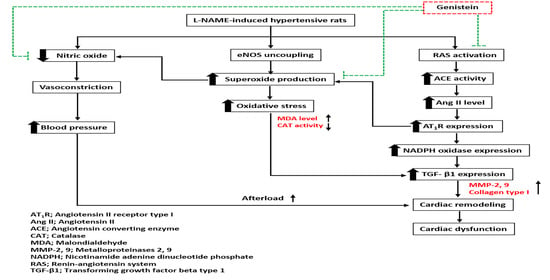
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Experimental Protocols
2.4. Indirect Measurement of Blood Pressure in Conscious Rats
2.5. Echocardiography
2.6. Hemodynamic Parameters Measurement
2.7. Immunofluorescence in Cardiac Tissue
2.8. Biochemical Measurements
2.8.1. Assays of Serum Angiotensin-Converting Enzyme Activity
2.8.2. Assay of Plasma Angiotensin II Concentration
2.9. Measurement of Oxidative Stress Markers
2.9.1. Assay of Vascular Superoxide (O2•−) Production
2.9.2. Assay of Plasma and Cardiac Malondialdehyde Levels
2.9.3. Assays of Serum and Cardiac Catalase Activities
2.10. Assay of Plasma Nitrate/Nitrite Concentrations
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
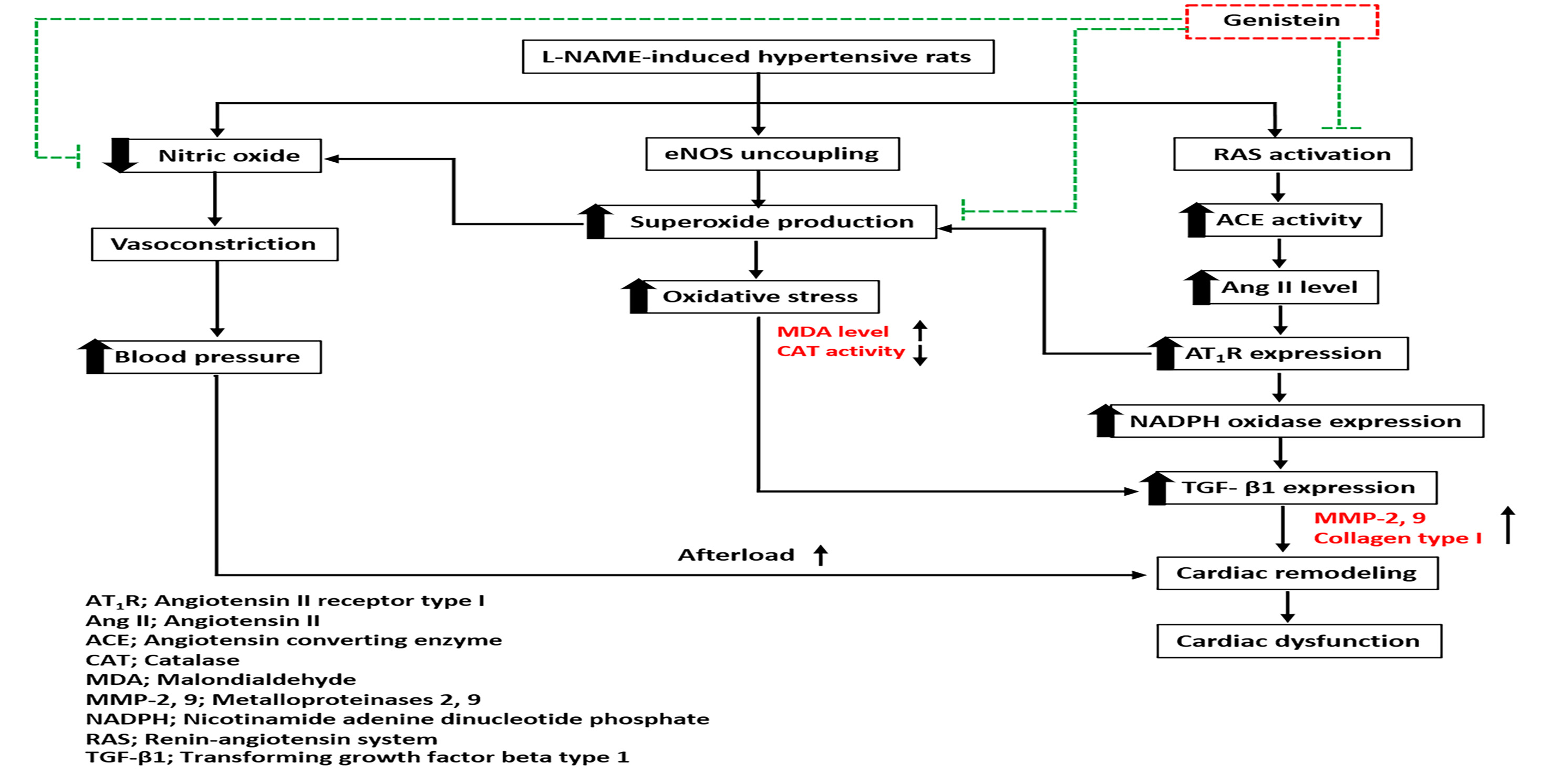
3.1. Effect of Genistein on Systolic Blood Pressure in a Weekly Context
3.2. Effect of Genistein on Heart Weights
3.3. Effect of Genistein on Hemodynamic Parameters
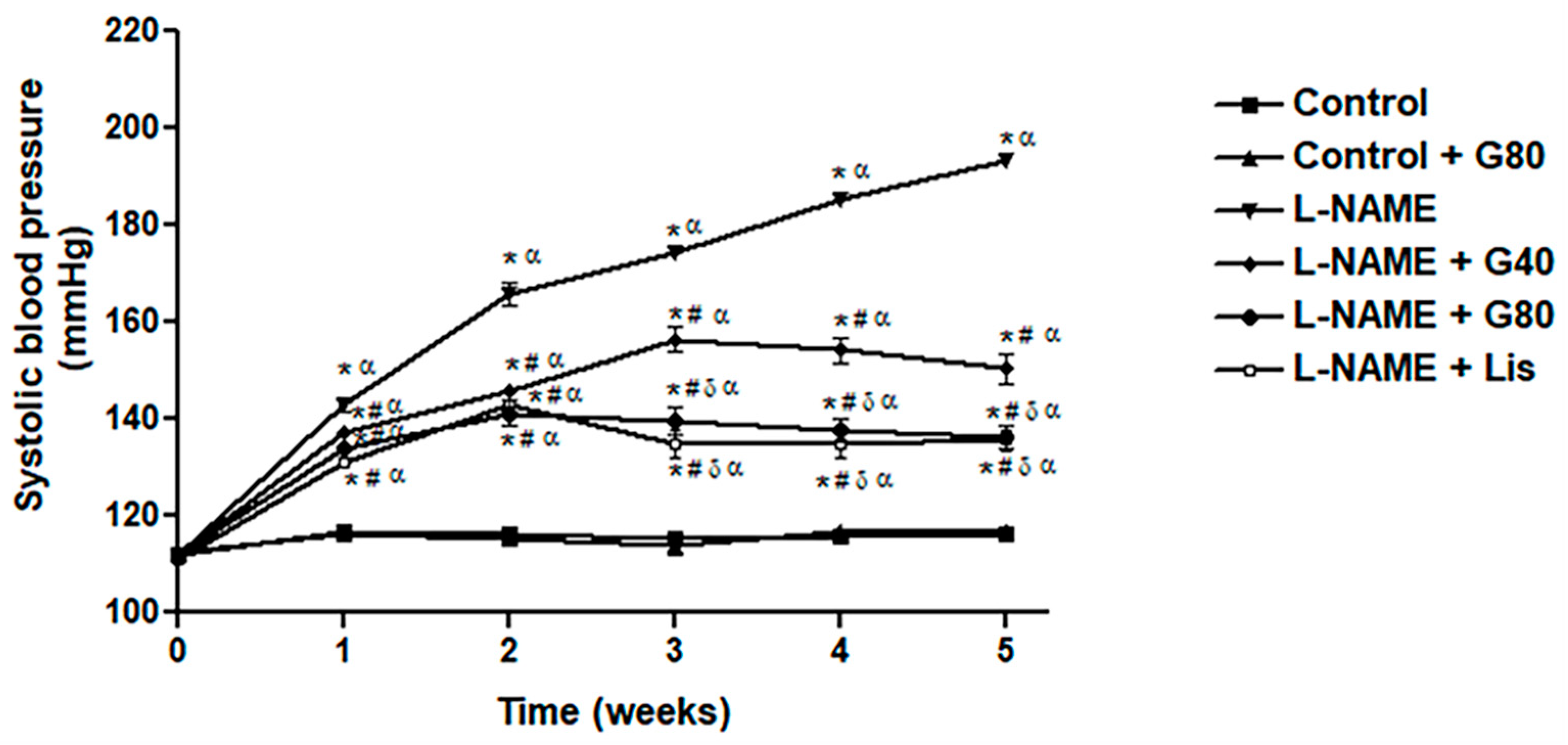
3.4. Effects of Genistein on Cardiac Function
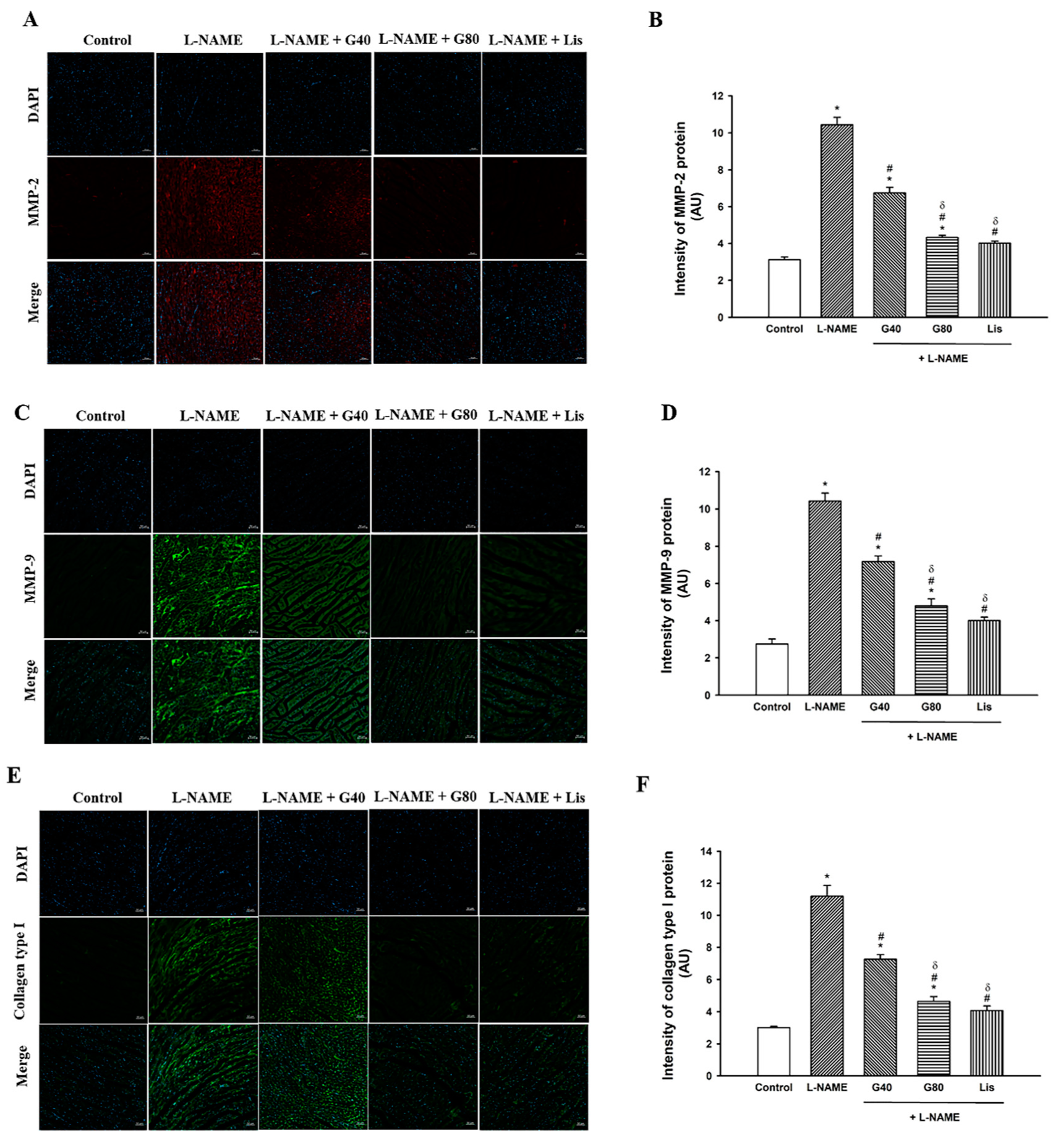
3.5. Effect of Genistein on MMP-2, MMP-9, and Collagen Type I Protein Expression in the Heart
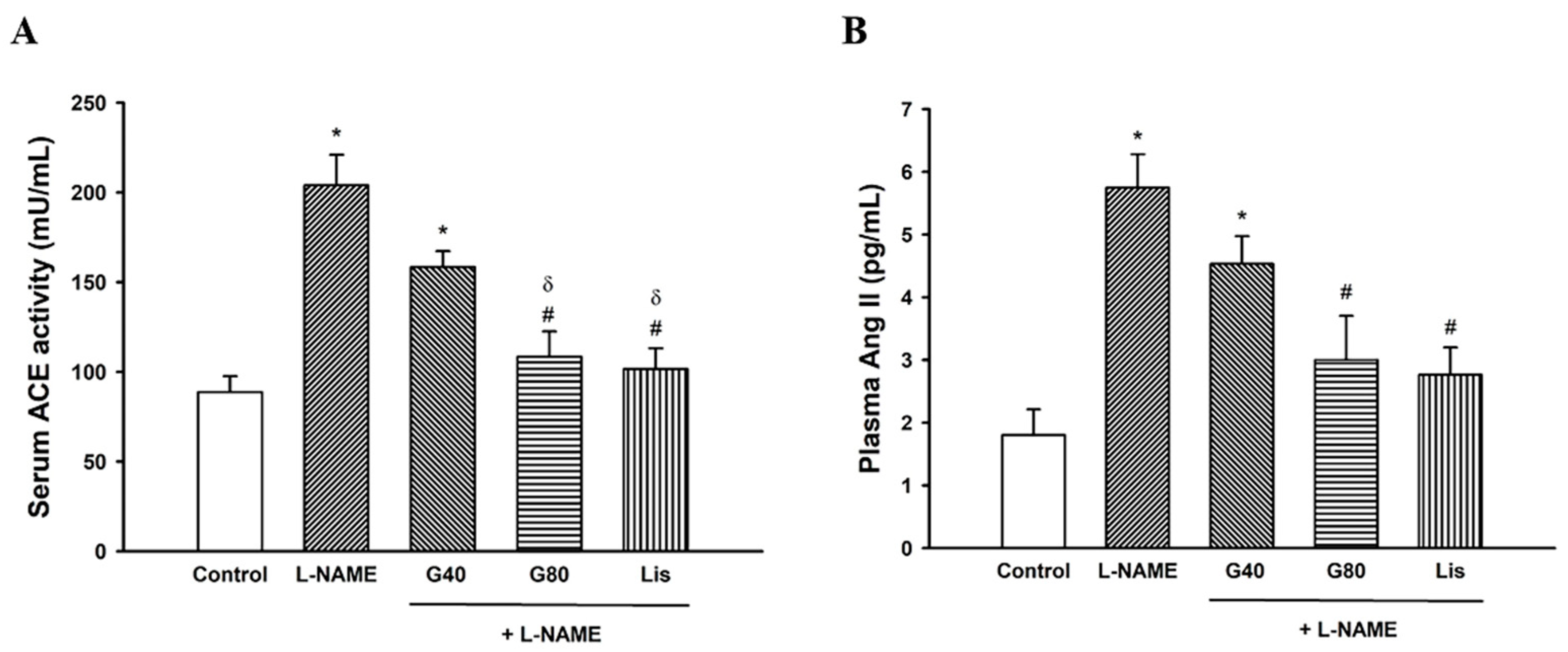
3.6. Effects of Genistein on Serum ACE Activity and Plasma Ang II Levels
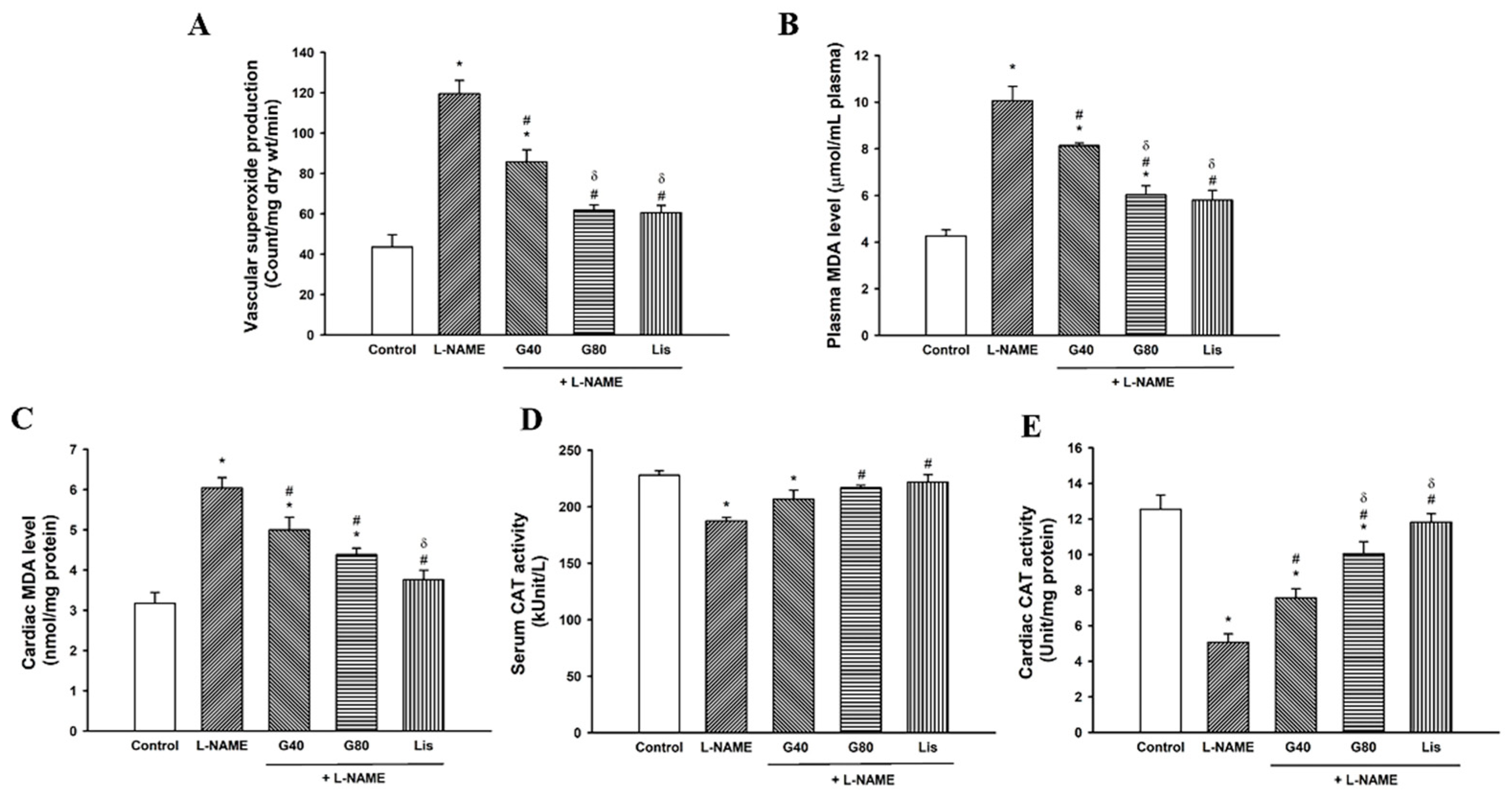
3.7. Effects of Genistein on Oxidative Stress Markers
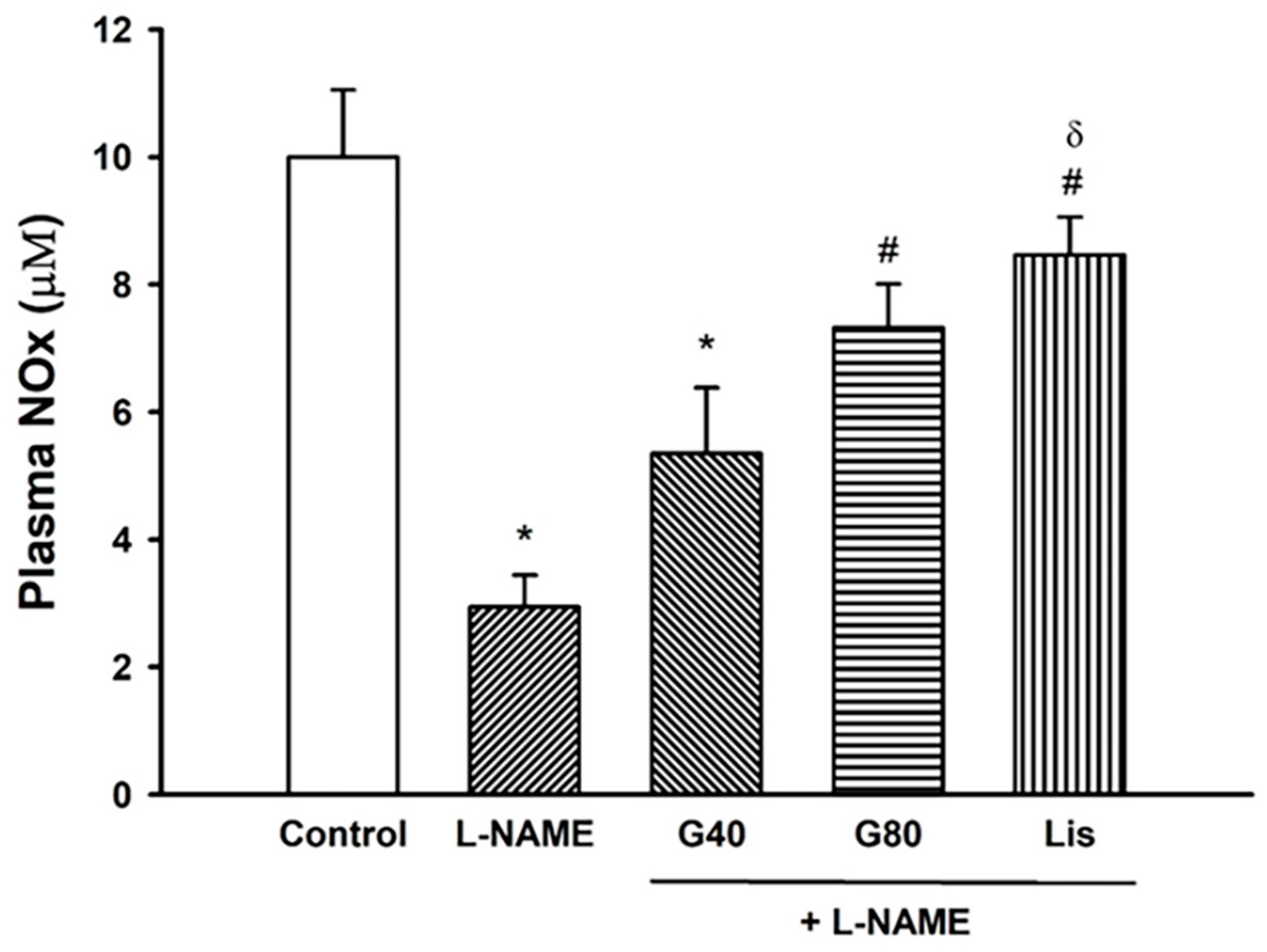
3.8. Effect of Genistein on Plasma NOx Concentration
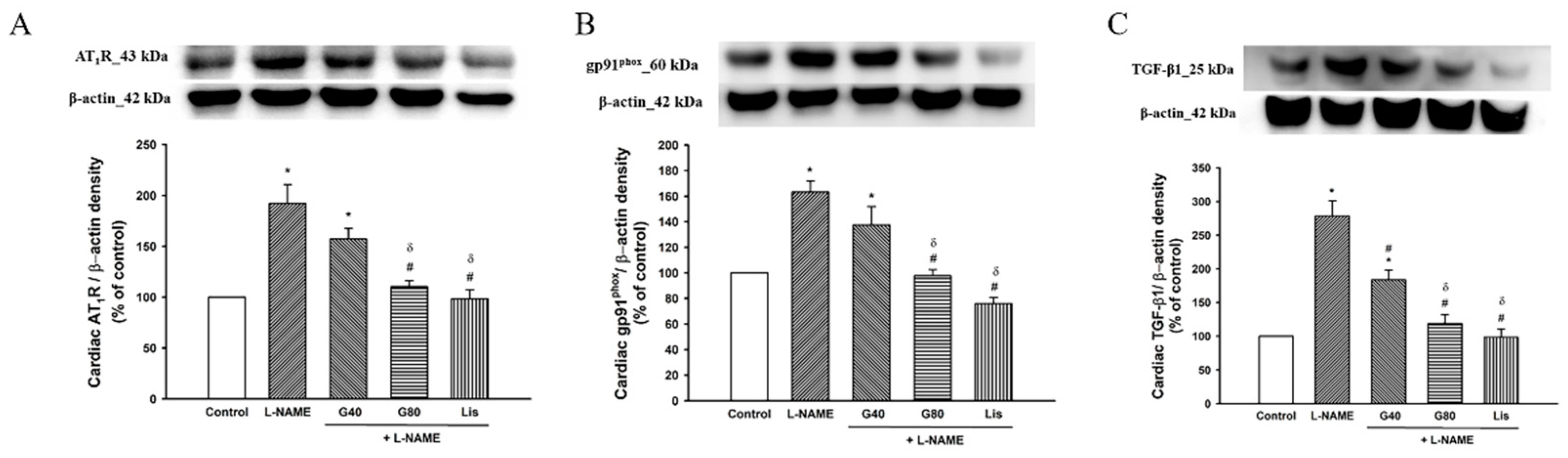
3.9. Effect of Genistein on AT1R, gp91phox, and TGF-β1 Protein Expressions in Cardiac Tissue
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Ganau, A.; Devereux, R.B.; Roman, M.J.; de Simone, G.; Pickering, T.G.; Saba, P.S.; Vargiu, P.; Simongini, I.; Laragh, J.H. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J. Am. Coll. Cardiol. 1992, 19, 1550–1558. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kramer, D.G.; Patel, A.R.; Maron, M.S.; Udelson, J.E. Left ventricular remodeling in heart failure: Current concepts in clinical significance and assessment. JACC Cardiovasc. Imaging 2011, 4, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Pechánová, O.; Bernátová, I.; Pelouch, V.; Babál, P. L-NAME-induced protein remodeling and fibrosis in the rat heart. Physiol. Res. 1999, 48, 353–362. [Google Scholar]
- Bernátová, I.; Pechánová, O.; Pelouch, V.; Simko, F. Regression of chronic L -NAME-treatment-induced left ventricular hypertrophy: Effect of captopril. J. Mol. Cell Cardiol. 2000, 32, 177–185. [Google Scholar] [CrossRef]
- Bunbupha, S.; Prachaney, P.; Kukongviriyapan, U.; Kukongviriyapan, V.; Welbat, J.U.; Pakdeechote, P. Asiatic acid alleviates cardiovascular remodelling in rats with L-NAME-induced hypertension. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1189–1197. [Google Scholar] [CrossRef]
- Kumar, S.; Prahalathan, P.; Raja, B. Vanillic acid: A potential inhibitor of cardiac and aortic wall remodeling in l-NAME induced hypertension through upregulation of endothelial nitric oxide synthase. Environ. Toxicol. Pharmacol. 2014, 38, 643–652. [Google Scholar] [CrossRef]
- Wunpathe, C.; Maneesai, P.; Rattanakanokchai, S.; Bunbupha, S.; Kukongviriyapan, U.; Tong-Un, T.; Pakdeechote, P. Tangeretin mitigates l-NAME-induced ventricular dysfunction and remodeling through the AT(1)R/pERK1/2/pJNK signaling pathway in rats. Food Funct. 2020, 11, 1322–1333. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, P.; Ramiro-Cortijo, D.; Reyes-Hernández, C.G.; López de Pablo, A.L.; González, M.C.; Arribas, S.M. Implication of Oxidative Stress in Fetal Programming of Cardiovascular Disease. Front. Physiol. 2018, 9, 602. [Google Scholar] [CrossRef]
- Mir, S.A.; Chatterjee, A.; Mitra, A.; Pathak, K.; Mahata, S.K.; Sarkar, S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem 2012, 287, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Maneesai, P.; Bunbupha, S.; Potue, P.; Berkban, T.; Kukongviriyapan, U.; Kukongviriyapan, V.; Prachaney, P.; Pakdeechote, P. Hesperidin Prevents Nitric Oxide Deficiency-Induced Cardiovascular Remodeling in Rats via Suppressing TGF-β1 and MMPs Protein Expression. Nutrients 2018, 10, 1549. [Google Scholar] [CrossRef]
- Yamazaki, T.; Komuro, I.; Yazaki, Y. Role of the renin-angiotensin system in cardiac hypertrophy. Am. J. Cardiol. 1999, 83, 53h–57h. [Google Scholar] [CrossRef]
- Zanchi, A.; Schaad, N.C.; Osterheld, M.C.; Grouzmann, E.; Nussberger, J.; Brunner, H.R.; Waeber, B. Effects of chronic NO synthase inhibition in rats on renin-angiotensin system and sympathetic nervous system. Am. J. Physiol. 1995, 268, H2267–H2273. [Google Scholar] [CrossRef] [PubMed]
- Rincón, J.; Correia, D.; Arcaya, J.L.; Finol, E.; Fernández, A.; Pérez, M.; Yaguas, K.; Talavera, E.; Chávez, M.; Summer, R.; et al. Role of Angiotensin II type 1 receptor on renal NAD(P)H oxidase, oxidative stress and inflammation in nitric oxide inhibition induced-hypertension. Life Sci. 2015, 124, 81–90. [Google Scholar] [CrossRef]
- Sonoda, K.; Ohtake, K.; Uchida, H.; Ito, J.; Uchida, M.; Natsume, H.; Tamada, H.; Kobayashi, J. Dietary nitrite supplementation attenuates cardiac remodeling in l-NAME-induced hypertensive rats. Nitric Oxide 2017, 67, 1–9. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Rababa’h, A.M.; Guillory, A.N.; Mustafa, R.; Hijjawi, T. Oxidative Stress and Cardiac Remodeling: An Updated Edge. Curr. Cardiol. Rev. 2018, 14, 53–59. [Google Scholar] [CrossRef]
- Pedro-Botet, J.; Covas, M.I.; Martín, S.; Rubiés-Prat, J. Decreased endogenous antioxidant enzymatic status in essential hypertension. J. Hum. Hypertens. 2000, 14, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Olivieri, O.; Girelli, D.; Faccini, G.; Zenari, M.L.; Lombardi, S.; Corrocher, R. Anti-oxidant status and lipid peroxidation in patients with essential hypertension. J. Hypertens. 1998, 16, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Chiangsaen, P.; Maneesai, P.; Kukongviriyapan, U.; Tong-Un, T.; Ishida, W.; Prachaney, P.; Pakdeechote, P. Tangeretin ameliorates erectile and testicular dysfunction in a rat model of hypertension. Reprod. Toxicol. 2020, 96, 1–10. [Google Scholar] [CrossRef]
- Maneesai, P.; Bunbupha, S.; Kukongviriyapan, U.; Senggunprai, L.; Kukongviriyapan, V.; Prachaney, P.; Pakdeechote, P. Effect of asiatic acid on the Ang II-AT1R-NADPH oxidase-NF-κB pathway in renovascular hypertensive rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 1073–1083. [Google Scholar] [CrossRef]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef]
- Deschamps, A.M.; Spinale, F.G. Pathways of matrix metalloproteinase induction in heart failure: Bioactive molecules and transcriptional regulation. Cardiovasc. Res. 2006, 69, 666–676. [Google Scholar] [CrossRef]
- Cau, S.B.; Guimaraes, D.A.; Rizzi, E.; Ceron, C.S.; Gerlach, R.F.; Tanus-Santos, J.E. The Nuclear Factor kappaB Inhibitor Pyrrolidine Dithiocarbamate Prevents Cardiac Remodelling and Matrix Metalloproteinase-2 Up-Regulation in Renovascular Hypertension. Basic Clin. Pharmacol. Toxicol. 2015, 117, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Luchtefeld, M.; Grote, K.; Grothusen, C.; Bley, S.; Bandlow, N.; Selle, T.; Struber, M.; Haverich, A.; Bavendiek, U.; Drexler, H.; et al. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem. Biophys. Res. Commun. 2005, 328, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, E.; Castro, M.M.; Ceron, C.S.; Neto-Neves, E.M.; Prado, C.M.; Rossi, M.A.; Tanus-Santos, J.E.; Gerlach, R.F. Tempol inhibits TGF-beta and MMPs upregulation and prevents cardiac hypertensive changes. Int. J. Cardiol. 2013, 165, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S. TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef]
- Wenzel, S.; Taimor, G.; Piper, H.M.; Schlüter, K.D. Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-beta expression in adult ventricular cardiomyocytes. FASEB J. 2001, 15, 2291–2293. [Google Scholar] [CrossRef]
- Liu, X.; Shan, X.; Chen, H.; Li, Z.; Zhao, P.; Zhang, C.; Guo, W.; Xu, M.; Lu, R. Stachydrine Ameliorates Cardiac Fibrosis Through Inhibition of Angiotensin II/Transformation Growth Factor β1 Fibrogenic Axis. Front. Pharmacol. 2019, 10, 538. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Díez, J. Targeting the Cardiac Myofibroblast Secretome to Treat Myocardial Fibrosis in Heart Failure. Circ. Heart Fail. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Chrysant, S.G. Antihypertensive effectiveness of low-dose lisinopril-hydrochlorothiazide combination. A large multicenter study. Lisinopril-Hydrochlorothiazide Group. Arch. Intern. Med. 1994, 154, 737–743. [Google Scholar] [CrossRef]
- Weinberger, M.H. Angiotensin converting enzyme inhibitors enhance the antihypertensive efficacy of diuretics and blunt or prevent adverse metabolic effects. J. Cardiovasc. Pharmacol. 1989, 13 (Suppl. 3), S1–S4. [Google Scholar] [CrossRef]
- Messerli, F.H.; Kaesser, U.R. Lisinopril in the treatment of hypertension. J. Hum. Hypertens 1989, 3 (Suppl. 1), 17–21. [Google Scholar]
- Saber, S.; Goda, R.; El-Tanbouly, G.S.; Ezzat, D. Lisinopril inhibits nuclear transcription factor kappa B and augments sensitivity to silymarin in experimental liver fibrosis. Int. ImmunoPharmacol. 2018, 64, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Messina, M. A brief historical overview of the past two decades of soy and isoflavone research. J. Nutr. 2010, 140, 1350s–1354s. [Google Scholar] [CrossRef] [PubMed]
- GolKhoo, S.; Ahmadi, A.R.; Hanachi, P.; Barantalab, F.; Vaziri, M. Determination of daidzein and genistein in soy milk in Iran by using HPLC analysis method. Pak. J. Biol. Sci. 2008, 11, 2254–2258. [Google Scholar] [CrossRef]
- Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe, S.; Itoh, N.; Shibuya, M.; Fukami, Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987, 262, 5592–5595. [Google Scholar] [CrossRef]
- Ruiz-Larrea, M.B.; Mohan, A.R.; Paganga, G.; Miller, N.J.; Bolwell, G.P.; Rice-Evans, C.A. Antioxidant activity of phytoestrogenic isoflavones. Free Radic. Res. 1997, 26, 63–70. [Google Scholar] [CrossRef]
- Wei, H.; Wei, L.; Frenkel, K.; Bowen, R.; Barnes, S. Inhibition of tumor promoter-induced hydrogen peroxide formation in vitro and in vivo by genistein. Nutr. Cancer 1993, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Tao, L.; Liu, H.; Christopher, T.A.; Lopez, B.L.; Ma, X.L. Role of ERK1/2 in the anti-apoptotic and cardioprotective effects of nitric oxide after myocardial ischemia and reperfusion. Apoptosis 2006, 11, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Jia, Q. Genistein ameliorates inflammation and insulin resistance through mediation of gut microbiota composition in type 2 diabetic mice. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Yang, R.; Liu, X.F.; Ma, S.F.; Wang, L. Genistein attenuates renal fibrosis in streptozotocin-induced diabetic rats. Mol. Med. Rep. 2019, 19, 423–431. [Google Scholar] [CrossRef]
- Jia, Q.; Yang, R.; Liu, X.F.; Ma, S.F. Protective Effects of Genistein on Myocardial Injury in Diabetic Rats. Sichuan Da Xue Xue Bao Yi Xue Ban 2018, 49, 706–711. [Google Scholar]
- Kondo, K.; Suzuki, Y.; Ikeda, Y.; Umemura, K. Genistein, an isoflavone included in soy, inhibits thrombotic vessel occlusion in the mouse femoral artery and in vitro platelet aggregation. Eur. J. Pharmacol. 2002, 455, 53–57. [Google Scholar] [CrossRef]
- Kokubo, Y.; Iso, H.; Ishihara, J.; Okada, K.; Inoue, M.; Tsugane, S. Association of dietary intake of soy, beans, and isoflavones with risk of cerebral and myocardial infarctions in Japanese populations: The Japan Public Health Center-based (JPHC) study cohort I. Circulation 2007, 116, 2553–2562. [Google Scholar] [CrossRef]
- Singh, S.; Dwivedi, R.; Chaturvedi, V. Influence of Vehicles Used for Oral Dosing of Test Molecules on the Progression of Mycobacterium tuberculosis Infection in Mice. Antimicrob. Agents Chemother. 2012, 56, 6026–6028. [Google Scholar] [CrossRef]
- Givvimani, S.; Narayanan, N.; Pushpakumar, S.B.; Tyagi, S.C. Anti-Parstatin Promotes Angiogenesis and Ameliorates Left Ventricular Dysfunction during Pressure Overload. Int. J. Biomed. Sci. 2014, 10, 1–7. [Google Scholar]
- Lu, F.J.; Lin, J.T.; Wang, H.P.; Huang, W.C. A simple, sensitive, non-stimulated photon counting system for detection of superoxide anion in whole blood. Experientia 1996, 52, 141–144. [Google Scholar] [CrossRef]
- Joukar, S.; Najafipour, H.; Haddad, M.; Sepehri, G.; Shahrokhi, N.; Dabiri, S.; Gholamhoseinian, A.; Hasanzadeh, S. The Effect of Saffron Consumption on Biochemical and Histopathological Heart Indices of Rats with Myocardial Infarction. Cardiovasc. Toxicol. 2010, 10, 66–71. [Google Scholar] [CrossRef]
- Nakmareong, S.; Kukongviriyapan, U.; Pakdeechote, P.; Donpunha, W.; Kukongviriyapan, V.; Kongyingyoes, B.; Sompamit, K.; Phisalaphong, C. Antioxidant and vascular protective effects of curcumin and tetrahydrocurcumin in rats with L-NAME-induced hypertension. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 519–529. [Google Scholar] [CrossRef]
- Góth, L. A simple method for determination of serum catalase activity and revision of reference range. Clin. Chim. Acta. 1991, 196, 143–151. [Google Scholar] [CrossRef]
- Ming, M.; Guanhua, L.; Zhanhai, Y.; Guang, C.; Xuan, Z. Effect of the Lycium barbarum polysaccharides administration on blood lipid metabolism and oxidative stress of mice fed high-fat diet in vivo. Food Chem. 2009, 113, 872–877. [Google Scholar] [CrossRef]
- Pakdeechote, P.; Bunbupha, S.; Kukongviriyapan, U.; Prachaney, P.; Khrisanapant, W.; Kukongviriyapan, V. Asiatic acid alleviates hemodynamic and metabolic alterations via restoring eNOS/iNOS expression, oxidative stress, and inflammation in diet-induced metabolic syndrome rats. Nutrients 2014, 6, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Bank, N.; Aynedjian, H.S.; Khan, G.A. Mechanism of vasoconstriction induced by chronic inhibition of nitric oxide in rats. Hypertension 1994, 24, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, H.; Kihara, Y.; Iwanaga, Y.; Onozawa, Y.; Toyokuni, S.; Kita, T. Angiotensin II, oxidative stress, and extracellular matrix degradation during transition to LV failure in rats with hypertension. J. Mol. Cell Cardiol. 2006, 41, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Vellaichamy, E.; Khurana, M.L.; Fink, J.; Pandey, K.N. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J. Biol. Chem. 2005, 280, 19230–19242. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991, 83, 1849–1865. [Google Scholar] [CrossRef]
- Biwer, L.A.; Broderick, T.L.; Xu, H.; Carroll, C.; Hale, T.M. Protection against L-NAME-induced reduction in cardiac output persists even after cessation of angiotensin-converting enzyme inhibitor treatment. Acta Physiol. (Oxf.) 2013, 207, 156–165. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Manabe, I.; Shindo, T.; Nagai, R. Gene expression in fibroblasts and fibrosis: Involvement in cardiac hypertrophy. Circ. Res. 2002, 91, 1103–1113. [Google Scholar] [CrossRef]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef] [PubMed]
- Bunbupha, S.; Pakdeechote, P.; Kukongviriyapan, U.; Prachaney, P.; Kukongviriyapan, V. Asiatic acid reduces blood pressure by enhancing nitric oxide bioavailability with modulation of eNOS and p47phox expression in L-NAME-induced hypertensive rats. Phytother. Res. 2014, 28, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Berkban, T.; Boonprom, P.; Bunbupha, S.; Welbat, J.U.; Kukongviriyapan, U.; Kukongviriyapan, V.; Pakdeechote, P.; Prachaney, P. Ellagic Acid Prevents L-NAME-Induced Hypertension via Restoration of eNOS and p47phox Expression in Rats. Nutrients 2015, 7, 5265–5280. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef]
- Godos, J.; Bergante, S.; Satriano, A.; Pluchinotta, F.R.; Marranzano, M. Dietary Phytoestrogen Intake is Inversely Associated with Hypertension in a Cohort of Adults Living in the Mediterranean Area. Molecules 2018, 23, 368. [Google Scholar] [CrossRef]
- Al-Nakkash, L.; Markus, B.; Bowden, K.; Batia, L.M.; Prozialeck, W.C.; Broderick, T.L. Effects of acute and 2-day genistein treatment on cardiac function and ischemic tolerance in ovariectomized rats. Gend. Med. 2009, 6, 488–497. [Google Scholar] [CrossRef]
- Liew, R.; Macleod, K.T.; Collins, P. Novel stimulatory actions of the phytoestrogen genistein: Effects on the gain of cardiac excitation-contraction coupling. FASEB J. 2003, 17, 1307–1309. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Yang, C.; Li, S.N. Effects of genistein on angiotensin-converting enzyme in rats. Life Sci. 2006, 79, 828–837. [Google Scholar] [CrossRef]
- Palanisamy, N.; Venkataraman, A.C. Beneficial effect of genistein on lowering blood pressure and kidney toxicity in fructose-fed hypertensive rats. Br. J. Nutr. 2013, 109, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Sanches Silva, A.; Sánchez-Machado, D.I.; López-Cervantes, J.; Daglia, M.; Nabavi, S.F.; Nabavi, S.M. Hypotensive effects of genistein: From chemistry to medicine. Chem. Biol. Interact. 2017, 268, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.W.; Ikeda, K.; Yamori, Y. Genistein inhibits expressions of NADPH oxidase p22phox and angiotensin II type 1 receptor in aortic endothelial cells from stroke-prone spontaneously hypertensive rats. Hypertens. Res. 2004, 27, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.Q.; Zhang, J.; Ramires, F.J. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J. Mol. Cell Cardiol. 1998, 30, 1559–1569. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Mishra, S.K.; Abbot, S.E.; Choudhury, Z.; Cheng, M.; Khatab, N.; Maycock, N.J.; Zavery, A.; Aaronson, P.I. Endothelium-dependent relaxation of rat aorta and main pulmonary artery by the phytoestrogens genistein and daidzein. Cardiovasc. Res. 2000, 46, 539–546. [Google Scholar] [CrossRef]
- Thind, G.S. Angiotensin converting enzyme inhibitors: Comparative structure, pharmacokinetics, and pharmacodynamics. Cardiovasc. Drugs Ther. 1990, 4, 199–206. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | Control + G80 | L-NAME | L-NAME + G40 | L-NAME + G80 | L-NAME + Lis |
|---|---|---|---|---|---|---|
| BW (g) | 380.13 ± 18.1 | 385 ± 12.26 | 382.88 ± 7.3 | 379.00 ± 10.3 | 372.46 ± 11.1 | 369.63 ± 7.6 |
| HW (g) | 1.02 ± 0.04 | 1.02 ± 0.01 | 1.14 ± 0.02 *α | 1.04 ± 0.02 | 1.02 ± 0.02 # | 0.91 ± 0.02 #δ |
| VW (g) | 0.89 ± 0.03 | 0.90 ± 0.02 | 1.02 ± 0.03 *α | 0.93 ± 0.03 | 0.92 ± 0.02 # | 0.83 ± 0.01 #δ |
| HW/BW (g/g) | 0.26 ± 0.00 | 0.27 ± 0.01 | 0.30 ± 0.01 *α | 0.28 ± 0.01 | 0.28 ± 0.01 | 0.26 ± 0.01 # |
| VW/BW (g/g) | 0.24 ± 0.00 | 0.24 ± 0.01 | 0.28 ± 0.01 *α | 0.26 ± 0.01 | 0.26 ± 0.01 | 0.24 ± 0.01 # |
| LVW/BW (g/g) | 0.18 ± 0.004 | 0.19 ± 0.00 | 0.22 ± 0.01 *α | 0.21 ± 0.01 * | 0.20 ± 0.01 | 0.19 ± 0.01 # |
| Parameters | Control | Control + G80 | L-NAME | L-NAME + G40 | L-NAME + G80 | L-NAME + Lis |
|---|---|---|---|---|---|---|
| SP (mmHg) | 122.14 ± 2.48 | 121.57 ± 2.62 | 202.96 ± 3.96 *α | 165.03 ± 2.17 *#α | 149.34 ± 3.76 *#δα | 134.77 ± 4.93 #δ |
| DP (mmHg) | 81.70 ± 3.66 | 77.99 ± 1.89 | 149.66 ± 5.26 *α | 114.65 ± 3.26 *#α | 102.52 ± 4.72 *#α | 90.65 ± 5.22 #δ |
| MAP (mmHg) | 94.97 ± 2.50 | 92.52 ± 2.09 | 168.11 ± 2.91 *α | 131.50 ± 2.23 *#α | 117.94 ± 2.87 *#δα | 106.36 ± 3.74 #δα |
| PP (mmHg) | 40.28 ± 0.60 | 43.58 ± 1.22 | 56.88 ± 2.73 *α | 50.72 ±1.53 * | 46.18 ± 1.60 # | 42.09 ± 1.95 #δ |
| HR (beats/min) | 356.05 ± 5.56 | 355.92 ± 8.34 | 442.24 ± 11.1 *α | 383.00 ± 10.4 # | 372.85 ± 10.9 # | 374.38 ± 6.16 # |
| Parameters | Control | L-NAME | L-NAME + G40 | L-NAME + G80 | L-NAME + Lis |
|---|---|---|---|---|---|
| IVSd (cm) | 0.17 ± 0.01 | 0.21 ± 0.01 * | 0.17 ± 0.01 # | 0.15 ± 0.01 # | 0.14 ± 0.01 # |
| IVSs (cm) | 0.25 ± 0.01 | 0.29 ± 0.01 * | 0.25 ± 0.01 # | 0.24 ± 0.01 # | 0.23 ± 0.01 # |
| LVIDd (cm) | 0.66 ± 0.02 | 0.57 ± 0.01 * | 0.63 ± 0.01 # | 0.64 ± 0.02 # | 0.65 ± 0.02 # |
| LVIDs (cm) | 0.38 ± 0.01 | 0.33 ± 0.01 * | 0.35 ± 0.01 | 0.38 ± 0.01 # | 0.40 ± 0.01 # |
| LVPWd (cm) | 0.19 ± 0.00 | 0.24 ± 0.01 * | 0.19 ± 0.00 # | 0.19 ± 0.01 # | 0.17 ± 0.00 # |
| LVPWs (cm) | 0.27 ± 0.00 | 0.26 ± 0.01 | 0.27 ± 0.00 | 0.26 ± 0.01 | 0.25 ± 0.01 |
| EDV (mL) | 0.71 ± 0.04 | 0.47 ± 0.02 * | 0.55 ± 0.03 * | 0.60 ± 0.03 # | 0.63 ± 0.04 # |
| ESV (mL) | 0.15 ± 0.01 | 0.14 ± 0.01 | 0.13 ± 0.01 | 0.15 ± 0.02 | 0.16 ± 0.01 |
| SV (mL) | 0.56 ± 0.03 | 0.33 ± 0.02 * | 0.42 ± 0.03 * | 0.45 ± 0.02 *# | 0.47 ± 0.03 # |
| EF (%) | 80.80 ± 0.45 | 70.15 ± 1.50 * | 76.47 ± 1.94 # | 77.78 ± 0.88 # | 78.02 ± 0.65 # |
| FS (%) | 44.83 ± 0.97 | 36.29 ± 1.34 * | 40.32 ± 1.63 | 42.17 ± 0.93 # | 43.38 ± 1.26 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poasakate, A.; Maneesai, P.; Rattanakanokchai, S.; Bunbupha, S.; Tong-Un, T.; Pakdeechote, P. Genistein Prevents Nitric Oxide Deficiency-Induced Cardiac Dysfunction and Remodeling in Rats. Antioxidants 2021, 10, 237. https://doi.org/10.3390/antiox10020237
Poasakate A, Maneesai P, Rattanakanokchai S, Bunbupha S, Tong-Un T, Pakdeechote P. Genistein Prevents Nitric Oxide Deficiency-Induced Cardiac Dysfunction and Remodeling in Rats. Antioxidants. 2021; 10(2):237. https://doi.org/10.3390/antiox10020237
Chicago/Turabian StylePoasakate, Anuson, Putcharawipa Maneesai, Siwayu Rattanakanokchai, Sarawoot Bunbupha, Terdthai Tong-Un, and Poungrat Pakdeechote. 2021. "Genistein Prevents Nitric Oxide Deficiency-Induced Cardiac Dysfunction and Remodeling in Rats" Antioxidants 10, no. 2: 237. https://doi.org/10.3390/antiox10020237
APA StylePoasakate, A., Maneesai, P., Rattanakanokchai, S., Bunbupha, S., Tong-Un, T., & Pakdeechote, P. (2021). Genistein Prevents Nitric Oxide Deficiency-Induced Cardiac Dysfunction and Remodeling in Rats. Antioxidants, 10(2), 237. https://doi.org/10.3390/antiox10020237