Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle—Do Myokines Play a Role?


Abstract
1. Introduction
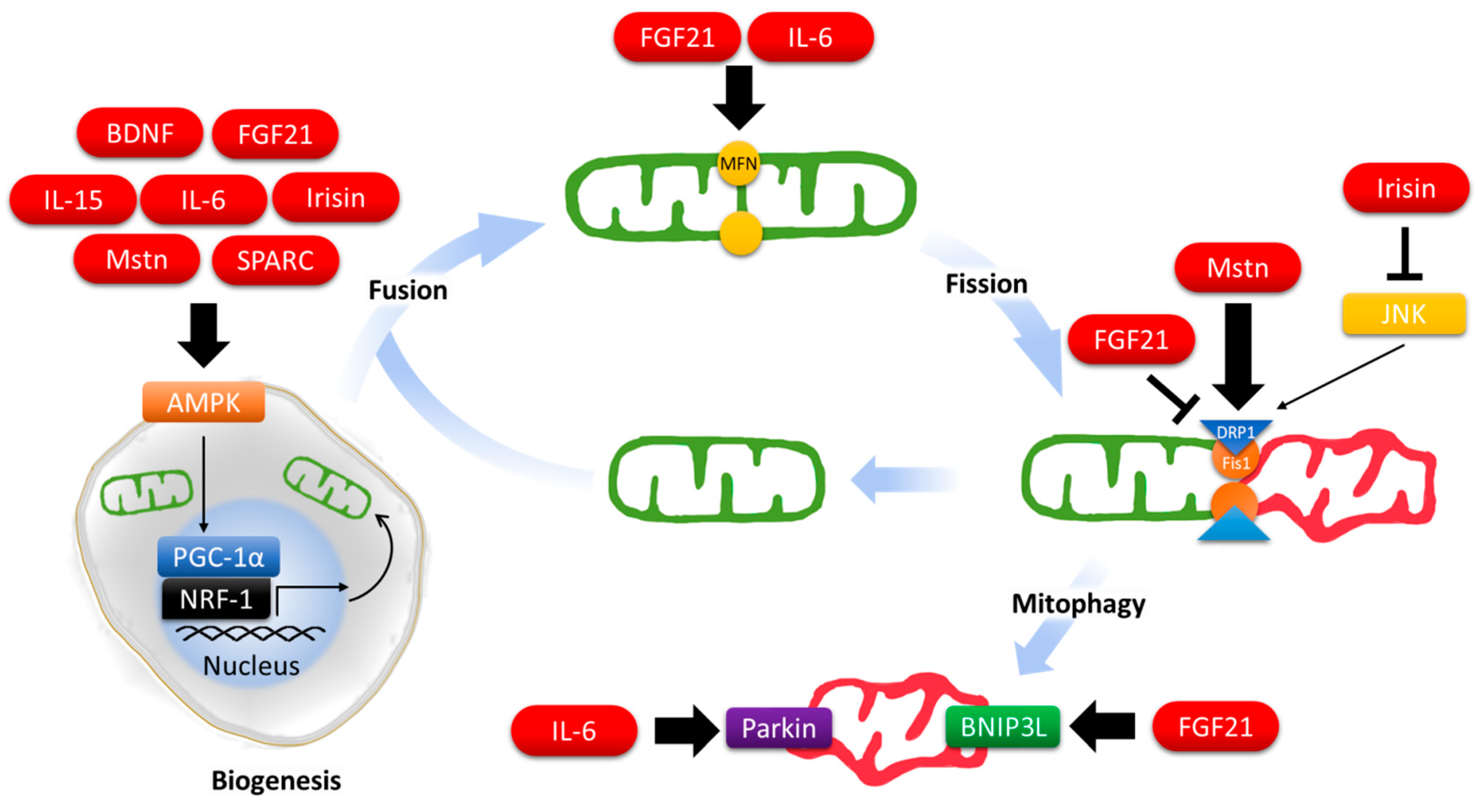
2. Myokines and Mitochondrial Dynamics
2.1. Brain-Derived Neurotrophic Factor (BDNF)
2.2. Fibroblast Growth Factor 21 (FGF21)
2.3. Interleukin 6 (IL-6)
2.4. Interleukin 15 (IL-15)
2.5. Irisin
2.6. Myostatin (Mstn)
2.7. Osteonectin/Secreted Protein Acidic and Rich in Cysteine (SPARC)
3. Myokines and Oxidative Stress
3.1. BDNF
3.2. FGF21
3.3. IL-6
3.4. IL-15
3.5. Irisin
3.6. Mstn
4. Therapeutic Potential of Myokines in Mitochondria/Ros ROS Dysregulation
4.1. Can Myokines Be Used as an Exercise Mimetic?
4.2. Can Myokines Be Used as Rejuvenation Agents?
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ferrer, J.C.; Favre, C.; Gomis, R.R.; Fernández-Novell, J.M.; García-Rocha, M.; de La Iglesia, N.; Cid, E.; Guinovart, J.J. Control of glycogen deposition. FEBS Lett. 2003, 546, 127–132. [Google Scholar] [CrossRef]
- Wagenmakers, A.J. Muscle amino acid metabolism at rest and during exercise: Role in human physiology and metabolism. Exerc. Sport Sci. Rev. 1998, 26, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.S. Humoral Nature of the Hypoglycemic Factor of Muscular Work. Diabetes 1961, 10, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, K.; Rohde, T.; Zacho, M.; Asp, S.; Pedersen, B.K. Evidence that interleukin-6 is produced in human skeletal muscle during prolonged running. J. Physiol. 1998, 508, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Myokines and Metabolism. In Metabolic Syndrome; Ahima, R.S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 541–554. [Google Scholar]
- Delezie, J.; Handschin, C. Endocrine Crosstalk between Skeletal Muscle and the Brain. Front. Neurol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Ghafoor, S. Myokines: Discovery Challenges and Therapeutic Impediments. J. Pak. Med. Assoc. 2019, 69, 1014–1017. [Google Scholar]
- Calvani, R.; Joseph, A.-M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Marzetti, E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol. Chem. 2013, 394, 393–414. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Barbieri, E.; Sestili, P. Reactive Oxygen Species in Skeletal Muscle Signaling. J. Signal Transduct. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Dominy, J.E.; Puigserver, P. Mitochondrial Biogenesis through Activation of Nuclear Signaling Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase–An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. BioEssays 2001, 23, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109. [Google Scholar] [CrossRef]
- Biswas, G.; Adebanjo, O.A.; Freedman, B.D.; Anandatheerthavarada, H.K.; Vijayasarathy, C.; Zaidi, M.; Kotlikoff, M.; Avadhani, N.G. Retrograde Ca2+ signaling in c2c12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: A novel mode of inter-organelle crosstalk. EMBO J. 1999, 18, 522–533. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.; Abudu, Y.P.; Larsen, K.B.; Brech, A.; Hansen, T.E.; Brenne, H.; Hansen, T.; et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Biol. 2018, 217, 3640–3655. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnóczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion–fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Van Der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2010, 18, 20–26. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Dulac, M.; Leduc-Gaudet, J.; Reynaud, O.; Ayoub, M.; Guérin, A.; Finkelchtein, M.; Na Hussain, S.; Gouspillou, G. Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation. J. Physiol. 2020, 598, 3691–3710. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Desbats, M.A.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nat. Cell Biol. 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent Anion Channels (VDACs) Recruit Parkin to Defective Mitochondria to Promote Mitochondrial Autophagy. J. Biol. Chem. 2012, 287, 40652–40660. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-K.; Lee, J.-A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a Ubiquitin-like Protein Required for Autophagosome Formation, Mediates Membrane Tethering and Hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef]
- Park, J.-S.; Koentjoro, B.; Sue, C.M. Commentary: Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Front. Mol. Neurosci. 2017, 10, 297. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Goberdhan, D.C.; Wilson, C.; Harris, A.L. Amino Acid Sensing by mTORC1: Intracellular Transporters Mark the Spot. Cell Metab. 2016, 23, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016, 26, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y.-J. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.R.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2010, 331, 456–461. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. Ampkalpha2 protects against the development of heart failure by enhancing mitophagy via pink1 phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Diniz, B.S.; Teixeira, A.L. Brain-Derived Neurotrophic Factor and Alzheimer’s Disease: Physiopathology and Beyond. NeuroMolecular Med. 2011, 13, 217–222. [Google Scholar] [CrossRef]
- Cuppini, R.; Sartini, S.; Agostini, D.; Guescini, M.; Ambrogini, P.; Betti, M.; Bertini, L.; Vallasciani, M.; Stocchi, V. Bdnf expression in rat skeletal muscle after acute or repeated exercise. Arch. Ital. Biol. 2007, 145, 99–110. [Google Scholar] [PubMed]
- Matthews, V.B.; Åström, M.-B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Brobst, D.; Chan, W.S.; Tse, M.C.L.; Herlea-Pana, O.; Ahuja, P.; Bi, X.; Zaw, A.M.; Kwong, Z.S.W.; Jia, W.-H.; et al. Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal. 2019, 12, eaau1468. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; Croix, B.S.; Wess, J.; et al. Novel metabolic role for bdnf in pancreatic beta-cell insulin secretion. Nat. Commun. 2020, 11, 1950. [Google Scholar] [CrossRef]
- Wood, J.; Tse, M.C.L.; Yang, X.; Brobst, D.; Liu, Z.; Pang, B.P.S.; Chan, W.S.; Zaw, A.M.; Chow, B.K.; Ye, K.; et al. BDNF mimetic alleviates body weight gain in obese mice by enhancing mitochondrial biogenesis in skeletal muscle. Metabolism 2018, 87, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef]
- Colitti, M.; Montanari, T. Brain-derived neurotrophic factor modulates mitochondrial dynamics and thermogenic phenotype on 3T3-L1 adipocytes. Tissue Cell 2020, 66, 101388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.-P.; Shao, Q.; Li, P.-F.; Sun, Y.; Luo, L.-Z.; Yan, X.-Q.; Fan, Z.-Y.; Hu, J.; Du, Z.-M.; et al. Brain-derived neurotrophic factor mimetic, 7,8-dihydroxyflavone, protects against myocardial ischemia by rebalancing optic atrophy 1 processing. Free. Radic. Biol. Med. 2019, 145, 187–197. [Google Scholar] [CrossRef]
- Cawley, M. The Role of Skeletal Muscle-Synthesized Brain Derived Neurotrophic Factor in the Maintenance of Motor Neuron Mitochondrial Populations. Master’s Thesis, Northern Michigan University, Marquette, MI, USA, June 2020. [Google Scholar]
- Shim, M.S.; Kim, K.-Y.; Noh, M.; Ko, J.Y.; Ahn, S.; An, M.A.; Iwata, T.; Perkins, G.A.; Weinreb, R.N.; Ju, W.-K. Optineurin E50K triggers BDNF deficiency-mediated mitochondrial dysfunction in retinal photoreceptor cell line. Biochem. Biophys. Res. Commun. 2018, 503, 2690–2697. [Google Scholar] [CrossRef]
- Oost, L.J.; Kustermann, M.; Armani, A.; Blaauw, B.; Romanello, V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J. Cachexia Sarcopenia Muscle 2019, 10, 630–642. [Google Scholar] [CrossRef]
- Li, X.; Hong, Y.; He, H.; Jiang, G.; You, W.; Liang, X.; Fu, Q.; Han, S.; Lian, Q.; Zhang, Y. FGF21 Mediates Mesenchymal Stem Cell Senescence via Regulation of Mitochondrial Dynamics. Oxidative Med. Cell. Longev. 2019, 2019, 4915149-13. [Google Scholar] [CrossRef] [PubMed]
- Forsström, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.-M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, L.; Wang, Q.; Zhan, S.; Wang, L.; Zhong, T.; Guo, J.; Zhang, H. Fibroblast growth factor 21 induces lipolysis more efficiently than it suppresses lipogenesis in goat adipocytes. Cytotechnology 2018, 70, 1423–1433. [Google Scholar] [CrossRef]
- Chen, X.-L.; Wang, Y.; Peng, W.-W.; Zheng, Y.-J.; Zhang, T.-N.; Wang, P.-J.; Huang, J.-D.; Zeng, Q. Effects of interleukin-6 and IL-6/AMPK signaling pathway on mitochondrial biogenesis and astrocytes viability under experimental septic condition. Int. Immunopharmacol. 2018, 59, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, Q.; Huang, Q.; Chen, Y.; Sun, X.; He, L.; Zhan, L.; Guo, X.; Yin, C.; Fang, Y.; et al. Upregulation of mtSSB by interleukin-6 promotes cell growth through mitochondrial biogenesis-mediated telomerase activation in colorectal cancer. Int. J. Cancer 2019, 144, 2516–2528. [Google Scholar] [CrossRef]
- Tomasz, A.B.; Magdalena, D.; Magdalena, S.; Beata, S.; Ewa, W.; Maria, M.W.; Karol, A.K. Interleukin-6 affects aging-related changes of the ppar[alpha]-pgc-1[alpha] axis in the myocardium. J. Interferon Cytokine Res. 2017, 37, 513. [Google Scholar]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; E Matesic, L.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef]
- Fix, D.K.; Hardee, J.P.; Gao, S.; Vanderveen, B.N.; Velázquez, K.T.; Carson, J.A. Role of gp130 in basal and exercise-trained skeletal muscle mitochondrial quality control. J. Appl. Physiol. 2018, 124, 1456–1470. [Google Scholar] [CrossRef]
- Fäldt, J.; Wernstedt, I.; Fitzgerald, S.M.; Wallenius, K.; Bergström, G.R.; Jansson, J.-O. Reduced exercise endurance in interleukin-6-deficient mice. Endocrinology 2004, 145, 2680–2686. [Google Scholar] [CrossRef]
- Wojewoda, M.; Kmiecik, K.; Majerczak, J.; Ventura-Clapier, R.; Fortin, D.; Onopiuk, M.; Róg, J.; Kamiński, K.A.; Chlopicki, S.; Zoladz, J. Skeletal Muscle Response to Endurance Training in IL-6−/− Mice. Int. J. Sports Med. 2015, 36, 1163–1169. [Google Scholar] [CrossRef]
- Hingorjo, M.R.; Zehra, S.; Saleem, S.; Qureshi, M.A. Serum Interleukin-15 and its relationship with adiposity Indices before and after short-term endurance exercise. Pak. J. Med. Sci. 2018, 34, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.; Carbó, N.; López-Soriano, J.; Drivdahl, R.H.; Busquets, S.; López-Soriano, F.J.; Argilés, J.M.; Quinn, L.S. Effects of interleukin-15 (IL-15) on adipose tissue mass in rodent obesity models: Evidence for direct IL-15 action on adipose tissue. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2002, 1570, 33–37. [Google Scholar] [CrossRef]
- Quinn, L.S.; Anderson, B.G.; Strait-Bodey, L.; Stroud, A.M.; Argilés, J.M. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am. J. Physiol. Metab. 2009, 296, E191–E202. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, L.; Aguer, C. Interleukin-15 as a myokine: Mechanistic insight into its effect on skeletal muscle metabolism. Appl. Physiol. Nutr. Metab. 2019, 44, 229–238. [Google Scholar] [CrossRef]
- Almendro, V.; Busquets, S.; Ametller, E.; Carbo, N.; Figueras, M.; Fuster, G.; Argiles, J.M.; Lopez-Soriano, F.J. Effects of interleukin-15 on lipid oxidation: Disposal of an oral [(14)c]-triolein load. Biochim. Biophys. Acta 2006, 1761, 37–42. [Google Scholar] [CrossRef]
- Thornton, S.M.; Krolopp, J.E.; Abbott, M.J. IL-15 Mediates Mitochondrial Activity through a PPARδ-Dependent-PPARα-Independent Mechanism in Skeletal Muscle Cells. PPAR Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Almendro, V.; Fuster, G.; Busquets, S.; Ametller, E.; Figueras, M.; Argilés, J.M.; López-Soriano, F.J. Effects of il-15 on rat brown adipose tissue: Uncoupling proteins and ppars. Obesity 2008, 16, 285–289. [Google Scholar] [CrossRef]
- O’Connell, G.C.; Pistilli, E.E. Interleukin-15 directly stimulates pro-oxidative gene expression in skeletal muscle in-vitro via a mechanism that requires interleukin-15 receptor alpha. Biochem. Biophys. Res. Commun. 2015, 458, 614–619. [Google Scholar] [CrossRef]
- Loro, E.; Bisetto, S.; Khurana, T.S. Mitochondrial ultrastructural adaptations in fast muscles of mice lacking IL15RA. J. Cell Sci. 2018, 131, jcs218313. [Google Scholar] [CrossRef]
- O’Connell, G.C.; Nichols, C.; Guo, G.; Croston, T.L.; Thapa, D.; Hollander, J.M.; Pistilli, E.E. IL-15Rα deficiency in skeletal muscle alters respiratory function and the proteome of mitochondrial subpopulations independent of changes to the mitochondrial genome. Mitochondrion 2015, 25, 87–97. [Google Scholar] [CrossRef]
- Pistilli, E.E.; Bogdanovich, S.; Garton, F.; Yang, N.; Gulbin, J.P.; Conner, J.D.; Anderson, B.G.; Quinn, L.S.; North, K.; Ahima, R.S.; et al. Loss of IL-15 receptor α alters the endurance, fatigability, and metabolic characteristics of mouse fast skeletal muscles. J. Clin. Investig. 2011, 121, 3120–3132. [Google Scholar] [CrossRef] [PubMed]
- Arhire, L.I.; Mihalache, L.; Covasa, M. Irisin: A Hope in Understanding and Managing Obesity and Metabolic Syndrome. Front. Endocrinol. 2019, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Mougios, V.; Kabasakalis, A.; Fatouros, I.; Siopi, A.; Douroudos, I.I.; Filippaios, A.; Panagiotou, G.; Park, K.H.; Mantzoros, C.S. Exercise-Induced Irisin Secretion Is Independent of Age or Fitness Level and Increased Irisin May Directly Modulate Muscle Metabolism Through AMPK Activation. J. Clin. Endocrinol. Metab. 2014, 99, E2154–E2161. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Gannon, N.P.; Mermier, C.M.; Conn, C.A. Irisin, a unique non-inflammatory myokine in stimulating skeletal muscle metabolism. J. Physiol. Biochem. 2015, 71, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, R.; Liu, X.; Wang, L.; Hui, P.; Chan, L.; Saha, P.K.; Hu, Z. ROCK1 reduces mitochondrial content and irisin production in muscle suppressing adipocyte browning and impairing insulin sensitivity. Sci. Rep. 2016, 6, 29669. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-S.; Kuo, C.-W.; Ko, J.-Y.; Chen, Y.-S.; Wang, S.-Y.; Ke, H.-J.; Kuo, P.-C.; Lee, C.-H.; Wu, J.-C.; Lu, W.-B.; et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants 2020, 9, 810. [Google Scholar] [CrossRef]
- Tan, Y.; Ouyang, H.; Xiao, X.; Zhong, J.; Dong, M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones 2019, 24, 595–608. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new tgf-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Girgenrath, S.; Song, K.; Whittemore, L.A. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow-and fast-type skeletal muscle. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2005, 31, 34–40. [Google Scholar] [CrossRef]
- Hennebry, A.; Berry, C.; Siriett, V.; O’Callaghan, P.; Chau, L.; Watson, T.; Sharma, M.; Kambadur, R. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am. J. Physiol. Physiol. 2009, 296, C525–C534. [Google Scholar] [CrossRef] [PubMed]
- Ploquin, C.; Chabi, B.; Fouret, G.; Vernus, B.; Feillet-Coudray, C.; Coudray, C.; Bonnieu, A.; Ramonatxo, C. Lack of myostatin alters intermyofibrillar mitochondria activity, unbalances redox status, and impairs tolerance to chronic repetitive contractions in muscle. Am. J. Physiol. Metab. 2012, 302, E1000–E1008. [Google Scholar] [CrossRef] [PubMed]
- Helge, A.; Raymond, M.; Roberto, N.; Markus, S.; Susan, C.B.; Anthony, O.; Thomas, V.; Francesco, M.; Gerta, V.; Terence, P.; et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA 2007, 104, 1835–1840. [Google Scholar]
- Ge, X.; Vajjala, A.; McFarlane, C.; Wahli, W.; Sharma, M.; Kambadur, R. Lack of Smad3 signaling leads to impaired skeletal muscle regeneration. Am. J. Physiol. Metab. 2012, 303, E90–E102. [Google Scholar] [CrossRef]
- Manfredi, L.H.; Ang, J.; Peker, N.; Dagda, R.K.; McFarlane, C. G protein-coupled receptor kinase 2 regulates mitochondrial bioenergetics and impairs myostatin-mediated autophagy in muscle cells. Am. J. Physiol. Physiol. 2019, 317, C674–C686. [Google Scholar] [CrossRef]
- Lokireddy, S.; Wijesoma, I.W.; Sze, S.K.; McFarlane, C.; Kambadur, R.; Sharma, M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am. J. Physiol. Physiol. 2012, 303, C512–C529. [Google Scholar] [CrossRef]
- Aoi, W.; Naito, Y.; Takagi, T.; Tanimura, Y.; Takanami, Y.; Kawai, Y.; Sakuma, K.; Hang, L.P.; Mizushima, K.; Hirai, Y.; et al. A novel myokine, secreted protein acidic and rich in cysteine (SPARC), suppresses colon tumorigenesis via regular exercise. Gut 2012, 62, 882–889. [Google Scholar] [CrossRef]
- Melouane, A.; Carbonell, A.; Yoshioka, M.; Puymirat, J.; St-Amand, J. Implication of SPARC in the modulation of the extracellular matrix and mitochondrial function in muscle cells. PLoS ONE 2018, 13, e0192714. [Google Scholar] [CrossRef]
- Melouane, A.; Yoshioka, M.; Kanzaki, M.; St-Amand, J. Sparc, an EPS-induced gene, modulates the extracellular matrix and mitochondrial function via ILK/AMPK pathways in C2C12 cells. Life Sci. 2019, 229, 277–287. [Google Scholar] [CrossRef]
- Hayyan, M.; Hashim, M.A.; Alnashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. NADPH Oxidases, Reactive Oxygen Species, and Hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31, S170–S180. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Tulard, A.; Hoffschir, F.; de Boisferon, F.H.; Luccioni, C.; Bravard, A. Persistent oxidative stress after ionizing radiation is involved in inherited radiosensitivity. Free. Radic. Biol. Med. 2003, 35, 68–77. [Google Scholar] [CrossRef]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef]
- Mao, X.; Yu, C.R.; Li, W.H. Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 2008, 18, 879–888. [Google Scholar] [CrossRef]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Gaetani, G.; Ferraris, A.; Rolfo, M.; Mangerini, R.; Arena, S.; Kirkman, H. Predominant role of catalase in the disposal of hydrogen peroxide within human erythrocytes. Blood 1996, 87, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Malys, N.; Gaskell, S.J.; Barber, J. Partition and Turnover of Glutathione Reductase from Saccharomyces cerevisiae: A Proteomic Approach. J. Proteome Res. 2013, 12, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Janczuk, A.; Wang, P.G. Current trends in the development of nitric oxide donors. Curr. Pharm. Des. 1999, 5, 417–441. [Google Scholar] [PubMed]
- Linley, J.E.; Ooi, L.; Pettinger, L.; Kirton, H.; Boyle, J.P.; Peers, C.; Gamper, N. Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc. Natl. Acad. Sci. USA 2012, 109, E1578–E1586. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling: H2O2, a Necessary Evil for Cell Signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free. Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Fan, X.; Hussien, R.; Brooks, G.A. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free. Radic. Biol. Med. 2010, 49, 1646–1654. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Wu, C.L.; Chen, S.D.; Yin, J.H.; Hwang, C.S.; Yang, D.I. Nuclear factor-kappab-dependent sestrin2 induction mediates the antioxidant effects of bdnf against mitochondrial inhibition in rat cortical neurons. Mol. Neurobiol. 2016, 53, 4126–4142. [Google Scholar] [CrossRef]
- Molinari, C.; Morsanuto, V.; Ruga, S.; Notte, F.; Farghali, M.; Galla, R.; Uberti, F. The Role of BDNF on Aging-Modulation Markers. Brain Sci. 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Psotta, L.; Brigadski, T.; Endres, T.; Lessmann, V. Chronic BDNF deficiency leads to an age-dependent impairment in spatial learning. Neurobiol. Learn. Mem. 2015, 120, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hachem, L.D.; Mothe, A.J.; Tator, C.H. Effect of BDNF and Other Potential Survival Factors in Models of In Vitro Oxidative Stress on Adult Spinal Cord–Derived Neural Stem/Progenitor Cells. BioResearch Open Access 2015, 4, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.C.; Jiao, C.H.; Gao, Z.M. Silencing of lncrna bdnf-as attenuates abeta25-35-induced neurotoxicity in pc12 cells by suppressing cell apoptosis and oxidative stress. Neurol Res. 2018, 40, 795–804. [Google Scholar] [CrossRef]
- Jin, H.; Zhu, Y.; Li, Y.; Ding, X.; Ma, W.; Han, X.; Wang, B. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis 2019, 24, 511–528. [Google Scholar] [CrossRef]
- Rosenblum, S.; Smith, T.N.; Wang, N.; Chua, J.Y.; Westbroek, E.; Wang, K.; Guzman, R. BDNF Pretreatment of Human Embryonic-Derived Neural Stem Cells Improves Cell Survival and Functional Recovery after Transplantation in Hypoxic–Ischemic Stroke. Cell Transplant. 2015, 24, 2449–2461. [Google Scholar] [CrossRef]
- Jin, H.; Zhu, Y.; Wang, X.-D.; Luo, E.-F.; Li, Y.-P.; Wang, B.-L.; Chen, Y.-F. BDNF corrects NLRP3 inflammasome-induced pyroptosis and glucose metabolism reprogramming through KLF2/HK1 pathway in vascular endothelial cells. Cell. Signal. 2021, 78, 109843. [Google Scholar] [CrossRef]
- Guo, D.; Xiao, L.; Hu, H.; Liu, M.; Yang, L.; Lin, X. FGF21 protects human umbilical vein endothelial cells against high glucose-induced apoptosis via PI3K/Akt/Fox3a signaling pathway. J. Diabetes Complicat. 2018, 32, 729–736. [Google Scholar] [CrossRef]
- Zeng, Z.; Zheng, Q.; Chen, J.; Tan, X.; Li, Q.; Ding, L.; Zhang, R.; Xiaolong, L. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp. Cell Res. 2020, 393, 112108. [Google Scholar] [CrossRef]
- Xu, M.; Wang, L.; Wang, M.; Wang, H.; Zhang, H.; Chen, Y.; Wang, X.; Gong, J.; Zhang, J.; Adcock, I.M.; et al. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free. Radic. Res. 2019, 53, 780–790. [Google Scholar] [CrossRef]
- Planavila, A.; Redondo-Angulo, I.; Ribas, F.; Garrabou, G.; Casademont, J.; Giralt, M.; Villarroya, F. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc. Res. 2014, 106, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Jiang, X.; Zhang, Y.; Wang, J.; Xie, J.; Wang, Y.-Q.; Li, Y.-H. FGF21 Protects Against Hypoxia Injury Through Inducing HSP72 in Cerebral Microvascular Endothelial Cells. Front. Pharmacol. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ma, L.; Wu, Y.; Ye, X.; Zhang, T.; Zhang, Q.; Rasoul, L.M.; Liu, Y.; Guo, M.; Zhou, B.; et al. FGF21 treatment ameliorates alcoholic fatty liver through activation of AMPK-SIRT1 pathway. Acta Biochim. Biophys. Sin. 2014, 46, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Abid, H.; Hart, C.; Lanza, I. Effects of local interleukin-6 on skeletal muscle mitochondrial physiology. FASEB J. 2018, 32, 603–607. [Google Scholar]
- Forcina, L.; Miano, C.; Scicchitano, B.M.; Rizzuto, E.; Berardinelli, M.G.; De Benedetti, F.; Pelosi, L.; Musarò, A. Increased Circulating Levels of Interleukin-6 Affect the Redox Balance in Skeletal Muscle. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wu, Z.; He, M.; Chen, Y.; Chen, Y.; Shen, X.; Zhao, X.; Zhang, L.; Yuan, B.; Zeng, Z.-C. ADAM9 mediates the interleukin-6-induced Epithelial–Mesenchymal transition and metastasis through ROS production in hepatoma cells. Cancer Lett. 2018, 421, 1–14. [Google Scholar] [CrossRef]
- Ding, W.; You, H.; Dang, H.; Leblanc, F.; Galicia, V.; Lu, S.C.; Stiles, B.; Rountree, C.B. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology 2010, 52, 945–953. [Google Scholar] [CrossRef]
- Ji, C.; Chen, X.; Gao, C.; Jiao, L.; Wang, J.; Xu, G.; Fu, H.; Guo, X.; Zhao, Y. IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J. Bioenerg. Biomembr. 2011, 43, 367–375. [Google Scholar] [CrossRef]
- Kojima, H.; Kunimoto, H.; Inoue, T.; Nakajima, K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle 2012, 11, 730–739. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; Murphy, R.P.; Cummins, P.M. Downregulation of Blood-Brain Barrier Phenotype by Proinflammatory Cytokines Involves NADPH Oxidase-Dependent ROS Generation: Consequences for Interendothelial Adherens and Tight Junctions. PLoS ONE 2014, 9, e101815. [Google Scholar] [CrossRef]
- Marasco, M.R.; Conteh, A.M.; Reissaus, C.A.; Cupit, J.E.T.; Appleman, E.M.; Mirmira, R.G.; Linnemann, A.K. Interleukin-6 reduces beta-cell oxidative stress by linking autophagy with the antioxidant response. Diabetes 2018, 67, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Naga, O.S.; Norell, H.; Al-Khami, A.A.; Scheffel, M.J.; Chakraborty, N.G.; Voelkel-Johnson, C.; Mukherji, B.; Mehrotra, S. T cells expanded in presence of IL-15 exhibit increased antioxidant capacity and innate effector molecules. Cytokine 2011, 55, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Neo, S.Y.; Chen, Z.; Cui, W.; Chen, Y.; Guo, M.; Wang, Y.; Xu, H.; Kurzay, A.; Alici, E.; et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J. Clin. Investig. 2020, 130, 5508–5522. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, Y.; Tang, Y.; Lin, B.; Kong, X.; Oladele, O.A.; Yin, Y. Protective effect of myokine IL-15 against H2O2-mediated oxidative stress in skeletal muscle cells. Mol. Biol. Rep. 2014, 41, 7715–7722. [Google Scholar] [CrossRef]
- Ho, M.-Y.; Wen, M.-S.; Yeh, J.-K.; Hsieh, I.-C.; Chen, C.-C.; Hsieh, M.-J.; Tsai, M.-L.; Yang, C.-H.; Wu, V.C.-C.; Hung, K.-C.; et al. Excessive irisin increases oxidative stress and apoptosis in murine heart. Biochem. Biophys. Res. Commun. 2018, 503, 2493–2498. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, K.; Han, Y.; Zhu, H.; Zhou, X.; Tan, T.; Zeng, J.; Zhang, J.; Liu, Y.; Li, Y.; et al. Irisin Protects Heart Against Ischemia-Reperfusion Injury Through a SOD2-Dependent Mitochondria Mechanism. J. Cardiovasc. Pharmacol. 2018, 72, 259–269. [Google Scholar] [CrossRef]
- Chen, K.; Xu, Z.; Liu, Y.; Wang, Z.; Li, Y.; Xu, X.; Chen, C.; Xia, T.; Liao, Q.; Yao, Y.; et al. Irisin protects mitochondria function during pulmonary ischemia/reperfusion injury. Sci. Transl. Med. 2017, 9, eaao6298. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.; Jiang, L. Irisin attenuates oxidized low-density lipoprotein impaired angiogenesis through AKT/mTOR/S6K1/Nrf2 pathway. J. Cell. Physiol. 2019, 234, 18951–18962. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.O.; Kim, N.; Kim, J.K.; Kim, H.I.; Lee, Y.W.; Kim, S.J.; Choi, J.-I.; Oh, Y.; Kim, J.H.; et al. Irisin, a Novel Myokine, Regulates Glucose Uptake in Skeletal Muscle Cells via AMPK. Mol. Endocrinol. 2015, 29, 873–881. [Google Scholar] [CrossRef]
- Aravena, J.; Abrigo, J.; Gonzalez, F.; Aguirre, F.; Gonzalez, A.; Simon, F.; Cabello-Verrugio, C. Angiotensin (1–7) decreases myostatin-induced nf-kappab signaling and skeletal muscle atrophy. Int. J. Mol. Sci. 2020, 21, 1167. [Google Scholar] [CrossRef]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an nf-kb independent mechanism in smad3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-Q.; Ming, S.-L.; Wu, H.-T.; Zeng, L.; Ba, G.; Li, J.; Lu, W.-F.; Han, J.; Du, Q.-J.; Sun, M.-M.; et al. Myostatin knockout induces apoptosis in human cervical cancer cells via elevated reactive oxygen species generation. Redox Biol. 2018, 19, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Milanesi, S.; Viazzi, F.; Ansaldo, F.; Saio, M.; Garibaldi, S.; Carta, A.; Costigliolo, F.; Salvidio, G.; Barisione, C.; et al. Enhanced myostatin expression and signalling promote tubulointerstitial inflammation in diabetic nephropathy. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Geto, Z.D.; Molla, M.D.; Challa, F.; Belay, Y.; Getahun, T. Mitochondrial Dynamic Dysfunction as a Main Triggering Factor for Inflammation Associated Chronic Non-Communicable Diseases. J. Inflamm. Res. 2020, 13, 97–107. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, B.; Li, T.; Zhu, Y.; Luo, G.; Jiang, Y.; Tang, F.; Jian, Z.; Xiao, Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. Int. J. Mol. Med. 2017, 41, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef] [PubMed]
- Eckel, J. Myokines in metabolic homeostasis and diabetes. Diabetologia 2019, 62, 1523–1528. [Google Scholar] [CrossRef]
- Bostroem, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostroem, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nat. Cell Biol. 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Görgens, S.W.; Eckardt, K.; Jensen, J.D.; Drevon, C.A.; Eckel, J. Exercise and Regulation of Adipokine and Myokine Production. Progress Mol. Biol. Transl. Sci. 2015, 135, 313–336. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef]
- WHO. Global Recommendations on Physical Activity for Health; WHO: Geneva, Switzerland, 2010. [Google Scholar]
- Powers, S.K.; Bomkamp, M.; Ozdemir, M.; Hyatt, H. Mechanisms of exercise-induced preconditioning in skeletal muscles. Redox Biol. 2020, 35, 101462. [Google Scholar] [CrossRef]
- Drake, J.C.; Wilson, R.J.; Yan, Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 2016, 30, 13–22. [Google Scholar] [CrossRef]
- Leek, B.T.; Mudaliar, S.R.D.; Henry, R.; Mathieu-Costello, O.; Richardson, R.S. Effect of acute exercise on citrate synthase activity in untrained and trained human skeletal muscle. Am. J. Physiol. Integr. Comp. Physiol. 2001, 280, R441–R447. [Google Scholar] [CrossRef] [PubMed]
- Menshikova, E.V.; Ritov, V.B.; Fairfull, L.; Ferrell, R.E.; Kelley, D.E.; Goodpaster, B.H. Effects of Exercise on Mitochondrial Content and Function in Aging Human Skeletal Muscle. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2006, 61, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Talanian, J.L.; Galloway, S.D.R.; Heigenhauser, G.J.F.; Bonen, A.; Spriet, L.L. Two weeks of high-intensity aerobic interval training increases the capacity for fat oxidation during exercise in women. J. Appl. Physiol. 2007, 102, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, M.J.; Zacharewicz, E.; Martin, B.J.; Haikalis, M.E.; Skelly, L.E.; Tarnopolsky, M.A.; Murphy, R.M.; Gibala, M.J. Superior mitochondrial adaptations in human skeletal muscle after interval compared to continuous single-leg cycling matched for total work. J. Physiol. 2017, 595, 2955–2968. [Google Scholar] [CrossRef] [PubMed]
- Arribat, Y.; Broskey, N.T.; Greggio, C.; Boutant, M.; Alonso, S.C.; Kulkarni, S.S.; Lagarrigue, S.; Carnero, E.A.; Besson, C.; Canto, C.; et al. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol. 2018, 225, e13179. [Google Scholar] [CrossRef]
- Brandt, N.; Gunnarsson, T.P.; Bangsbo, J.; Pilegaard, H. Exercise and exercise training-induced increase in autophagy markers in human skeletal muscle. Physiol. Rep. 2018, 6, e13651. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopes, M.A.; Batista, J.M.L. Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. SpringerPlus 2014, 3, 1–12. [Google Scholar] [CrossRef]
- Fan, W.; Evans, R.M. Exercise Mimetics: Impact on Health and Performance. Cell Metab. 2017, 25, 242–247. [Google Scholar] [CrossRef]
- Colaianni, G.; Mongelli, T.; Cuscito, C.; Pignataro, P.; Lippo, L.; Spiro, G.; Notarnicola, A.; Severi, I.; Passeri, G.; Mori, G.; et al. Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Trovato, E.; Di Felice, V.; Barone, R. Extracellular Vesicles: Delivery Vehicles of Myokines. Front. Physiol. 2019, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Huertas, J.R.; Casuso, R.A.; Agustín, P.H.; Cogliati, S. Stay Fit, Stay Young: Mitochondria in Movement: The Role of Exercise in the New Mitochondrial Paradigm. Oxidative Med. Cell. Longev. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jubrias, S.A.; Esselman, P.C.; Price, L.B.; Cress, M.E.; Conley, K.E. Large energetic adaptations of elderly muscle to resistance and endurance training. J. Appl. Physiol. 2001, 90, 1663–1670. [Google Scholar] [CrossRef]
- Ekaterina, S.; Ana, S.; Zhiyong, Z.; Zhan, G.; Siva Rama Krishna, K.; Santiago, R.; Elizabeth, S.; Susan, A.W.; Michael, R.A.; Carmen, M.P.-T.; et al. Musclin is an activity-stimulated myokine that enhances physical endurance. Proc. Natl. Acad. Sci. USA 2015, 112, 16042–16047. [Google Scholar]
- Wolsk, E.; Mygind, H.; Grøndahl, T.S.; Pedersen, B.K.; van Hall, G. IL-6 selectively stimulates fat metabolism in human skeletal muscle. Am. J. Physiol. Metab. 2010, 299, E832–E840. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Anderson, B.G.; Conner, J.D.; Wolden-Hanson, T. IL-15 Overexpression Promotes Endurance, Oxidative Energy Metabolism, and Muscle PPARδ, SIRT1, PGC-1α, and PGC-1β Expression in Male Mice. Endocrinology 2013, 154, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Dillard, C.J.; Litov, R.E.; Savin, W.M.; Dumelin, E.E.; Tappel, A.L. Effects of exercise, vitamin E, and ozone on pulmonary function and lipid peroxidation. J. Appl. Physiol. 1978, 45, 927–932. [Google Scholar] [CrossRef]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Venditti, P.; di Meo, S. Effect of Training on Antioxidant Capacity, Tissue Damage, and Endurance of Adult Male Rats. Int. J. Sports Med. 1997, 18, 497–502. [Google Scholar] [CrossRef]
- Saborido, A.; Naudí, A.; Portero-Otin, M.; Pamplona, R.; Megías, A. Stanozolol treatment decreases the mitochondrial ROS generation and oxidative stress induced by acute exercise in rat skeletal muscle. J. Appl. Physiol. 2011, 110, 661–669. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Olguín, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.E.; Schjerling, P.; Viollet, B.; Wojtaszewski, J.F.P.; Richter, E.A. AMPK α1 Activation Is Required for Stimulation of Glucose Uptake by Twitch Contraction, but Not by H2O2, in Mouse Skeletal Muscle. PLoS ONE 2008, 3, e2102. [Google Scholar] [CrossRef] [PubMed]
- Santos-Silva, A.; Rebelo, M.I.; Castro, E.M.B.; Belo, L.; Guerra, A.; Rego, C.; Quintanilha, A. Leukocyte activation, erythrocyte damage, lipid profile and oxidative stress imposed by high competition physical exercise in adolescents. Clin. Chim. Acta 2001, 306, 119–126. [Google Scholar] [CrossRef]
- Sun, F.; Norman, I.J.; While, A.E. Physical activity in older people: A systematic review. BMC Public Health 2013, 13, 449. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylavsky, F.A.; Visser, M.; Newman, A.B.; et al. The Loss of Skeletal Muscle Strength, Mass, and Quality in Older Adults: The Health, Aging and Body Composition Study. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef]
- Baumann, C.W.; Kwak, D.; Liu, H.M.; Thompson, L.V. Age-induced oxidative stress: How does it influence skeletal muscle quantity and quality? J. Appl. Physiol. 2016, 121, 1047–1052. [Google Scholar] [CrossRef]
- Kim, J.-S.; Lee, Y.-H.; Yi, H.-K. Gradual downhill running improves age-related skeletal muscle and bone weakness: Implication of autophagy and bone morphogenetic proteins. Exp. Physiol. 2016, 101, 1528–1540. [Google Scholar] [CrossRef]
- Kim, H.-J.; So, B.; Choi, M.; Kang, D.; Song, W. Resistance exercise training increases the expression of irisin concomitant with improvement of muscle function in aging mice and humans. Exp. Gerontol. 2015, 70, 11–17. [Google Scholar] [CrossRef]
- Piccirillo, R. Exercise-Induced Myokines With Therapeutic Potential for Muscle Wasting. Front. Physiol. 2019, 10, 287. [Google Scholar] [CrossRef]
- Nakamura, K.; Yamanouchi, K.; Nishihara, M. Secreted protein acidic and rich in cysteine internalization and its age-related alterations in skeletal muscle progenitor cells. Aging Cell 2013, 13, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Moon, K.M.; Min, K.-W. Exercise-Induced Myokines can Explain the Importance of Physical Activity in the Elderly: An Overview. Healthcare 2020, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jun, H.-S. Role of Myokines in Regulating Skeletal Muscle Mass and Function. Front. Physiol. 2019, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Gianni, P.; Jan, K.J.; Douglas, M.J.; Stuart, P.M.; Tarnopolsky, M.A. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp. Gerontol. 2004, 39, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Gunn, M.J.; Lewandowski, P.A. Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr. 2013, 13, 104. [Google Scholar] [CrossRef]
- Liverini, G.; Bianco, F.; Mazzoli, A.; Giacco, A.; Liverini, G.; Iossa, S. Alterations in proton leak, oxidative status and uncoupling protein 3 content in skeletal muscle subsarcolemmal and intermyofibrillar mitochondria in old rats. BMC Geriatr. 2014, 14, 79. [Google Scholar] [CrossRef]
- Joseph, A.-M.; Adhihetty, P.J.; Leeuwenburgh, C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J. Physiol. 2016, 594, 5105–5123. [Google Scholar] [CrossRef]
- Carter, H.N.; Chen, C.C.W.; Hood, D.A. Mitochondria, Muscle Health, and Exercise with Advancing Age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Ji, L.L. Aging alters acetylation status in skeletal and cardiac muscles. GeroScience 2020, 42, 963–976. [Google Scholar] [CrossRef]
- Pansarasa, O.; Castagna, L.; Colombi, B.; Vecchiet, J.; Felzani, G.; Marzatico, F. Age and sex differences in human skeletal muscle: Role of reactive oxygen species. Free. Radic. Res. 2000, 33, 287–293. [Google Scholar] [CrossRef]
- Siu, P.M.; Pistilli, E.E.; Alway, S.E. Age-dependent increase in oxidative stress in gastrocnemius muscle with unloading. J. Appl. Physiol. 2008, 105, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Bertorelli, L.; Vecchiet, J.; Felzani, G.; Marzatico, F. Age-dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free. Radic. Biol. Med. 1999, 27, 617–622. [Google Scholar] [CrossRef]
- Safdar, A.; Hamadeh, M.J.; Kaczor, J.J.; Raha, S.; Debeer, J.; Tarnopolsky, M.A. Aberrant Mitochondrial Homeostasis in the Skeletal Muscle of Sedentary Older Adults. PLoS ONE 2010, 5, e10778. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; van Wittenberghe, L.; Georger, C.; Scherman, D.; Kichler, A. Identification of decorin derived peptides with a zinc dependent anti-myostatin activity. Neuromuscul. Disord. 2012, 22, 1057–1068. [Google Scholar]
- Reza, M.M.; Subramaniyam, N.; Sim, C.M.; Ge, X.; Sathiakumar, D.; McFarlane, C.; Sharma, M.; Kambadur, R. Irisin is a pro-myogenic factor that induces skeletal muscle hypertrophy and rescues denervation-induced atrophy. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Jang, Y.C.; Sinha, M.; Cerletti, M.; Dall’Osso, C.; Wagers, A.J. Skeletal Muscle Stem Cells: Effects of Aging and Metabolism on Muscle Regenerative Function. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 101–111. [Google Scholar] [CrossRef]
- Belli, R.; Bonato, A.; De Angelis, L.; Mirabilii, S.; Ricciardi, M.R.; Tafuri, A.; Molfino, A.; Leigheb, M.; Costelli, P.; Caruso, M.; et al. Metabolic Reprogramming Promotes Myogenesis During Aging. Front. Physiol. 2019, 10, 897. [Google Scholar] [CrossRef]
- Camporez, J.-P.G.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc. Natl. Acad. Sci. USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef]
- Delanaye, P.; Bataille, S.; Quinonez, K.; Buckinx, F.; Warling, X.; Krzesinski, J.-M.; Pottel, H.; Burtey, S.; Bruyère, O.; Cavalier, E. Myostatin and Insulin-Like Growth Factor 1 Are Biomarkers of Muscle Strength, Muscle Mass, and Mortality in Patients on Hemodialysis. J. Ren. Nutr. 2019, 29, 511–520. [Google Scholar] [CrossRef]
- Coelho-Junior, H.; Picca, A.; Calvani, R.; Uchida, M.C.; Marzetti, E. If my muscle could talk: Myokines as a biomarker of frailty. Exp. Gerontol. 2019, 127, 110715. [Google Scholar] [CrossRef]
- Park, H.-S.; Kim, H.C.; Zhang, D.; Yeom, H.; Kil Lim, S. The novel myokine irisin: Clinical implications and potential role as a biomarker for sarcopenia in postmenopausal women. Endocrine 2019, 64, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Iino, N.; Koda, R.; Narita, I.; Kaneko, Y. Brain-derived neurotrophic factor is associated with sarcopenia and frailty in Japanese hemodialysis patients. Geriatr. Gerontol. Int. 2021, 21, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.S.; Ho, C.; Tam, W.; Kua, E.H.; Ho, R. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Khan, I.U. Irisin: As a Therapeutic Target for Metabolic Disorders. J. Endocrinol. Metab. 2018, 8, 87–93. [Google Scholar] [CrossRef]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Muñoz-Fuentes, V.; Cacheiro, P.; Meehan, T.F.; Aguilar-Pimentel, J.A.; Brown, S.D.M.; Flenniken, A.M.; Flicek, P.; Galli, A.; Mashhadi, H.H.; Hrabě de Angelis, M.; et al. The International Mouse Phenotyping Consortium (IMPC): A functional catalogue of the mammalian genome that informs conservation. Conserv. Genet. 2018, 19, 995–1005. [Google Scholar] [CrossRef]
- Talukdar, S.; Zhou, Y.; Li, D.; Rossulek, M.; Dong, J.; Somayaji, V.; Weng, Y.; Clark, R.; Lanba, A.; Owen, B.M.; et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016, 23, 427–440. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Hsieh, L.-L.; Tang, R.-P.; Liao, S.-K.; Yeh, K.-Y. Interleukin-6 (IL-6) released by macrophages induces IL-6 secretion in the human colon cancer HT-29 cell line. Hum. Immunol. 2009, 70, 151–158. [Google Scholar] [CrossRef]
- Fukuno, N.; Matsui, H.; Kanda, Y.; Suzuki, O.; Matsumoto, K.; Sasaki, K.; Kobayashi, T.; Tamura, S. Tgf-beta-activated kinase 1 mediates mechanical stress-induced il-6 expression in osteoblasts. Biochem. Biophys. Res. Commun. 2011, 408, 202–207. [Google Scholar] [CrossRef]
- Zampetaki, A.; Zhang, Z.; Hu, Y.; Xu, Q. Biomechanical stress induces il-6 expression in smooth muscle cells via ras/rac1-p38 mapk-nf-kappab signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2946–H2954. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; White, A.; Perry, R.J.; Camporez, J.-P.; Hidalgo, J.; Shulman, G.I.; Davis, R.J. Regulation of adipose tissue inflammation by interleukin 6. Proc. Natl. Acad. Sci. USA 2020, 117, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Glund, S.; Deshmukh, A.; Long, Y.C.; Moller, T.; Koistinen, H.A.; Caidahl, K.; Zierath, J.R.; Krook, A. Interleukin-6 Directly Increases Glucose Metabolism in Resting Human Skeletal Muscle. Diabetes 2007, 56, 1630–1637. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Myokine | Cell/Tissue Type | Treatment/Pathological Condition | Oxidative Stress Regulation | Reference |
|---|---|---|---|---|
| BDNF | Cortical neurons | 3-Nitropropionic acid challenge | Alleviates ROS level | [122] |
| NSPCs | H2O2 stress | Enhances the activities of GR and SOD | [125] | |
| PC12 | / | Lowers ROS intensity and malondialdehyde content | [126] | |
| Brain microvascular endothelial cells | Hyperglycemia | Inhibits ROS generation and prevents apoptosis | [127] | |
| HUVECs | oxLDL incubation | Suppresses ROS surge | [129] | |
| FGF21 | HUVECs | High glucose concentration incubation | ROS elimination | [130] |
| Vascular endothelial cells | oxLDL incubation | Inhibits mtROS production | [131,132] | |
| Cardiomyocytes | / | Upregulates antioxidative genes | [133] | |
| Cerebral microvascular endothelial cells | Hypoxia | Inhibits ROS synthesis and restores antioxidants’ activities | [134] | |
| MSCs | / | Reduces mtROS level | [63] | |
| HepG2 cells/liver | Ethanol incubation/alcoholic fatty liver disease | Restores activities of SOD and glutathione peroxidase | [135] | |
| IL-6 | C2C12 myotubes | / | Increases mtROS production | [136] |
| Diaphragm muscles | IL-6 overexpression | Increases NOX2 expression, promotes ROS accumulation, and diminishes NRF2-related antioxidant responses | [137] | |
| Colorectal cancer cells | Cancer | Promotes mtROS synthesis | [67] | |
| Hepatocellular carcinoma cells | Cancer | Increases ROS generation | [138,139] | |
| 3T3-L1 adipocytes | / | Enhances ROS level | [140] | |
| TIG3 fibroblasts | / | Enhances ROS level | [141] | |
| Human brain microvascular endothelial cells | / | Enhances ROS level | [142] | |
| β-cells | / | Lowers ROS level, removes the antioxidant repressor KEAP1 | [143] | |
| IL-15 | Human T cells | / | Upregulates antioxidative genes | [144] |
| NK cells | / | Preserves TXN and increases thiol group density on cell surface | [145] | |
| Muscle cells | H2O2 incubation | Relieves oxidative stress | [146] | |
| Irisin | Cardiomyocytes | Irisin overexpression and ischemia/reperfusion injury | Increases superoxide level, promotes SOD activity and localization in mitochondria | [147,148] |
| Alveolar cells and hepatocytes | Ischemia/reperfusion injury | Lowers ROS content | [90,149] | |
| Vascular endothelial cells | oxLDL incubation | Reduces ROS generation | [150] | |
| L6 muscle cells | / | Induces ROS generation | [151] | |
| Mstn | C2C12 myotubes | / | Increases ROS content | [152,153] |
| Mstn knockout mice | [94] | |||
| Gastrocnemius and soleus muscles | Maintains the activities of several antioxidants | |||
| MSTN knockout | [154] | |||
| HeLa cells | Promotes mtROS generation | |||
| Proximal tubular epithelial cell | / | Raises ROS level and upregulates NOX expression | [155] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, B.P.S.; Chan, W.S.; Chan, C.B. Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle—Do Myokines Play a Role? Antioxidants 2021, 10, 179. https://doi.org/10.3390/antiox10020179
Pang BPS, Chan WS, Chan CB. Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle—Do Myokines Play a Role? Antioxidants. 2021; 10(2):179. https://doi.org/10.3390/antiox10020179
Chicago/Turabian StylePang, Brian Pak Shing, Wing Suen Chan, and Chi Bun Chan. 2021. "Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle—Do Myokines Play a Role?" Antioxidants 10, no. 2: 179. https://doi.org/10.3390/antiox10020179
APA StylePang, B. P. S., Chan, W. S., & Chan, C. B. (2021). Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle—Do Myokines Play a Role? Antioxidants, 10(2), 179. https://doi.org/10.3390/antiox10020179