Unique Cellular and Biochemical Features of Human Mitochondrial Peroxiredoxin 3 Establish the Molecular Basis for Its Specific Reaction with Thiostrepton

 , and
, and 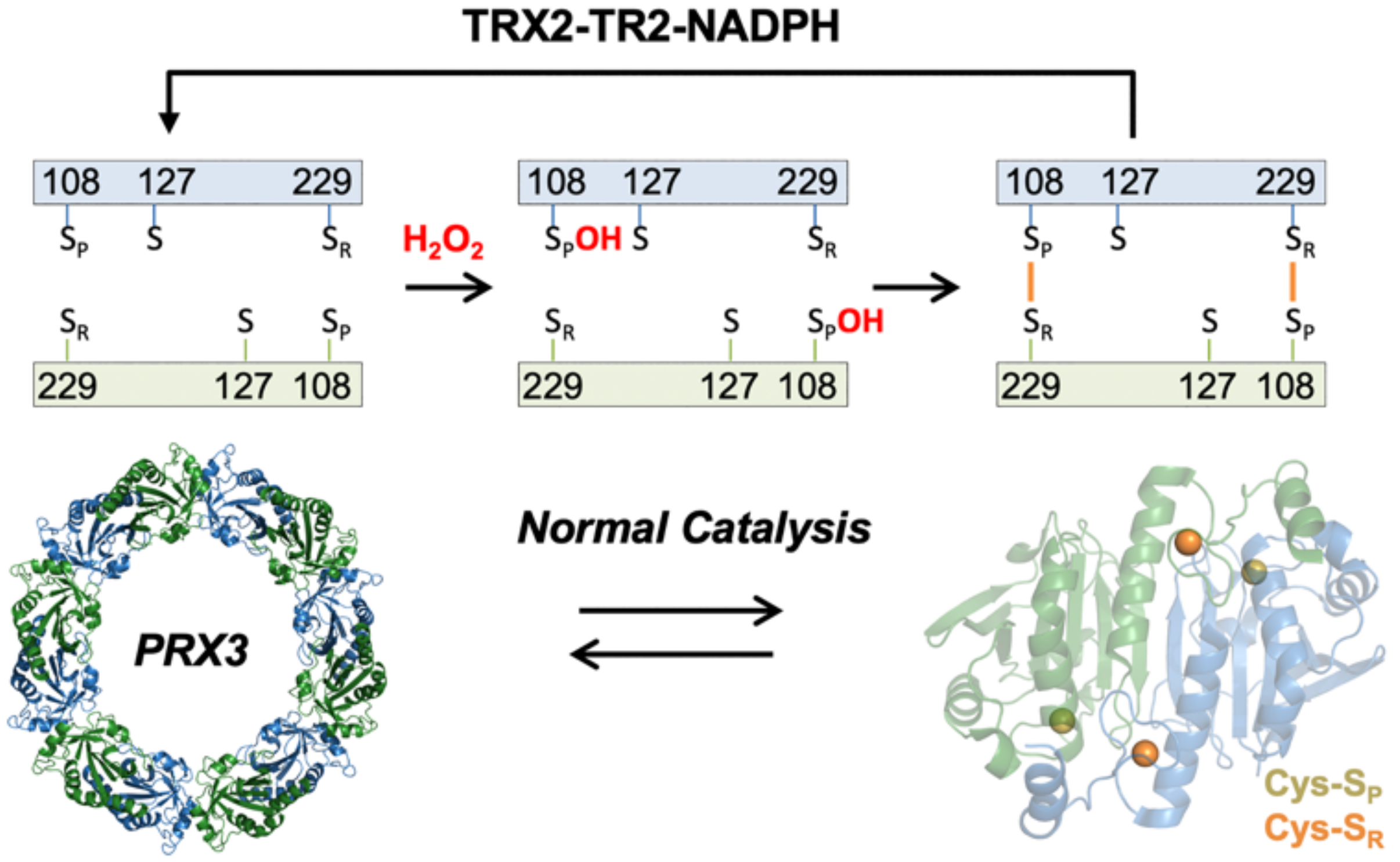{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cells and Cell Culture
2.3. Immunoblotting
2.4. Cell Viability Assays
2.5. Agilent Seahorse XF Cell Mito Stress Test Assay
2.6. MitoSOX Red Detection of Mitochondrial ROS
2.7. Expression and Purification of Recombinant Proteins
2.8. In Vitro Turnover Assays with TS and PRX
2.9. Reaction of TS with PRX3-SH vs. PRX3-SOH
2.10. SEC-MALS Analysis to Measure PRX Size Distribution
3. Results
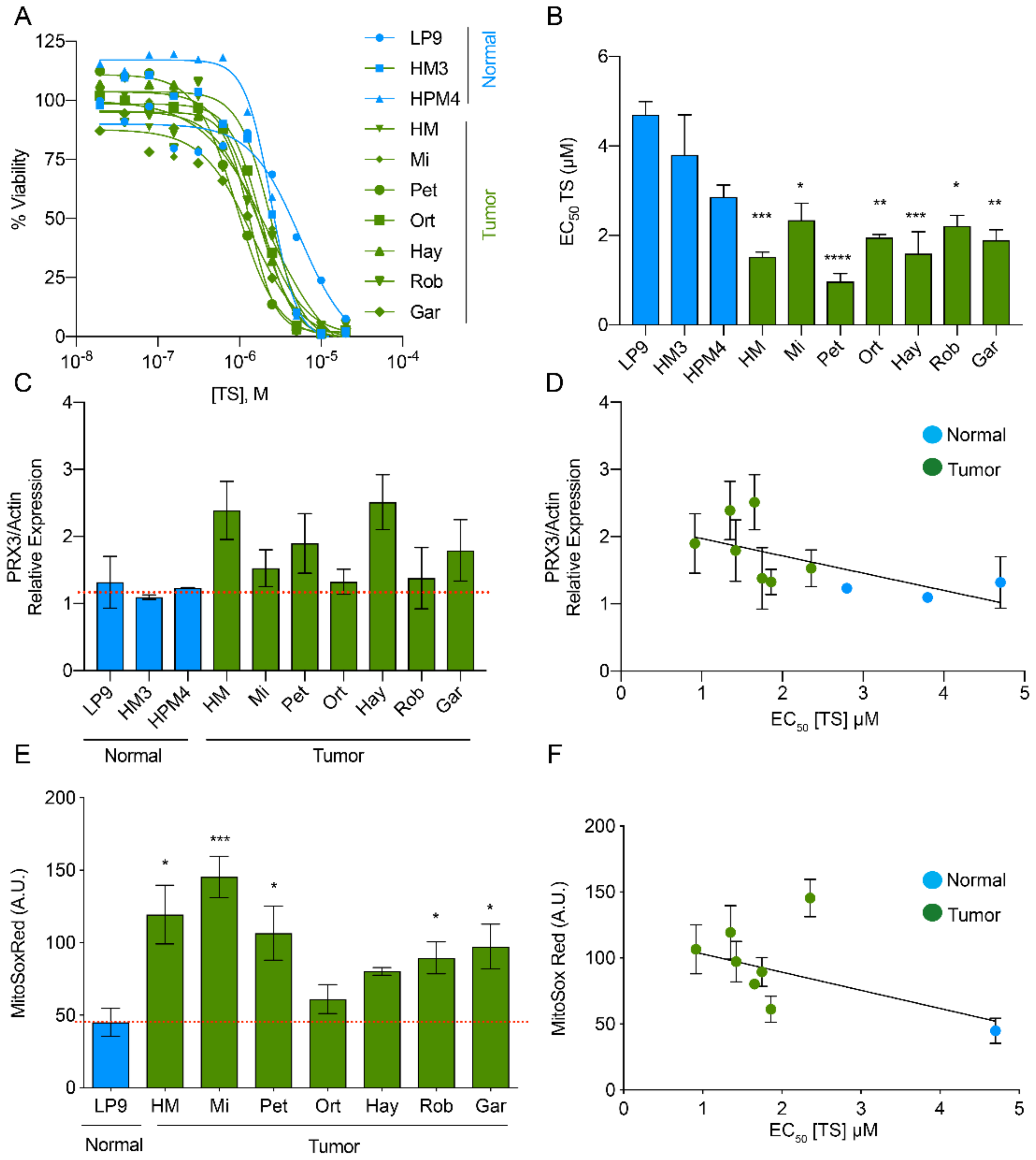
3.1. PRX3 Expression and Mitochondrial ROS Levels Correlate with Sensitivity to Thiostrepton
3.2. In Vitro (Cellular) Specificity of TS for Mitochondrial PRX3
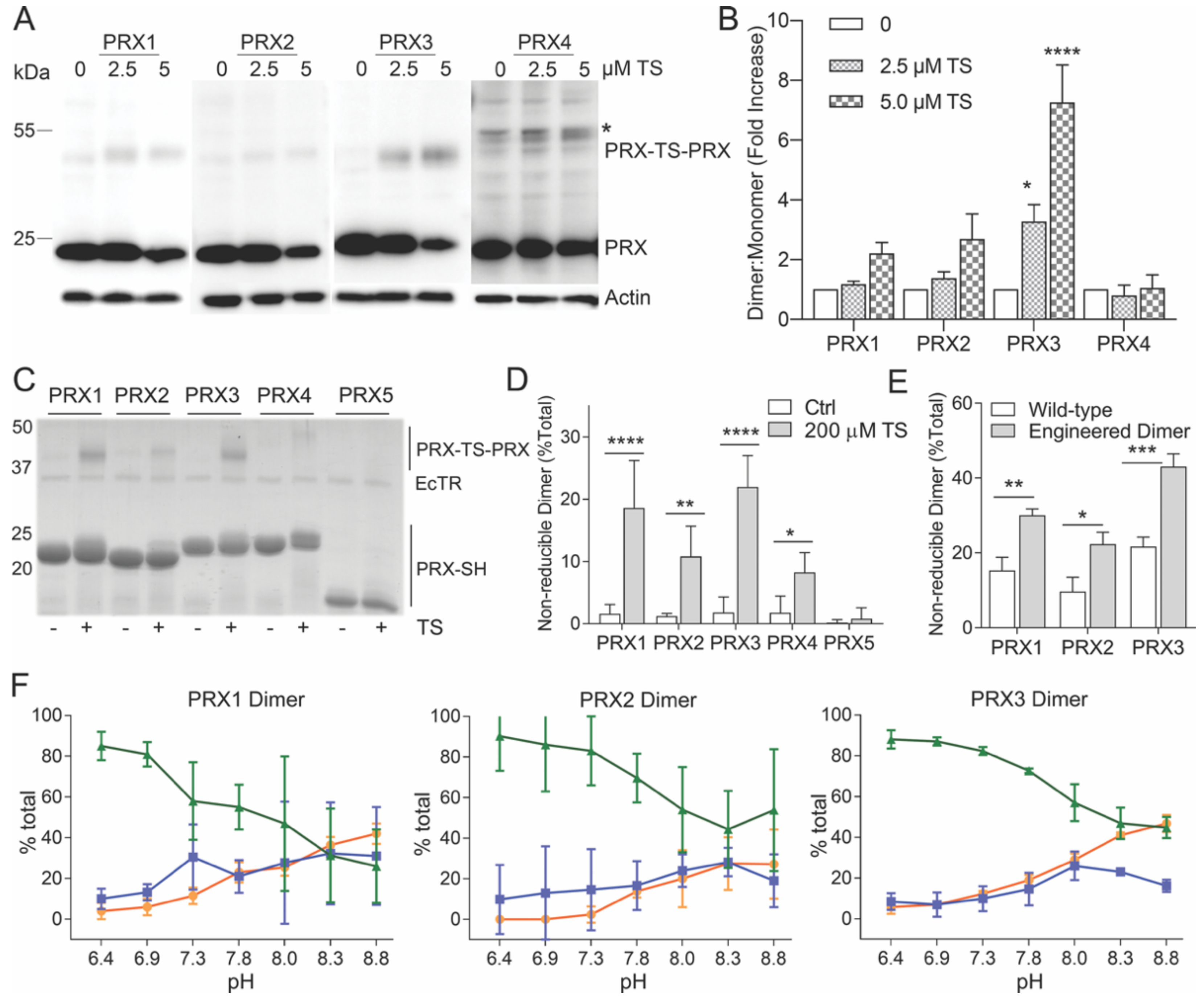
3.3. Specificity of TS for Recombinant PRX3
3.4. Thiostrepton Preferentially Adducts Dimeric PRX Species
3.5. TS Adducts PRX Dimers Only at Mitochondrial pH
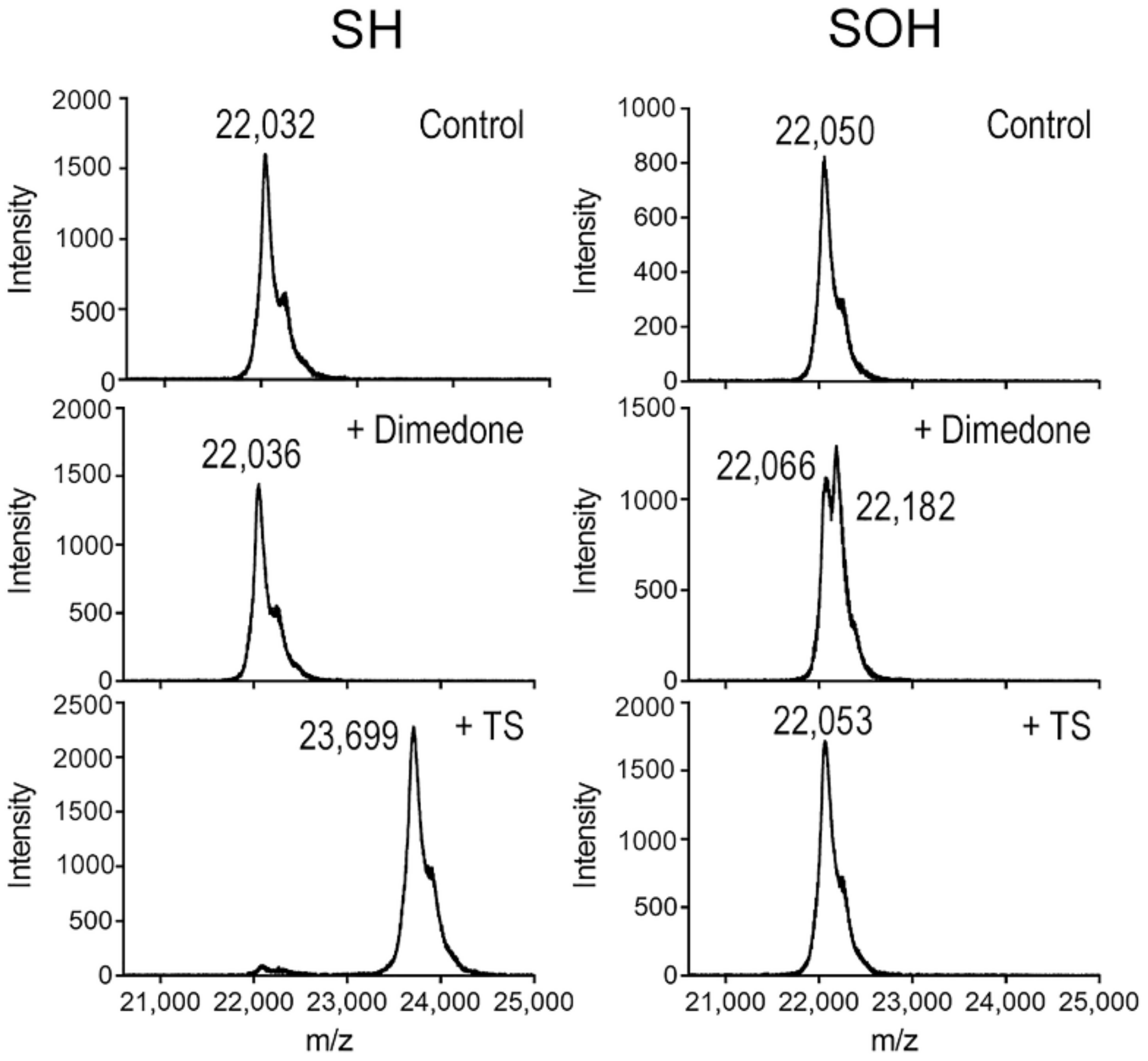
3.6. TS Reacts with the Reduced Cys Thiolate in PRX3, but Not the Sulfenic Acid Intermediate
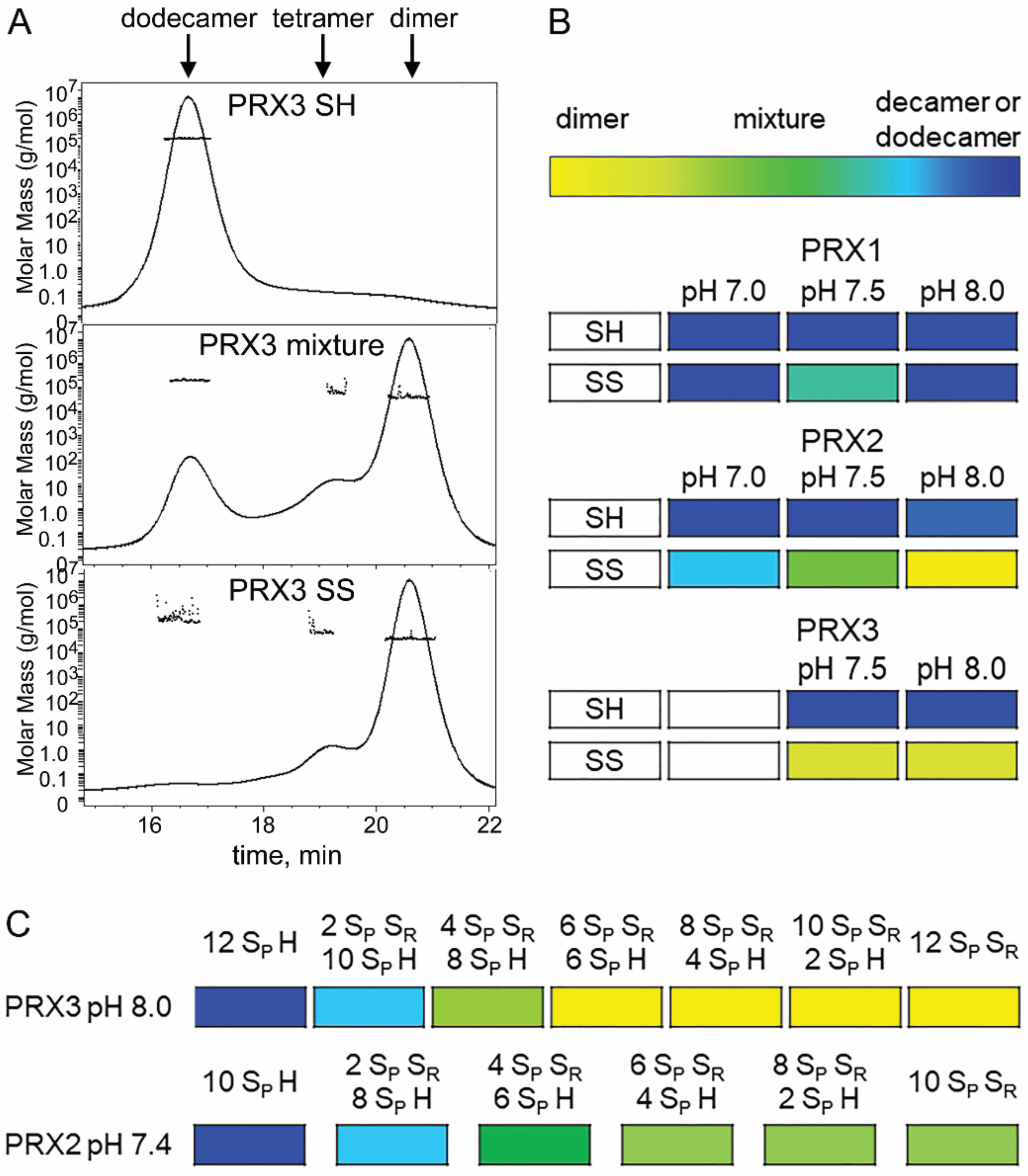
3.7. PRX3 Is More Likely to Be Found as Dimers than PRX1 or PRX2
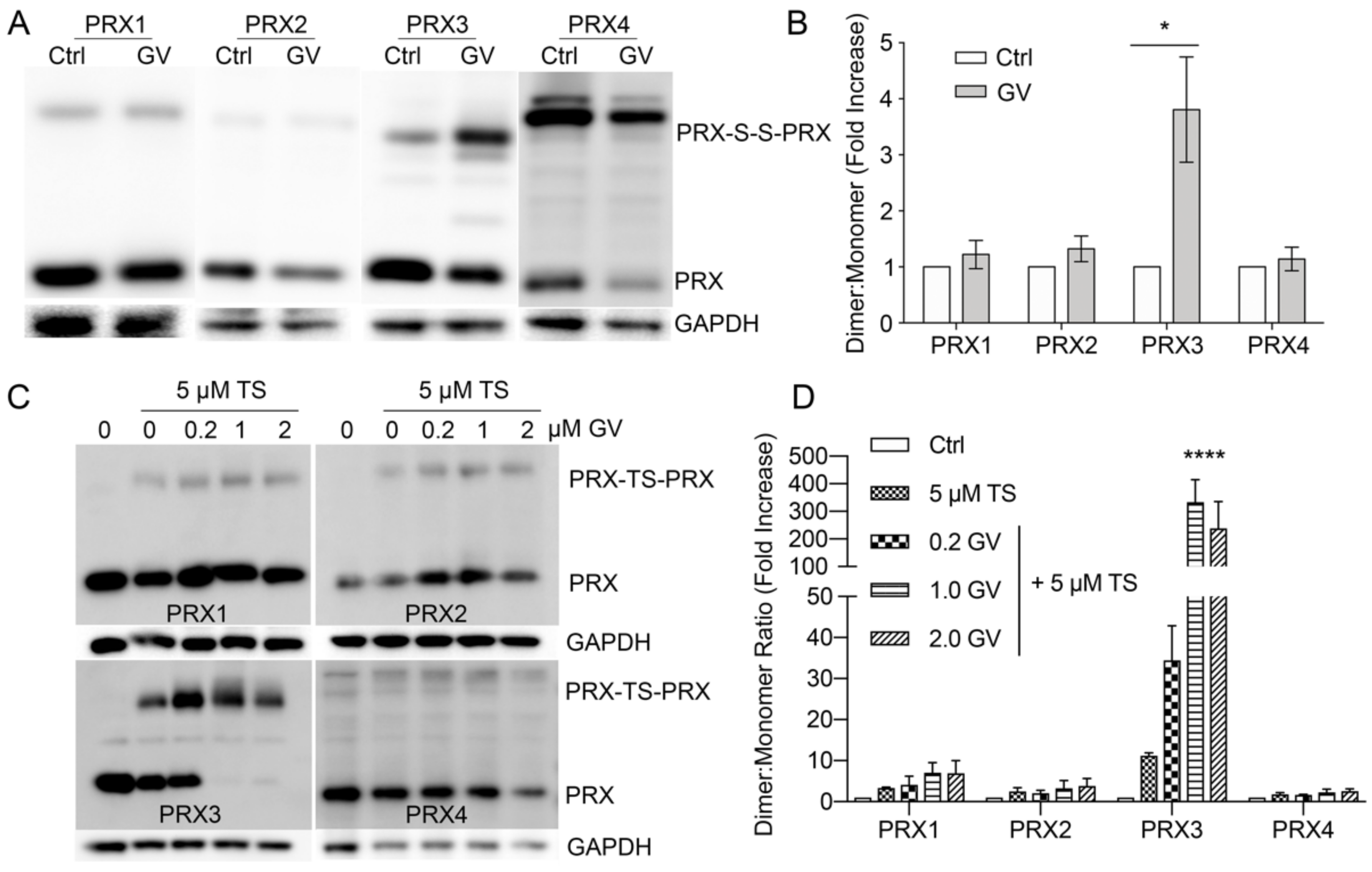
3.8. Gentian Violet Potentiates TS Adduction of PRX3 but Not PRX1 and PRX2
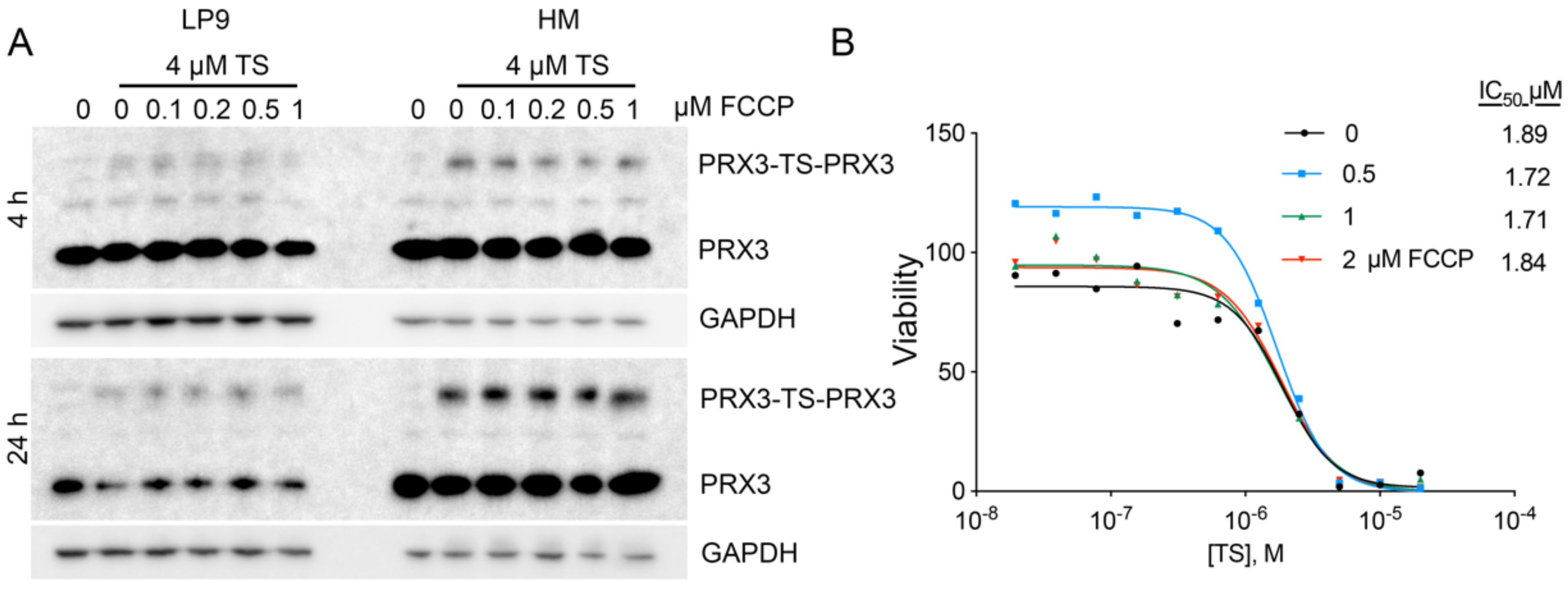
3.9. Disruption of the Mitochondrial Membrane Potential Does Not Affect TS-Mediated Crosslinking of PRX3
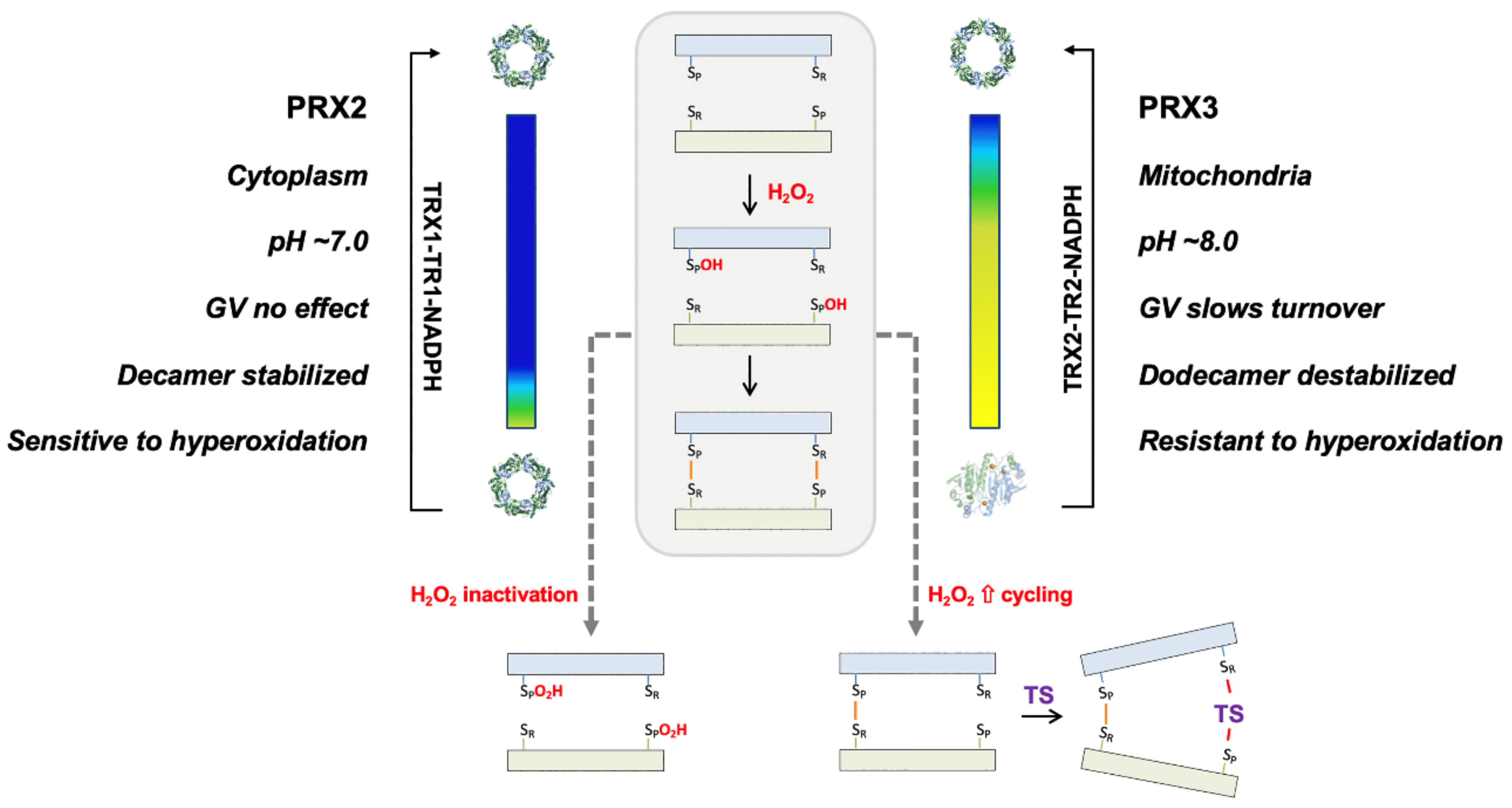
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; Newick, K.; Nelson, K.J.; Wozniak, A.N.; Beuschel, S.; Leavitt, B.; Bhave, A.; Butnor, K.; Koenig, A.; Chouchani, E.T.; et al. Disabling mitochondrial peroxide metabolism via combinatorial targeting of peroxiredoxin 3 as an effective therapeutic approach for malignant mesothelioma. PLoS ONE 2015, 10, e0127310. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; Cunniff, B.; Preston, K.; Held, P.; Arbiser, J.; Pass, H.; Mossman, B.; Shukla, A.; Heintz, N. Peroxiredoxin 3 is a redox-dependent target of thiostrepton in malignant mesothelioma cells. PLoS ONE 2012, 7, e39404. [Google Scholar] [CrossRef] [PubMed]
- Forshaw, T.E.; Holmila, R.; Nelson, K.J.; Lewis, J.E.; Kemp, M.L.; Tsang, A.W.; Poole, L.B.; Lowther, W.T.; Furdui, C.M. Peroxiredoxins in cancer and response to radiation therapies. Antioxidants 2019, 8, 11. [Google Scholar] [CrossRef]
- Scalcon, V.; Bindoli, A.; Rigobello, M.P. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radic. Biol. Med. 2018, 127, 62–79. [Google Scholar] [CrossRef]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef]
- Poynton, R.A.; Peskin, A.V.; Haynes, A.C.; Lowther, W.T.; Hampton, M.B.; Winterbourn, C.C. Kinetic analysis of structural influences on the susceptibility of peroxiredoxins 2 and 3 to hyperoxidation. Biochem. J. 2016, 473, 411–421. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Nelson, K.J.; Haynes, A.C.; Lee, J.; Reisz, J.A.; Graff, A.H.; Clodfelter, J.E.; Parsonage, D.; Poole, L.B.; Furdui, C.M.; et al. Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. [Google Scholar] [CrossRef]
- Haynes, A.C.; Qian, J.; Reisz, J.A.; Furdui, C.M.; Lowther, W.T. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J. Biol. Chem. 2013, 288, 29714–29723. [Google Scholar] [CrossRef]
- Ismail, T.; Kim, Y.; Lee, H.; Lee, D.S.; Lee, H.S. Interplay between mitochondrial peroxiredoxins and ROS in cancer development and progression. Int. J. Mol. Sci. 2019, 20, 4407. [Google Scholar] [CrossRef]
- Reeves, S.A.; Parsonage, D.; Nelson, K.J.; Poole, L.B. Kinetic and thermodynamic features reveal that Escherichia coli BCP is an unusually versatile peroxiredoxin. Biochemistry 2011, 50, 8970–8981. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, Z.; Johnson, L.C.; Weissbach, H.; Brot, N.; Lively, M.O.; Lowther, W.T. Free methionine-(R)-sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proc. Natl. Acad. Sci. USA 2007, 104, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 2010, 402, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.J.; Parsonage, D.; Hall, A.; Karplus, P.A.; Poole, L.B. Cysteine pK(a) values for the bacterial peroxiredoxin AhpC. Biochemistry 2008, 47, 12860–12868. [Google Scholar] [CrossRef]
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol. Life Sci. 2009, 66, 3663–3673. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Yewdall, N.A.; Peskin, A.V.; Hampton, M.B.; Goldstone, D.C.; Pearce, F.G.; Gerrard, J.A. Quaternary structure influences the peroxidase activity of peroxiredoxin 3. Biochem. Biophys. Res. Commun. 2018, 497, 558–563. [Google Scholar] [CrossRef]
- Cao, Z.; Roszak, A.W.; Gourlay, L.J.; Lindsay, J.G.; Isaacs, N.W. Bovine mitochondrial peroxiredoxin III forms a two-ring catenane. Structure 2005, 13, 1661–1664. [Google Scholar] [CrossRef]
- Jonsson, T.J.; Johnson, L.C.; Lowther, W.T. Structure of the sulphiredoxin-peroxiredoxin complex reveals an essential repair embrace. Nature 2008, 451, 98–101. [Google Scholar] [CrossRef]
- Jonsson, T.J.; Johnson, L.C.; Lowther, W.T. Protein engineering of the quaternary sulfiredoxin.peroxiredoxin enzyme.substrate complex reveals the molecular basis for cysteine sulfinic acid phosphorylation. J. Biol. Chem. 2009, 284, 33305–33310. [Google Scholar] [CrossRef]
- Loberg, M.A.; Hurtig, J.E.; Graff, A.H.; Allan, K.M.; Buchan, J.A.; Spencer, M.K.; Kelly, J.E.; Clodfelter, J.E.; Morano, K.A.; Lowther, W.T.; et al. Aromatic residues at the dimer-dimer interface in the peroxiredoxin Tsa1 facilitate decamer formation and biological function. Chem. Res. Toxicol. 2019, 32, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Parsonage, D.; Youngblood, D.S.; Sarma, G.N.; Wood, Z.A.; Karplus, P.A.; Poole, L.B. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 2005, 44, 10583–10592. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Cho, Y.Y.; Shim, M.S.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeted drug delivery in cancers. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165808. [Google Scholar] [CrossRef] [PubMed]
- Han, J.C.; Han, G.Y. A procedure for quantitative determination of tris(2-carboxyethyl)phosphine, an odorless reducing agent more stable and effective than dithiothreitol. Anal. Biochem. 1994, 220, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Setterdahl, A.T.; Chivers, P.T.; Hirasawa, M.; Lemaire, S.D.; Keryer, E.; Miginiac-Maslow, M.; Kim, S.K.; Mason, J.; Jacquot, J.P.; Longbine, C.C.; et al. Effect of pH on the oxidation-reduction properties of thioredoxins. Biochemistry 2003, 42, 14877–14884. [Google Scholar] [CrossRef] [PubMed]
- Reisz, J.A.; Bechtold, E.; King, S.B.; Poole, L.B.; Furdui, C.M. Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 2013, 280, 6150–6161. [Google Scholar] [CrossRef]
- Poole, L.B.; Furdui, C.M.; King, S.B. Introduction to approaches and tools for the evaluation of protein cysteine oxidation. Essays Biochem. 2020, 64, 1–17. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Drug delivery to mitochondria: The key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef]
- Gadelha, F.R.; Moreno, S.N.; De Souza, W.; Cruz, F.S.; Docampo, R. The mitochondrion of Trypanosoma cruzi is a target of crystal violet toxicity. Mol. Biochem. Parasitol. 1989, 34, 117–126. [Google Scholar] [CrossRef]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Lehtonen, S.; Sormunen, R.; Kaarteenaho-Wiik, R.; Kang, S.W.; Rhee, S.G.; Soini, Y. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J. Pathol. 2002, 196, 316–323. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell. Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Pearson, A.G.; Pullar, J.M.; Jonsson, T.J.; Lowther, W.T.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 2009, 421, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Stein, K.T.; Moon, S.J.; Nguyen, A.N.; Sikes, H.D. Kinetic modeling of H2O2 dynamics in the mitochondria of HeLa cells. PLoS Comput. Biol. 2020, 16, e1008202. [Google Scholar] [CrossRef]
- Pastor-Flores, D.; Talwar, D.; Pedre, B.; Dick, T.P. Real-time monitoring of peroxiredoxin oligomerization dynamics in living cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16313–16323. [Google Scholar] [CrossRef]
- Morais, M.A.; Giuseppe, P.O.; Souza, T.A.; Alegria, T.G.; Oliveira, M.A.; Netto, L.E.; Murakami, M.T. How pH modulates the dimer-decamer interconversion of 2-Cys peroxiredoxins from the Prx1 subfamily. J. Biol. Chem. 2015, 290, 8582–8590. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Cunniff, B.; Benson, K.; Stumpff, J.; Newick, K.; Held, P.; Taatjes, D.; Joseph, J.; Kalyanaraman, B.; Heintz, N.H. Mitochondrial-targeted nitroxides disrupt mitochondrial architecture and inhibit expression of peroxiredoxin 3 and FOXM1 in malignant mesothelioma cells. J. Cell Physiol. 2013, 228, 835–845. [Google Scholar] [CrossRef]
- Qiao, S.; Lamore, S.D.; Cabello, C.M.; Lesson, J.L.; Munoz-Rodriguez, J.L.; Wondrak, G.T. Thiostrepton is an inducer of oxidative and proteotoxic stress that impairs viability of human melanoma cells but not primary melanocytes. Biochem. Pharmacol. 2012, 83, 1229–1240. [Google Scholar] [CrossRef]
- Bowling, B.D.; Doudican, N.; Manga, P.; Orlow, S.J. Inhibition of mitochondrial protein translation sensitizes melanoma cells to arsenic trioxide cytotoxicity via a reactive oxygen species dependent mechanism. Cancer Chemother. Pharmacol. 2008, 63, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox paradox: A novel approach to therapeutics-resistant cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, K.J.; Messier, T.; Milczarek, S.; Saaman, A.; Beuschel, S.; Gandhi, U.; Heintz, N.; Smalley, T.L., Jr.; Lowther, W.T.; Cunniff, B. Unique Cellular and Biochemical Features of Human Mitochondrial Peroxiredoxin 3 Establish the Molecular Basis for Its Specific Reaction with Thiostrepton. Antioxidants 2021, 10, 150. https://doi.org/10.3390/antiox10020150
Nelson KJ, Messier T, Milczarek S, Saaman A, Beuschel S, Gandhi U, Heintz N, Smalley TL Jr., Lowther WT, Cunniff B. Unique Cellular and Biochemical Features of Human Mitochondrial Peroxiredoxin 3 Establish the Molecular Basis for Its Specific Reaction with Thiostrepton. Antioxidants. 2021; 10(2):150. https://doi.org/10.3390/antiox10020150
Chicago/Turabian StyleNelson, Kimberly J., Terri Messier, Stephanie Milczarek, Alexis Saaman, Stacie Beuschel, Uma Gandhi, Nicholas Heintz, Terrence L. Smalley, Jr., W. Todd Lowther, and Brian Cunniff. 2021. "Unique Cellular and Biochemical Features of Human Mitochondrial Peroxiredoxin 3 Establish the Molecular Basis for Its Specific Reaction with Thiostrepton" Antioxidants 10, no. 2: 150. https://doi.org/10.3390/antiox10020150
APA StyleNelson, K. J., Messier, T., Milczarek, S., Saaman, A., Beuschel, S., Gandhi, U., Heintz, N., Smalley, T. L., Jr., Lowther, W. T., & Cunniff, B. (2021). Unique Cellular and Biochemical Features of Human Mitochondrial Peroxiredoxin 3 Establish the Molecular Basis for Its Specific Reaction with Thiostrepton. Antioxidants, 10(2), 150. https://doi.org/10.3390/antiox10020150