
Cholesterol Dysmetabolism in Alzheimer’s Disease: A Starring Role for Astrocytes?

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Astrocyte Functions and Their Contribution to Alzheimer’s Disease
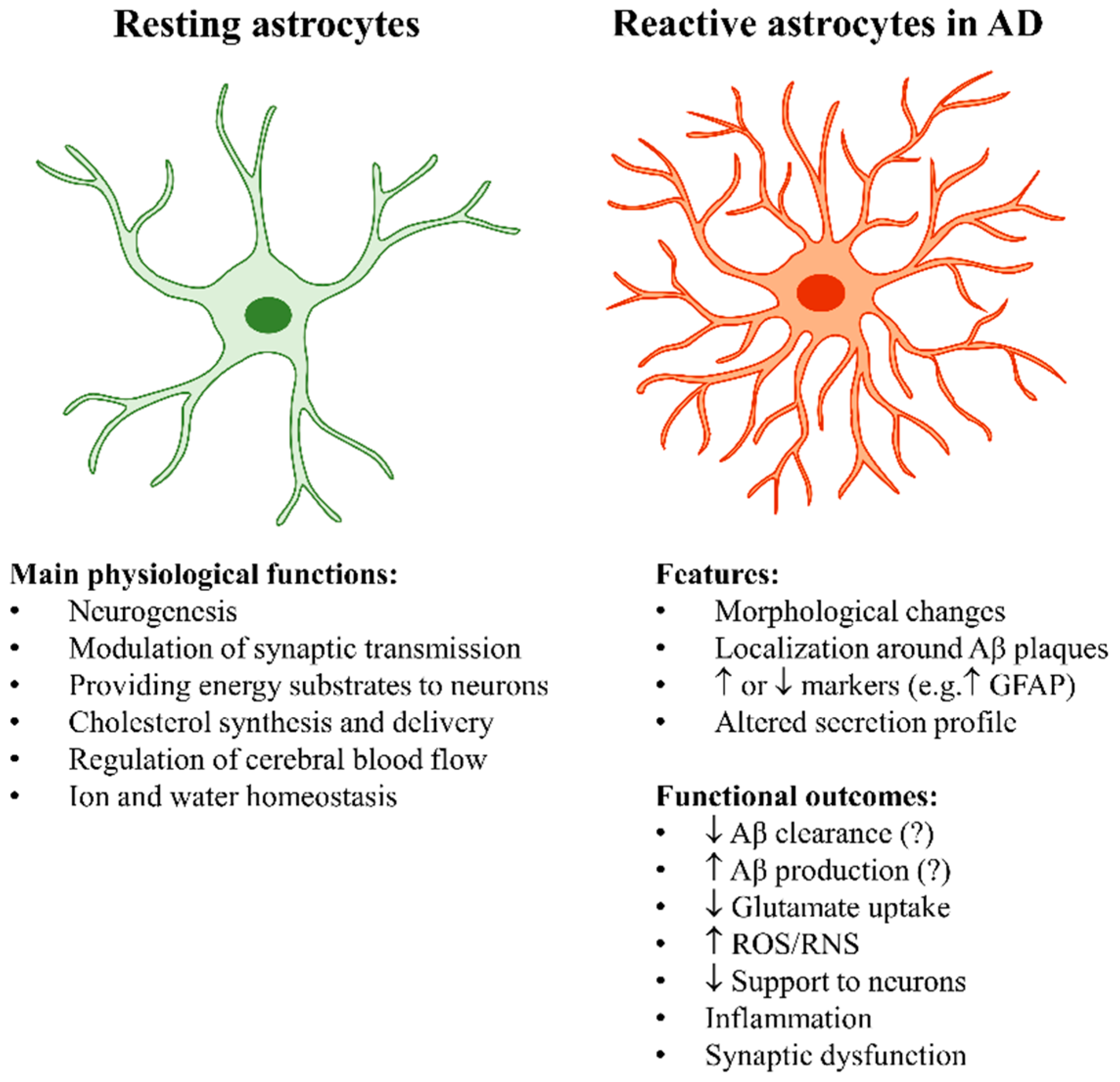
2.1. The “Stars” of the Brain: The Key Physiological Roles of Astrocytes
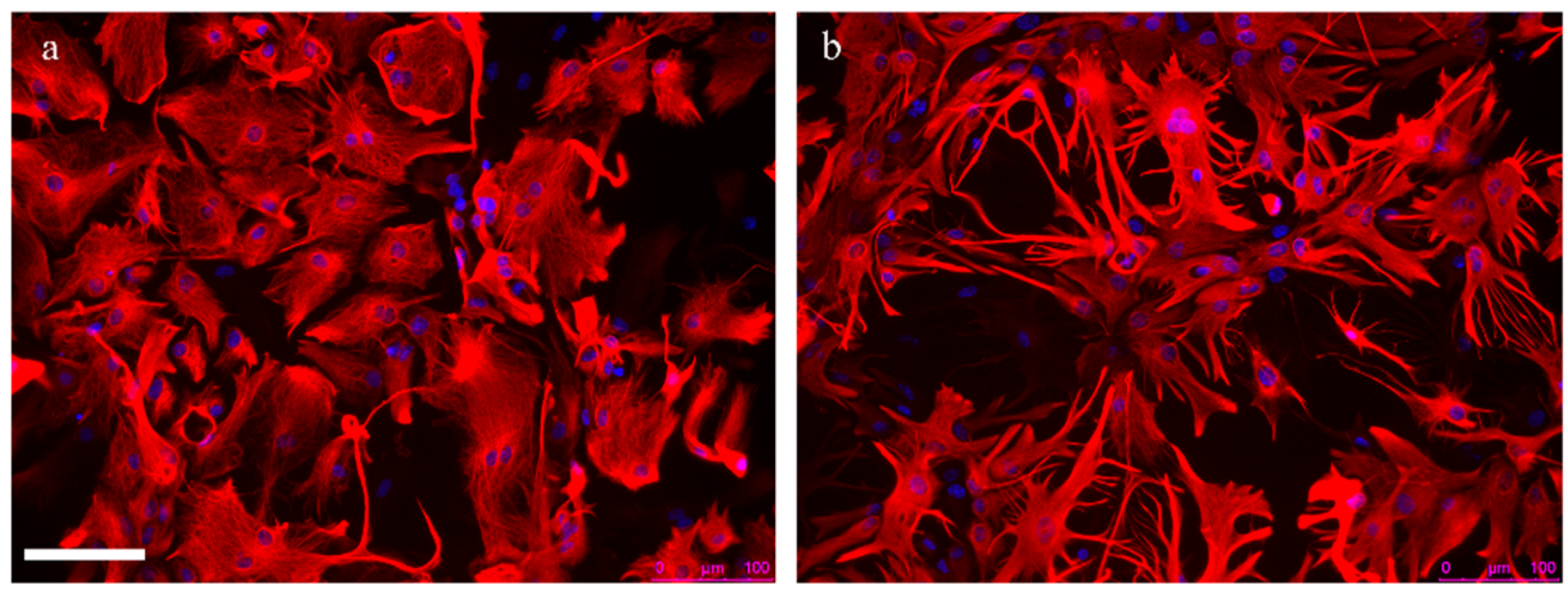
2.2. Astrocyte Reactivity in Alzheimer’s Disease
2.2.1. Astrocyte Reactivity and Its Complexity
2.2.2. The Main Outcomes of Astrocyte Reactivity in Alzheimer’s Disease
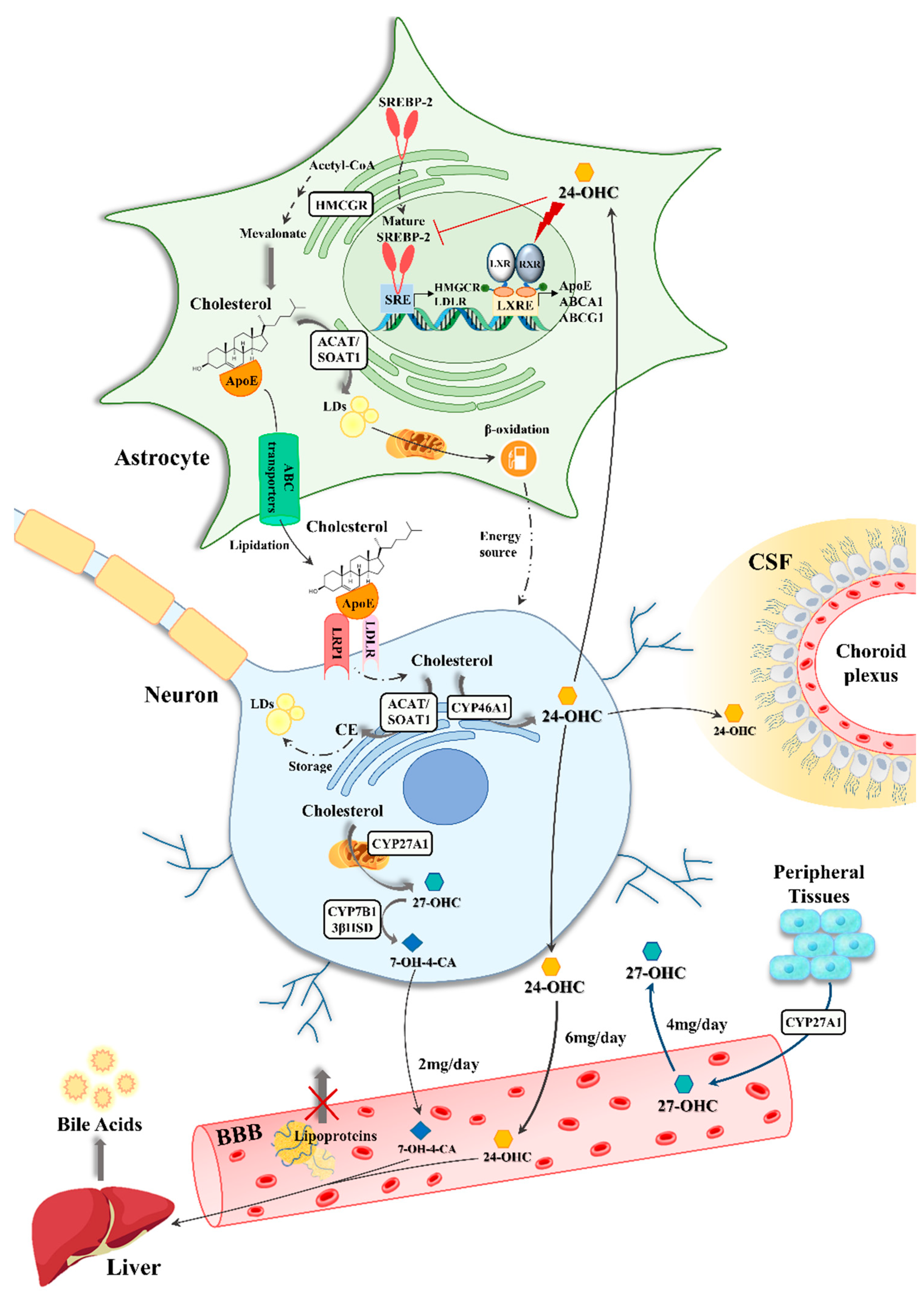
3. The Involvement of Astrocytes in Brain Cholesterol Dysmetabolism
3.1. Astrocyte-Neuron Interplay in Brain Cholesterol Metabolism
3.2. The Impact of Oxysterols on Astrocytes in Alzheimer’s Disease
3.3. The Role of ApoE4 Astrocytes in Alzheimer’s Disease
4. Conclusions
Funding
Conflicts of Interest
References
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimers Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Gamba, P.; Testa, G.; Gargiulo, S.; Staurenghi, E.; Poli, G.; Leonarduzzi, G. Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escartin, C.; Guillemaud, O.; Carrillo-de Sauvage, M.A. Questions and (some) answers on reactive astrocytes. Glia 2019, 67, 2221–2247. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zou, L.; Meng, L.; Qiang, G.; Yan, M.; Zhang, Z. Cholesterol Metabolism in Neurodegenerative Diseases: Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 2021, 58, 2183–2201. [Google Scholar] [CrossRef]
- Varma, V.R.; Busra Luleci, H.; Oommen, A.M.; Varma, S.; Blackshear, C.T.; Griswold, M.E.; An, Y.; Roberts, J.A.; O’Brien, R.; Pletnikova, O.; et al. Abnormal brain cholesterol homeostasis in Alzheimer’s disease-a targeted metabolomic and transcriptomic study. NPJ Aging Mech. Dis. 2021, 7, 11. [Google Scholar] [CrossRef]
- Saiz-Vazquez, O.; Puente-Martinez, A.; Ubillos-Landa, S.; Pacheco-Bonrostro, J.; Santabarbara, J. Cholesterol and Alzheimer’s Disease Risk: A Meta-Meta-Analysis. Brain Sci. 2020, 10, 386. [Google Scholar] [CrossRef]
- Marwarha, G.; Ghribi, O. Does the oxysterol 27-hydroxycholesterol underlie Alzheimer’s disease-Parkinson’s disease overlap? Exp. Gerontol. 2015, 68, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, I.H.; Polidori, M.C.; Griffiths, H.R. Hypercholesterolaemia-induced oxidative stress at the blood-brain barrier. Biochem. Soc. Trans. 2014, 42, 1001–1005. [Google Scholar] [CrossRef]
- Virchow, R. Die Cellularpathologie in Ihrer Begründung auf Physiologische und Pathologische Gewebelehre; Hirschwald: Berlin, Germany, 1858. [Google Scholar]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of astrocytic form and function. Methods Mol. Biol. 2012, 814, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549.e9. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.W.; Stiles, L.; et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat. Commun. 2021, 12, 3958. [Google Scholar] [CrossRef]
- Stogsdill, J.A.; Ramirez, J.; Liu, D.; Kim, Y.H.; Baldwin, K.T.; Enustun, E.; Ejikeme, T.; Ji, R.R.; Eroglu, C. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 2017, 551, 192–197. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2021, 590, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Valtcheva, S.; Venance, L. Control of Long-Term Plasticity by Glutamate Transporters. Front. Synaptic Neurosci. 2019, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Noriega-Prieto, J.A.; Araque, A. Sensing and Regulating Synaptic Activity by Astrocytes at Tripartite Synapse. Neurochem. Res. 2021, 46, 2580–2585. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.O.; Petravicz, J.; Rikhye, R.V.; Garcia, R.; Mellios, N.; Sur, M. Astrocyte glutamate uptake coordinates experience-dependent, eye-specific refinement in developing visual cortex. Glia 2021, 69, 1723–1735. [Google Scholar] [CrossRef]
- Jourdain, P.; Bergersen, L.H.; Bhaukaurally, K.; Bezzi, P.; Santello, M.; Domercq, M.; Matute, C.; Tonello, F.; Gundersen, V.; Volterra, A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007, 10, 331–339. [Google Scholar] [CrossRef]
- Papouin, T.; Dunphy, J.M.; Tolman, M.; Dineley, K.T.; Haydon, P.G. Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron 2017, 94, 840–854. [Google Scholar] [CrossRef] [Green Version]
- Corkrum, M.; Covelo, A.; Lines, J.; Bellocchio, L.; Pisansky, M.; Loke, K.; Quintana, R.; Rothwell, P.E.; Lujan, R.; Marsicano, G.; et al. Dopamine-Evoked Synaptic Regulation in the Nucleus Accumbens Requires Astrocyte Activity. Neuron 2020, 105, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesecker, K.R.; Srienc, A.I.; Shimoda, A.M.; Agarwal, A.; Bergles, D.E.; Kofuji, P.; Newman, E.A. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J. Neurosci. 2016, 36, 9435–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef] [PubMed]
- He, X.F.; Liu, D.X.; Zhang, Q.; Liang, F.Y.; Dai, G.Y.; Zeng, J.S.; Pei, Z.; Xu, G.Q.; Lan, Y. Voluntary Exercise Promotes Glymphatic Clearance of Amyloid Beta and Reduces the Activation of Astrocytes and Microglia in Aged Mice. Front. Mol. Neurosci. 2017, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Machler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martin, A.; Romero-Gomez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lachance, B.B.; Mattson, M.P.; Jia, X. Glucose metabolic crosstalk and regulation in brain function and diseases. Prog. Neurobiol. 2021, 204, 102089. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Bushong, E.A.; Price, D.L.; Smarr, B.L.; Phung, V.; Terada, M.; Ellisman, M.H.; Pekny, M. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl. Acad. Sci. USA 2006, 103, 17513–17518. [Google Scholar] [CrossRef] [Green Version]
- Frik, J.; Merl-Pham, J.; Plesnila, N.; Mattugini, N.; Kjell, J.; Kraska, J.; Gomez, R.M.; Hauck, S.M.; Sirko, S.; Gotz, M. Cross-talk between monocyte invasion and astrocyte proliferation regulates scarring in brain injury. EMBO Rep. 2018, 19, e45294. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, W.; Middeldorp, J.; Kooijman, L.; Sluijs, J.A.; Kooi, E.J.; Moeton, M.; Freriks, M.; Mizee, M.R.; Hol, E.M. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 492–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.H.; Yang, A.W.; Chang, C.H.; Perng, M.D. Elevated GFAP isoform expression promotes protein aggregation and compromises astrocyte function. FASEB J. 2021, 35, e21614. [Google Scholar] [CrossRef]
- Crupi, R.; Cambiaghi, M.; Spatz, L.; Hen, R.; Thorn, M.; Friedman, E.; Vita, G.; Battaglia, F. Reduced adult neurogenesis and altered emotional behaviors in autoimmune-prone B-cell activating factor transgenic mice. Biol. Psychiatry 2010, 67, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Munch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Tyzack, G.E.; Sitnikov, S.; Barson, D.; Adams-Carr, K.L.; Lau, N.K.; Kwok, J.C.; Zhao, C.; Franklin, R.J.; Karadottir, R.T.; Fawcett, J.W.; et al. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 2014, 5, 4294. [Google Scholar] [CrossRef] [Green Version]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; McDonald, J.; Cuyugan, L.; Aldrich, J.; Kurdoglu, A.; Adkins, J.; Serrano, G.; Beach, T.G.; Craig, D.W.; Valla, J.; et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 2015, 36, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; de Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Muzikansky, A.; Gomez-Isla, T.; Growdon, J.H.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2013, 72, 462–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, E.; Morrison, W.; Hudry, E.; Arbel-Ornath, M.; Bacskai, B.J.; Gomez-Isla, T.; Stanley, H.E.; Hyman, B.T. Topological analyses in APP/PS1 mice reveal that astrocytes do not migrate to amyloid-beta plaques. Proc. Natl. Acad. Sci. USA 2015, 112, 15556–15561. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, D.S.; Jones, E.V.; Quesseveur, G.; Davoli, M.A.; Ferreira, T.A.; Quirion, R.; Davoli, M.A.; Mechawar, N.; Murai, K.K. High Resolution Dissection of Reactive Glial Nets in Alzheimer’s Disease. Sci. Rep. 2016, 6, 24544. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Mielke, M.L.; Gomez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Begard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Kashon, M.L.; Ross, G.W.; O’Callaghan, J.P.; Miller, D.B.; Petrovitch, H.; Burchfiel, C.M.; Sharp, D.S.; Markesbery, W.R.; Davis, D.G.; Hardman, J.; et al. Associations of cortical astrogliosis with cognitive performance and dementia status. J. Alzheimers Dis. 2004, 6, 595–604; discussion 581–673. [Google Scholar] [CrossRef]
- Hartlage-Rubsamen, M.; Zeitschel, U.; Apelt, J.; Gartner, U.; Franke, H.; Stahl, T.; Gunther, A.; Schliebs, R.; Penkowa, M.; Bigl, V.; et al. Astrocytic expression of the Alzheimer’s disease beta-secretase (BACE1) is stimulus-dependent. Glia 2003, 41, 169–179. [Google Scholar] [CrossRef]
- Liang, Y.; Raven, F.; Ward, J.F.; Zhen, S.; Zhang, S.; Sun, H.; Miller, S.J.; Choi, S.H.; Tanzi, R.E.; Zhang, C. Upregulation of Alzheimer’s Disease Amyloid-beta Protein Precursor in Astrocytes Both in vitro and in vivo. J. Alzheimers Dis. 2020, 76, 1071–1082. [Google Scholar] [CrossRef]
- Chacon-Quintero, M.V.; Pineda-Lopez, L.G.; Villegas-Lanau, C.A.; Posada-Duque, R.; Cardona-Gomez, G.P. Beta-Secretase 1 Underlies Reactive Astrocytes and Endothelial Disruption in Neurodegeneration. Front. Cell. Neurosci. 2021, 15, 656832. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Hwang, E.M.; Sim, H.J.; Cho, H.J.; Boo, J.H.; Oh, S.S.; Kim, S.U.; Mook-Jung, I. Interferon gamma stimulates beta-secretase expression and sAPPbeta production in astrocytes. Biochem. Biophys. Res. Commun. 2003, 307, 922–927. [Google Scholar] [CrossRef]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Abeta production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Cho, H.J.; Kim, Y.W.; Hwang, J.Y.; Mook-Jung, I. Abeta-induced Ca(2+) influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem. Biophys. Res. Commun. 2012, 425, 649–655. [Google Scholar] [CrossRef]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Kayed, R. Astrocytes contain amyloid-beta annular protofibrils in Alzheimer’s disease brains. FEBS Lett. 2011, 585, 3052–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihlaja, R.; Koistinaho, J.; Malm, T.; Sikkila, H.; Vainio, S.; Koistinaho, M. Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 2008, 56, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.M.; Veerhuis, R.; Holmqvist, B.; Janciauskiene, S. Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia 2009, 57, 978–988. [Google Scholar] [CrossRef]
- Sollvander, S.; Nikitidou, E.; Brolin, R.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.F.; Turk, J.; et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Abeta Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Basak, J.M.; Verghese, P.B.; Yoon, H.; Kim, J.; Holtzman, D.M. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J. Biol. Chem. 2012, 287, 13959–13971. [Google Scholar] [CrossRef] [Green Version]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef] [Green Version]
- Li, M.Z.; Zheng, L.J.; Shen, J.; Li, X.Y.; Zhang, Q.; Bai, X.; Wang, Q.S.; Ji, J.G. SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen. Res. 2018, 13, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Davey, K.; Tsartsalis, S.; Khozoie, C.; Fancy, N.; Tang, S.S.; Liaptsi, E.; Weinert, M.; McGarry, A.; Muirhead, R.C.J.; et al. Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Staurenghi, E.; Cerrato, V.; Gamba, P.; Testa, G.; Giannelli, S.; Leoni, V.; Caccia, C.; Buffo, A.; Noble, W.; Perez-Nievas, B.G.; et al. Oxysterols present in Alzheimer’s disease brain induce synaptotoxicity by activating astrocytes: A major role for lipocalin-2. Redox Biol. 2021, 39, 101837. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Takanashi, Y.; Sugita, K.; Miyazawa, M.; Yanagihara, R.; Yasuda, K.; Onouchi, H.; Kawabe, N.; Nakata, M.; Yamamoto, Y.; et al. Endogenous reactive oxygen species cause astrocyte defects and neuronal dysfunctions in the hippocampus: A new model for aging brain. Aging Cell 2017, 16, 39–51. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Brahmachari, S.; Fung, Y.K.; Pahan, K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 2006, 26, 4930–4939. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Gupta, S.; Joshi, N.; Sharma, S.; Singh, S. Astrocyte activation and neurotoxicity: A study in different rat brain regions and in rat C6 astroglial cells. Environ. Toxicol. Pharmacol. 2015, 40, 122–139. [Google Scholar] [CrossRef]
- Liu, F.T.; Xu, S.M.; Xiang, Z.H.; Li, X.N.; Li, J.; Yuan, H.B.; Sun, X.J. Molecular hydrogen suppresses reactive astrogliosis related to oxidative injury during spinal cord injury in rats. CNS Neurosci. Ther. 2014, 20, 778–786. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta 2004, 1742, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.G.; Ransom, B.R.; Chen, X.C.; Ye, Z.C. Dual pathways mediate beta-amyloid stimulated glutathione release from astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Adzic, M.; Stevanovic, I.; Josipovic, N.; Laketa, D.; Lavrnja, I.; Bjelobaba, I.M.; Bozic, I.; Jovanovic, M.; Milosevic, M.; Nedeljkovic, N. Extracellular ATP induces graded reactive response of astrocytes and strengthens their antioxidative defense in vitro. J. Neurosci. Res. 2017, 95, 1053–1066. [Google Scholar] [CrossRef]
- Wang, J.L.; Xu, C.J. Astrocytes autophagy in aging and neurodegenerative disorders. Biomed. Pharmacother. 2020, 122, 109691. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Cho, K.S.; Koh, J.Y. Oxidative injury triggers autophagy in astrocytes: The role of endogenous zinc. Glia 2009, 57, 1351–1361. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; LeDue, J.; Ko, R.W.; Mahler, J.; Murphy, T.H.; MacVicar, B.A. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Abeta plaques by iGluSnFR two-photon imaging. Nat. Commun. 2016, 7, 13441. [Google Scholar] [CrossRef] [Green Version]
- Mookherjee, P.; Green, P.S.; Watson, G.S.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G.; et al. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J. Alzheimers Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Feng, A.; Rock, R.B. Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem. Res. 2013, 38, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1712. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; Consortium, U.K.B.E.; North American Brain Expression, C.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deijk, A.F.; Camargo, N.; Timmerman, J.; Heistek, T.; Brouwers, J.F.; Mogavero, F.; Mansvelder, H.D.; Smit, A.B.; Verheijen, M.H. Astrocyte lipid metabolism i.is critical for synapse development and function in vivo. Glia 2017, 65, 670–682. [Google Scholar] [CrossRef]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Wood, W.G.; Li, L.; Muller, W.E.; Eckert, G.P. Cholesterol as a causative factor in Alzheimer’s disease: A debatable hypothesis. J. Neurochem. 2014, 129, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Segatto, M.; Leboffe, L.; Trapani, L.; Pallottini, V. Cholesterol homeostasis failure in the brain: Implications for synaptic dysfunction and cognitive decline. Curr. Med. Chem. 2014, 21, 2788–2802. [Google Scholar] [CrossRef]
- Petrov, A.M.; Kasimov, M.R.; Zefirov, A.L. Cholesterol in the Pathogenesis of Alzheimer’s, Parkinson’s Diseases and Autism: Link to Synaptic Dysfunction. Acta Nat. 2017, 9, 26–37. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanson, C.E.; Stopa, E.G.; McMillan, P.N. The blood-cerebrospinal fluid barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 101–131. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [Green Version]
- Nieweg, K.; Schaller, H.; Pfrieger, F.W. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 2009, 109, 125–134. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. The impairment of cholesterol metabolism in Huntington disease. Biochim. Biophys. Acta 2015, 1851, 1095–1105. [Google Scholar] [CrossRef]
- Anchisi, L.; Dessi, S.; Pani, A.; Mandas, A. Cholesterol homeostasis: A key to prevent or slow down neurodegeneration. Front. Physiol. 2012, 3, 486. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.G.; Ahmed, T.; Korovaichuk, A.; Venero, C.; Menchon, S.A.; Salas, I.; Munck, S.; Herreras, O.; Balschun, D.; Dotti, C.G. Constitutive hippocampal cholesterol loss underlies poor cognition in old rodents. EMBO Mol. Med. 2014, 6, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Investig. 1985, 76, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Chaves, E.P.; Narayanaswami, V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008, 3, 505–530. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.S.; Weickert, C.S.; Garner, B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 2008, 104, 1145–1166. [Google Scholar] [CrossRef]
- Kim, W.S.; Guillemin, G.J.; Glaros, E.N.; Lim, C.K.; Garner, B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. NeuroReport 2006, 17, 891–896. [Google Scholar] [CrossRef]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Costet, P.; Luo, Y.; Wang, N.; Tall, A.R. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J. Biol. Chem. 2000, 275, 28240–28245. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Gamba, P.; Staurenghi, E.; Testa, G.; Giannelli, S.; Sottero, B.; Leonarduzzi, G. A Crosstalk Between Brain Cholesterol Oxidation and Glucose Metabolism in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 556. [Google Scholar] [CrossRef] [Green Version]
- Wustner, D.; Mondal, M.; Tabas, I.; Maxfield, F.R. Direct observation of rapid internalization and intracellular transport of sterol by macrophage foam cells. Traffic 2005, 6, 396–412. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, N.; Miyazaki, A.; Takeya, M.; Horiuchi, S.; Chang, C.C.; Chang, T.Y.; Takahashi, K. Localization of human acyl-coenzyme A: Cholesterol acyltransferase-1 (ACAT-1) in macrophages and in various tissues. Am. J. Pathol. 2000, 156, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Koch, S.; Donarski, N.; Goetze, K.; Kreckel, M.; Stuerenburg, H.J.; Buhmann, C.; Beisiegel, U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 2001, 42, 1143–1151. [Google Scholar] [CrossRef]
- Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M.; Russell, D.W. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 2003, 278, 22980–22988. [Google Scholar] [CrossRef] [Green Version]
- Bjorkhem, I. Crossing the barrier: Oxysterols as cholesterol transporters and metabolic modulators in the brain. J. Intern. Med. 2006, 260, 493–508. [Google Scholar] [CrossRef]
- Meaney, S.; Bodin, K.; Diczfalusy, U.; Bjorkhem, I. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: Critical importance of the position of the oxygen function. J. Lipid Res. 2002, 43, 2130–2135. [Google Scholar] [CrossRef] [Green Version]
- Iuliano, L.; Crick, P.J.; Zerbinati, C.; Tritapepe, L.; Abdel-Khalik, J.; Poirot, M.; Wang, Y.; Griffiths, W.J. Cholesterol metabolites exported from human brain. Steroids 2015, 99, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.W.; Zheng, L.; Wang, Q. Regulation of cholesterol homeostasis by liver X receptors. Clin. Chim. Acta 2010, 411, 617–625. [Google Scholar] [CrossRef]
- Abildayeva, K.; Jansen, P.J.; Hirsch-Reinshagen, V.; Bloks, V.W.; Bakker, A.H.; Ramaekers, F.C.; de Vente, J.; Groen, A.K.; Wellington, C.L.; Kuipers, F.; et al. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E.E-mediated cholesterol efflux. J. Biol. Chem. 2006, 281, 12799–12808. [Google Scholar] [CrossRef] [Green Version]
- Bjorkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Asp. Med. 2009, 30, 171–179. [Google Scholar] [CrossRef]
- Meaney, S.; Heverin, M.; Panzenboeck, U.; Ekstrom, L.; Axelsson, M.; Andersson, U.; Diczfalusy, U.; Pikuleva, I.; Wahren, J.; Sattler, W.; et al. Novel route for elimination of brain oxysterols across the blood-brain barrier: Conversion into 7alpha-hydroxy-3-oxo-4-cholestenoic acid. J. Lipid Res. 2007, 48, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Heverin, M.; Bogdanovic, N.; Lutjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Bjorkhem, I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diczfalusy, U. On the formation and possible biological role of 25-hydroxycholesterol. Biochimie 2013, 95, 455–460. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Zhang, D.D.; Yu, H.L.; Ma, W.W.; Lu, Y.H.; Liu, Q.R.; Xiao, R. 27-Hydroxycholesterol regulates cholesterol synthesis and transport in C6 glioma cells. Neurotoxicology 2017, 59, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA 2007, 104, 6511–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Alrosan, A.Z.; Sharpe, L.J.; Brown, A.J.; Callaghan, R.; Gelissen, I.C. Regulation of ABCG4 transporter expression by sterols and LXR ligands. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129769. [Google Scholar] [CrossRef]
- Leoni, V.; Masterman, T.; Patel, P.; Meaney, S.; Diczfalusy, U.; Bjorkhem, I. Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J. Lipid Res. 2003, 44, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fanto, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, M.A.; Scheff, S.W. Oxidative s.stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kundig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-beta and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Head, E.; Garzon-Rodriguez, W.; Johnson, J.K.; Lott, I.T.; Cotman, C.W.; Glabe, C. Oxidation of Abeta and plaque biogenesis in Alzheimer’s disease and Down syndrome. Neurobiol. Dis. 2001, 8, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. NeuroSci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, A.; Hammouda, S.; Ghzaiel, I.; Hammami, S.; Khamlaoui, W.; Ahmed, S.H.; Lizard, G.; Hammami, M. Association Between Oxidative Stress and Altered.d Cholesterol Metabolism in Alzheimer’s Disease Patients. Curr. Alzheimer Res. 2020, 17, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Brown, J., 3rd; Theisler, C.; Silberman, S.; Magnuson, D.; Gottardi-Littell, N.; Lee, J.M.; Yager, D.; Crowley, J.; Sambamurti, K.; Rahman, M.M.; et al. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J. Biol. Chem. 2004, 279, 34674–34681. [Google Scholar] [CrossRef] [Green Version]
- Bogdanovic, N.; Bretillon, L.; Lund, E.G.; Diczfalusy, U.; Lannfelt, L.; Winblad, B.; Russell, D.W.; Bjorkhem, I. On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci. Lett. 2001, 314, 45–48. [Google Scholar] [CrossRef]
- Zarrouk, A.; Debbabi, M.; Bezine, M.; Karym, E.M.; Badreddine, A.; Rouaud, O.; Moreau, T.; Cherkaoui-Malki, M.; El Ayeb, M.; Nasser, B.; et al. Lipid Biomarkers in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Staurenghi, E.; Giannelli, S.; Gargiulo, S.; Guglielmotto, M.; Tabaton, M.; Tamagno, E.; Gamba, P.; Leonarduzzi, G. A silver lining for 24-hydroxycholesterol in Alzheimer’s disease: The involvement of the neuroprotective enzyme sirtuin 1. Redox Biol. 2018, 17, 423–431. [Google Scholar] [CrossRef]
- Gamba, P.; Giannelli, S.; Staurenghi, E.; Testa, G.; Sottero, B.; Biasi, F.; Poli, G.; Leonarduzzi, G. The Controversial Role of 24-S-Hydroxycholesterol in Alzheimer’s Disease. Antioxidants 2021, 10, 740. [Google Scholar] [CrossRef]
- Ma, W.W.; Li, C.Q.; Yu, H.L.; Zhang, D.D.; Xi, Y.D.; Han, J.; Liu, Q.R.; Xiao, R. The oxysterol 27-hydroxycholesterol increases oxidative stress and regulate Nrf2 signaling pathway in astrocyte cells. Neurochem. Res. 2015, 40, 758–766. [Google Scholar] [CrossRef]
- Cigliano, L.; Spagnuolo, M.S.; Napolitano, G.; Iannotta, L.; Fasciolo, G.; Barone, D.; Venditti, P. 24S-hydroxycholesterol affects redox homeostasis in human glial U-87MG cells. Mol. Cell. Endocrinol. 2019, 486, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Nury, T.; Samadi, M.; Zarrouk, A.; Riedinger, J.M.; Lizard, G. Improved synthesis and in vitro evaluation of the cytotoxic profile of oxysterols oxidized at C4 (4alpha- and 4beta-hydroxycholesterol) and C7 (7-ketocholesterol, 7alpha- and 7beta-hydroxycholesterol) on cells of the central nervous system. Eur. J. Med. Chem. 2013, 70, 558–567. [Google Scholar] [CrossRef]
- Bochelen, D.; Langley, K.; Adamczyk, M.; Kupferberg, A.; Hor, F.; Vincendon, G.; Mersel, M. 7beta-hydroxysterol is cytotoxic to neonatal rat astrocytes in primary culture when cAMP levels are increased. J. Neurosci. Res. 2000, 62, 99–111. [Google Scholar] [CrossRef]
- Yao, Y.; Sun, S.; Kong, Q.; Tong, E. 7b.beta-hydroxycholesterol reduces the extent of reactive gliosis caused by iron deposition in the hippocampus but does not attenuate the iron-induced seizures in rats. Neuroscience 2006, 138, 1097–1103. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Ismail, M.A.; Goikolea, J.; Lodeiro, M.; Mateos, L.; Bjorkhem, I.; Puerta, E.; Romao, M.A.; Gomes, C.M.; Merino-Serrais, P.; et al. Hypercholesterolemia and 27-Hydroxycholesterol Increase S100A8 and RAGE Expression in the Brain: A Link Between Cholesterol, Alarmins, and Neurodegeneration. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hannedouche, S.; Zhang, J.; Yi, T.; Shen, W.; Nguyen, D.; Pereira, J.P.; Guerini, D.; Baumgarten, B.U.; Roggo, S.; Wen, B.; et al. Oxysterols direct immune cell migration via EBI2. Nature 2011, 475, 524–527. [Google Scholar] [CrossRef]
- Rutkowska, A.; Shimshek, D.R.; Sailer, A.W.; Dev, K.K. EBI2 regulates pro-inflammatory signalling and cytokine release in astrocytes. Neuropharmacology 2018, 133, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.A.; Mateos, L.; Maioli, S.; Merino-Serrais, P.; Ali, Z.; Lodeiro, M.; Westman, E.; Leitersdorf, E.; Gulyas, B.; Olof-Wahlund, L.; et al. 27-Hydroxycholesterol impairs neuronal glucose uptake through an IRAP/GLUT4 system dysregulation. J. Exp. Med. 2017, 214, 699–717. [Google Scholar] [CrossRef]
- Miners, S.; Ashby, E.; Baig, S.; Harrison, R.; Tayler, H.; Speedy, E.; Prince, J.A.; Love, S.; Kehoe, P.G. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl. Res. 2009, 1, 163–177. [Google Scholar]
- Mateos, L.; Ismail, M.A.; Gil-Bea, F.J.; Schule, R.; Schols, L.; Heverin, M.; Folkesson, R.; Bjorkhem, I.; Cedazo-Minguez, A. Side chain-oxidized oxysterols regulate the brain renin-angiotensin system through a liver X receptor-dependent mechanism. J. Biol. Chem. 2011, 286, 25574–25585. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Hu, J.; Tsai, C.W.; Yue, M.; Melrose, H.L.; Kanekiyo, T.; Bu, G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015, 35, 5851–5859. [Google Scholar] [CrossRef] [Green Version]
- Brooks, S.W.; Dykes, A.C.; Schreurs, B.G. A High-Cholesterol Diet Increases 27-Hydroxycholesterol and Modifies Estrogen Receptor Expression and Neurodegeneration in Rabbit Hippocampus. J. Alzheimers Dis. 2017, 56, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Wang, L.M.; Chen, Y.; Gao, J.Y.; Marshall, C.; Cai, Z.Y.; Hu, G.; Xiao, M. Changes in astrocyte functional markers and beta-amyloid metabolism-related proteins in the early stages of hypercholesterolemia. Neuroscience 2016, 316, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Roe, C.M.; Xiong, C.; Fagan, A.M.; Goate, A.M.; Holtzman, D.M.; Mintun, M.A. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosta, F.; Vossel, K.A.; Miller, B.L.; Migliaccio, R.; Bonasera, S.J.; Filippi, M.; Boxer, A.L.; Karydas, A.; Possin, K.L.; Gorno-Tempini, M.L. Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2009, 106, 2018–2022. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Gong, Y.; Gan, W.; Beach, T.; Holtzman, D.M.; Wisniewski, T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience 2003, 122, 305–315. [Google Scholar] [CrossRef]
- Shinohara, M.; Murray, M.E.; Frank, R.D.; Shinohara, M.; DeTure, M.; Yamazaki, Y.; Tachibana, M.; Atagi, Y.; Davis, M.D.; Liu, C.C.; et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Elias-Sonnenschein, L.S.; Viechtbauer, W.; Ramakers, I.H.; Verhey, F.R.; Visser, P.J. Predictive value of APOE-epsilon4 allele for progression from MCI to AD-type dementia: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1149–1156. [Google Scholar] [CrossRef]
- Liraz, O.; Boehm-Cagan, A.; Michaelson, D.M. ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol. Neurodegener. 2013, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wilson, W.A.; Moore, S.D.; Mace, B.E.; Maeda, N.; Schmechel, D.E.; Sullivan, P.M. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol. Dis. 2005, 18, 390–398. [Google Scholar] [CrossRef]
- Rodriguez, G.A.; Burns, M.P.; Weeber, E.J.; Rebeck, G.W. Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learn. Mem. 2013, 20, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef] [Green Version]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J. Neuroinflamm. 2014, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294. [Google Scholar] [CrossRef] [Green Version]
- Prasad, H.; Rao, R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. USA 2018, 115, E6640–E6649. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Liu, C.C.; Chen, X.F.; Zhang, Y.W.; Xu, H.; Bu, G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol. Neurodegener 2015, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [Green Version]
- Rawat, V.; Wang, S.; Sima, J.; Bar, R.; Liraz, O.; Gundimeda, U.; Parekh, T.; Chan, J.; Johansson, J.O.; Tang, C.; et al. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J. Neurosci. 2019, 39, 9611–9622. [Google Scholar] [CrossRef]
- Tai, L.M.; Mehra, S.; Shete, V.; Estus, S.; Rebeck, G.W.; Bu, G.; LaDu, M.J. Soluble apoE/Abeta complex: Mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 2014, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Li, D.; He, C.; He, K.; Xue, T.; Wan, L.; Zhang, C.; Liu, Q. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron 2021, 109, 957–970.e8. [Google Scholar] [CrossRef]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; LaDu, M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Ceyzeriat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Sadick, J.S.; Liddelow, S.A. Don’t forget astrocytes when targeting Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3585–3598. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staurenghi, E.; Giannelli, S.; Testa, G.; Sottero, B.; Leonarduzzi, G.; Gamba, P. Cholesterol Dysmetabolism in Alzheimer’s Disease: A Starring Role for Astrocytes? Antioxidants 2021, 10, 1890. https://doi.org/10.3390/antiox10121890
Staurenghi E, Giannelli S, Testa G, Sottero B, Leonarduzzi G, Gamba P. Cholesterol Dysmetabolism in Alzheimer’s Disease: A Starring Role for Astrocytes? Antioxidants. 2021; 10(12):1890. https://doi.org/10.3390/antiox10121890
Chicago/Turabian StyleStaurenghi, Erica, Serena Giannelli, Gabriella Testa, Barbara Sottero, Gabriella Leonarduzzi, and Paola Gamba. 2021. "Cholesterol Dysmetabolism in Alzheimer’s Disease: A Starring Role for Astrocytes?" Antioxidants 10, no. 12: 1890. https://doi.org/10.3390/antiox10121890
APA StyleStaurenghi, E., Giannelli, S., Testa, G., Sottero, B., Leonarduzzi, G., & Gamba, P. (2021). Cholesterol Dysmetabolism in Alzheimer’s Disease: A Starring Role for Astrocytes? Antioxidants, 10(12), 1890. https://doi.org/10.3390/antiox10121890