
The Blood–Brain Barrier, Oxidative Stress, and Insulin Resistance



{kind=link}
{kind=link}
Abstract
:1. Introduction
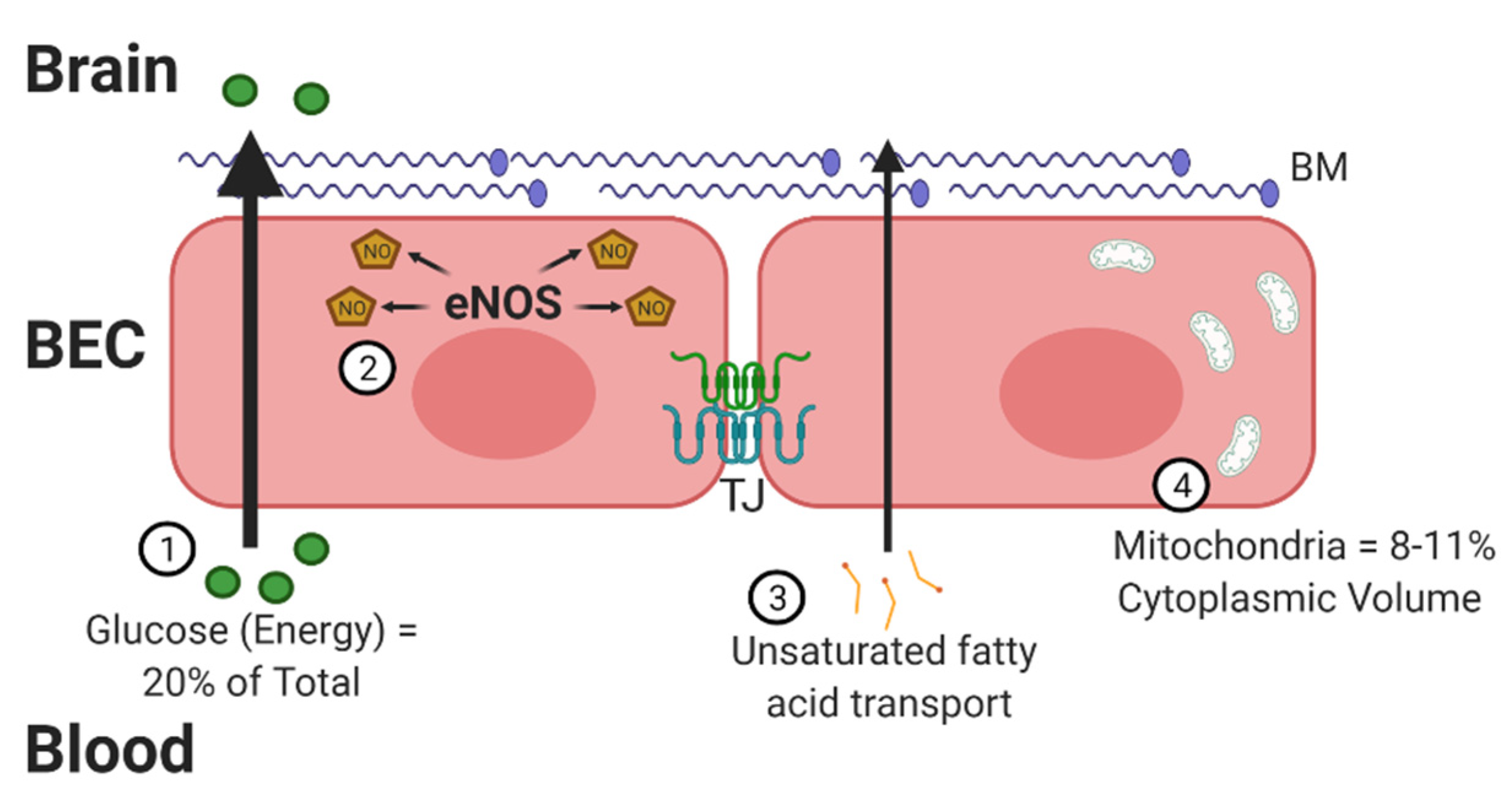
2. Oxidative Stress at the BBB
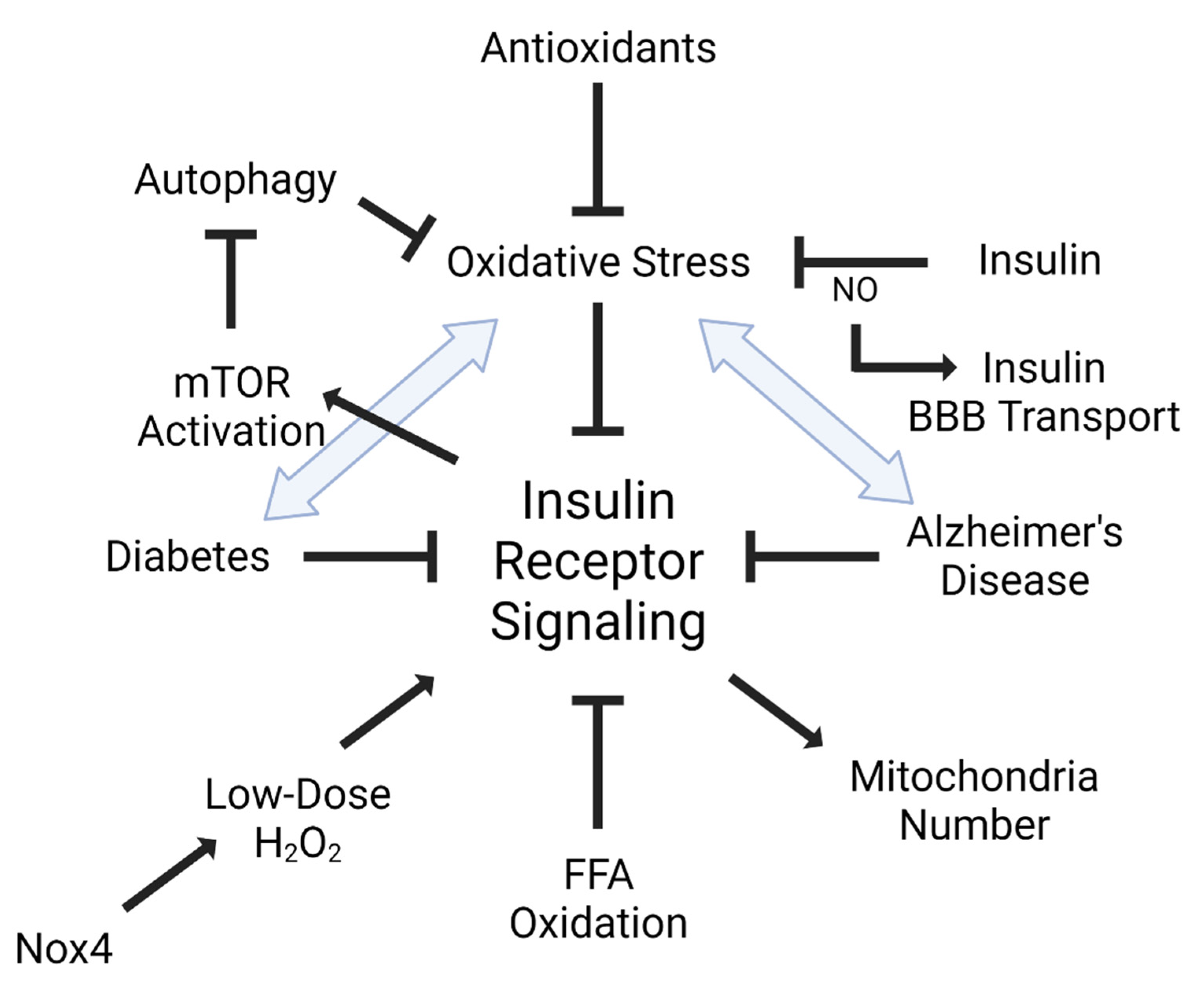
3. Insulin, Oxidative Stress, and the BBB
4. Apolipoprotein E Impact on BBB Oxidative Stress
5. Diseases Associated with Insulin and Oxidative Stress
5.1. Diabetes Mellitus (DM)
5.2. Alzheimer’s Disease (AD)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| AGE | advanced glycation end product |
| ApoE | apolipoprotein E |
| BBB | blood–brain barrier |
| BEC | brain endothelial cell |
| BM | basement membrane |
| BRB | blood–retinal barrier |
| CNS | central nervous system |
| DHA | docosahexaenoic acid |
| DM | diabetes mellitus |
| FATP | fatty acid transport protein |
| FFA | free fatty acid |
| LRP-1 | low density lipoprotein (LDL) receptor-related protein 1 |
| MMP | matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NO | nitric oxide |
| NOS | nitric oxide synthase (i- inducible, e- endothelial, n- neuronal) |
| Nox | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| NVU | neurovascular unit |
| Pgp | P-glycoprotein |
| RAGE | receptor for advanced glycation end product |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| TJ | tight junction |
| VEGF | vascular endothelial growth factor |
References
- Tayarani, I.; Chaudiere, J.; Lefauconnier, J.M.; Bourre, J.M. Enzymatic protection against peroxidative damage in isolated brain capillaries. J. Neurochem. 1987, 48, 1399–1402. [Google Scholar] [CrossRef]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo(-/-) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta 2012, 1822, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Aljofan, M.; Triggle, C.R. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J. Cell Physiol. 2007, 212, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Banks, W.A. Interactions of Lipids, Lipoproteins, and Apolipoproteins with the Blood-Brain Barrier. Pharm. Res. 2021, 38, 1469–1475. [Google Scholar] [CrossRef]
- Rizzo, M.T.; Saquib, M.; Leaver, H.A. Oxidative Stress and Brain Endothelial Cells. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1959–1977. [Google Scholar]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Martinez, J.; Miller, B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev. Mol. Med. 2011, 13, e19. [Google Scholar] [CrossRef]
- Mooradian, A.D. Effect of aging on the blood-brain barrier. Neurobiol. Aging 1988, 9, 31–39. [Google Scholar] [CrossRef]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood-brain barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharmacol. 2018, 175, 1279–1292. [Google Scholar] [CrossRef]
- Anasooya Shaji, C.; Robinson, B.D.; Yeager, A.; Beeram, M.R.; Davis, M.L.; Isbell, C.L.; Huang, J.H.; Tharakan, B. The Tri-phasic Role of Hydrogen Peroxide in Blood-Brain Barrier Endothelial cells. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, J.; Qi, J.; Jin, Y.; Tong, L. Activation of NADPH/ROS pathway contributes to angiogenesis through JNK signaling in brain endothelial cells. Microvasc. Res. 2020, 131, 104012. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreibelt, G.; Kooij, G.; Reijerkerk, A.; van Doorn, R.; Gringhuis, S.I.; van der Pol, S.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Piontek, J.; et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21, 3666–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, W.H.; Rhea, E.M.; Qu, Z.C.; Hecker, M.R.; May, J.M. Intracellular ascorbate tightens the endothelial permeability barrier through Epac1 and the tubulin cytoskeleton. Am. J. Physiol. Cell Physiol. 2016, 311, C652–C662. [Google Scholar] [CrossRef] [Green Version]
- Meredith, M.E.; Qu, Z.C.; May, J.M. Ascorbate reverses high glucose- and RAGE-induced leak of the endothelial permeability barrier. Biochem. Biophys. Res. Commun. 2014, 445, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190. [Google Scholar] [CrossRef]
- Bresgen, N.; Karlhuber, G.; Krizbai, I.; Bauer, H.; Bauer, H.C.; Eckl, P.M. Oxidative stress in cultured cerebral endothelial cells induces chromosomal aberrations, micronuclei, and apoptosis. J. Neurosci. Res. 2003, 72, 327–333. [Google Scholar] [CrossRef]
- May, J.M.; Jayagopal, A.; Qu, Z.C.; Parker, W.H. Ascorbic acid prevents high glucose-induced apoptosis in human brain pericytes. Biochem. Biophys. Res. Commun. 2014, 452, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdev, U.; Lotze, M.T. Perpetual change: Autophagy, the endothelium, and response to vascular injury. J. Leukoc. Biol. 2017, 102, 221–235. [Google Scholar] [CrossRef]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef]
- Carresi, C.; Mollace, R.; Macri, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Coppoletta, A.R.; Guarnieri, L.; Ruga, S.; Zito, M.C.; et al. Oxidative Stress Triggers Defective Autophagy in Endothelial Cells: Role in Atherothrombosis Development. Antioxidants 2021, 10, 387. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Breton-Romero, R.; Linder, E.A.; Berk, B.D.; Duess, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamu, O.; Rado, M.; Ekpo, O.; Fisher, D. Differential Sensitivity of Two Endothelial Cell Lines to Hydrogen Peroxide Toxicity: Relevance for In Vitro Studies of the Blood-Brain Barrier. Cells 2020, 9, 403. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, M.L.; Mertsch, K.; Giese, H.; Muller, S.; Sporbert, A.; Hickel, B.; Blasig, I.E. Astrocytes enhance radical defence in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett. 1999, 449, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhea, E.M.; Banks, W.A. A historical perspective on the interactions of insulin at the blood-brain barrier. J. Neuroendocr. 2021, 33, e12929. [Google Scholar] [CrossRef]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Investig. 2006, 116, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Costantino, S.; Cosentino, F. Role of oxidative stress in endothelial insulin resistance. World J. Diabetes 2015, 6, 326–332. [Google Scholar] [CrossRef]
- Lanzillotta, C.; Tramutola, A.; Di Giacomo, G.; Marini, F.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Insulin resistance, oxidative stress and mitochondrial defects in Ts65dn mice brain: A harmful synergistic path in down syndrome. Free Radic. Biol. Med. 2021, 165, 152–170. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Sounding the alarm: Protein kinase cascades activated by stress and inflammation. J. Biol. Chem. 1996, 271, 24313–24316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jacob, S.; Lehmann, R.; Rett, K.; Häring, H.-U. Oxidative stress and insulin action: A role for antioxidants. In Antioxidants in Diabetes Management; Packer, L., Rosen, P., Tritschler, H.J., King, G.L., Eds.; Marcel Dekker: New York, NY, USA, 2000; pp. 319–338. [Google Scholar]
- Gonzalez, M.; Rojas, S.; Avila, P.; Cabrera, L.; Villalobos, R.; Palma, C.; Aguayo, C.; Pena, E.; Gallardo, V.; Guzman-Gutierrez, E.; et al. Insulin reverses D-glucose-increased nitric oxide and reactive oxygen species generation in human umbilical vein endothelial cells. PLoS ONE 2015, 10, e0122398. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [Green Version]
- Xaio, H.; Banks, W.A.; Niehoff, M.L.; Morley, J.E. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001, 896, 36–42. [Google Scholar] [CrossRef]
- Banks, W.A.; Dohgu, S.; Lynch, J.L.; Fleegal-DeMotta, M.A.; Erickson, M.A.; Nakaoke, R.; Vo, T.Q. Nitric oxide isoenzymes regulate lipopolysaccharide-enhanced insulin transport across the blood-brain barrier. Endocrinology 2008, 149, 1514–1523. [Google Scholar] [CrossRef] [Green Version]
- Livingston, J.N.; Gurny, P.A.; Lockwood, D.H. Insulin-like effects of polyamines in fat cells. Mediation by H2O2 formation. J. Biol. Chem. 1977, 252, 560–562. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Ochiai, Y.; Uchida, Y.; Ohtsuki, S.; Tachikawa, M.; Aizawa, S.; Terasaki, T. The blood-brain barrier fatty acid transport protein 1 (FATP1/SLC27A1) supplies docosahexaenoic acid to the brain, and insulin facilitates transport. J. Neurochem. 2017, 141, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Richey, P.L.; Siedlak, S.L.; Smith, M.A.; Perry, G. Apolipoprotein E interaction with the neurofibrillary tangles and senile plaques in Alzheimer disease: Implications for disease pathogenesis. Biochem. Biophys. Res. Commun. 1995, 208, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [Green Version]
- Ramassamy, C.; Averill, D.; Beffert, U.; Theroux, L.; Lussier-Cacan, S.; Cohn, J.S.; Christen, Y.; Schoofs, A.; Davignon, J.; Poirier, J. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol. Dis. 2000, 7, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Boyle, P.A.; Nag, S.; Leurgans, S.; Buchman, A.S.; Wilson, R.S.; Arvanitakis, Z.; Farfel, J.M.; De Jager, P.L.; Bennett, D.A.; et al. APOE and cerebral amyloid angiopathy in community-dwelling older persons. Neurobiol. Aging 2015, 36, 2946–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox. Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Lloret, M.A.; Monllor, P.; López, B.; León, J.L.; Cervera-Ferri, A. Is Oxidative Stress the Link Between Cerebral Small Vessel Disease, Sleep Disruption, and Oligodendrocyte Dysfunction in the Onset of Alzheimer’s Disease? Front. Physiol. 2021, 12, 708061. [Google Scholar] [CrossRef]
- Brown, C.M.; Wright, E.; Colton, C.A.; Sullivan, P.M.; Laskowitz, D.T.; Vitek, M.P. Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radic. Biol. Med. 2002, 32, 1071–1075. [Google Scholar] [CrossRef]
- Colton, C.A.; Brown, C.M.; Cook, D.; Needham, L.K.; Xu, Q.; Czapiga, M.; Saunders, A.M.; Schmechel, D.E.; Rasheed, K.; Vitek, M.P. APOE and the regulation of microglial nitric oxide production: A link between genetic risk and oxidative stress. Neurobiol. Aging 2002, 23, 777–785. [Google Scholar] [CrossRef]
- Hayek, T.; Oiknine, J.; Brook, J.G.; Aviram, M. Increased plasma and lipoprotein lipid peroxidation in apo E-deficient mice. Biochem. Biophys. Res. Commun. 1994, 201, 1567–1574. [Google Scholar] [CrossRef]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Rhea, E.M.; Raber, J.; Banks, W.A. ApoE and cerebral insulin: Trafficking, receptors, and resistance. Neurobiol. Dis. 2020, 137, 104755. [Google Scholar] [CrossRef]
- Chan, E.S.; Chen, C.; Soong, T.W.; Wong, B.S. Differential Binding of Human ApoE Isoforms to Insulin Receptor is Associated with Aberrant Insulin Signaling in AD Brain Samples. Neuromol. Med. 2018, 20, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Chen, C.; Cole, G.M.; Wong, B.S. Differential interaction of Apolipoprotein-E isoforms with insulin receptors modulates brain insulin signaling in mutant human amyloid precursor protein transgenic mice. Sci. Rep. 2015, 5, 13842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G.J. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toborek, M.; Lee, Y.W.; Flora, G.; Pu, H.; Andreeff, M.; Wylegala, E.; Henning, B.; Nath, A. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell. Mol. Neurobiol. 2005, 25, 181–199. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Potula, R.; Fan, S.; Eidem, T.; Papugani, A.; Reichenbach, N.; Dykstra, H.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; et al. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J. Cereb. Blood Flow Metab. 2009, 29, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.; Hasko, J.; Krizbai, I.A. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Toborek, M.; Lee, Y.W.; Pu, H.; Malecki, A.; Flora, G.; Garrido, R.; Hennig, B.; Bauer, H.C.; Nath, A. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J. Neurochem. 2003, 84, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Manda, K.R.; Banerjee, A.; Banks, W.A.; Ercal, N. Highly active antiretroviral therapy drug combination induces oxidative stress and mitochondrial dysfunction in immortalized human blood-brain barrier endothelial cells. Free Radic. Biol. Med. 2011, 50, 801–810. [Google Scholar] [CrossRef]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 84–96. [Google Scholar] [CrossRef]
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef]
- Xu, Z.; Zeng, W.; Sun, J.; Chen, W.; Zhang, R.; Yang, Z.; Yao, Z.; Wang, L.; Song, L.; Chen, Y.; et al. The quantification of blood-brain barrier disruption using dynamic contrast-enhanced magnetic resonance imaging in aging rhesus monkeys with spontaneous type 2 diabetes mellitus. Neuroimage 2017, 158, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; VanGilder, R.L.; Houser, K.A. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am. J. Physiol. 2006, 291, H2660–H2668. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Mortell, W.G.; Logsdon, A.F.; Butterfield, D.A.; Banks, W.A. Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: Prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 2019, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Salameh, T.; Shah, G.N.; Price, T.O.; Hayden, M.R.; Banks, W.A. Blood-brain barrier disruption and neurovascular unit dysfunction in diabetic mice: Protection with the mitochondrial carbonic anhydrase inhibitor topiramate. J. Pharmacol. Exp. Ther. 2016, 359, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. The Blood-Brain Barrier Interface in Diabetes Mellitus: Dysfunctions, Mechanisms and Approaches to Treatment. Curr. Pharm. Des. 2020, 26, 1438–1447. [Google Scholar] [CrossRef]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Hammes, H.P.; Lin, J.H.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Diabetic retinopathy: Mitochondrial dysfunction and retinal capillary cell death. Antioxid. Redox Signal. 2005, 7, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P.; Katychev, A.; Wang, X.; Van Buren, E. CNS microvascular pericytes exhibit multipotential stem cell activity. J. Cereb. Blood Flow Metab. 2006, 26, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.O.; Eranki, V.; Banks, W.A.; Ercal, N.; Shah, G.N. Topiramate treatment protects blood-brain barrier pericytes from hyperglycemia-induced oxidative damage in diabetic mice. Endocrinology 2012, 153, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, G.N.; Price, T.O.; Banks, W.A.; Morofuji, Y.; Kovac, A.; Ercal, N.; Sorenson, C.M.; Shin, E.S.; Sheibai, N. Pharmacological inhibition of mitochondrial carbonic anhydrases protects mouse cerebral pericytes from high glucose-induced oxidative stress and apoptosis. J. Pharmacol. Exp. Ther. 2013, 344, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobi, A.; Rosanaly, S.; Devin, A.; Baret, P.; Meilhac, O.; Harry, G.J.; d’Hellencourt, C.L.; Rondeau, P. Advanced glycation end-products disrupt brain microvascular endothelial cell barrier: The role of mitochondria and oxidative stress. Microvasc. Res. 2021, 133, 104098. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Maloney, R.E.; Circu, M.L.; Alexander, J.S.; Aw, T.Y. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: Role for glutathione-dependent metabolism of methylglyoxal. Free Radic. Biol. Med. 2013, 54, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Maloney, R.E.; Aw, T.Y. High glucose, glucose fluctuation and carbonyl stress enhance brain microvascular endothelial barrier dysfunction: Implications for diabetic cerebral microvasculature. Redox Biol. 2015, 5, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Y.; Fan, Z.; Chen, Q.; Chen, J.; Sun, Y.; Jiang, X.; Xiao, Q. Soluble epoxide hydrolase inhibitor protects against blood-brain barrier dysfunction in a mouse model of type 2 diabetes via the AMPK/HO-1 pathway. Biochem. Biophys. Res. Commun. 2020, 524, 354–359. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Shudou, M.; Jing, F.; Tsukuda, K.; Ohshima, K.; Iwanami, J.; Horiuchi, M. Peroxisome proliferator-activated receptor-gamma activation with angiotensin II type 1 receptor blockade is pivotal for the prevention of blood-brain barrier impairment and cognitive decline in type 2 diabetic mice. Hypertension 2012, 59, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Poon, H.F.; Joshi, G.; Sultana, R.; Farr, S.A.; Banks, W.A.; Morley, J.E.; Calabrese, V.; Butterfield, D.A. Antisense directed at the Abeta region of APP decreases brain oxidative markers in aged senescence accelerated mice. Brain Res. 2004, 1018, 86–96. [Google Scholar] [CrossRef]
- Butterfield, D.A. The 2013 SFRBM Discovery Award: Selected discoveries from the Butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radic. Biol. Med. 2014, 74, 157–174. [Google Scholar] [CrossRef] [Green Version]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Morley, J.E. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Dore-Duffy, P. Pericytes: Pluripotent cells of the blood brain barrier. Curr. Pharm. Des. 2008, 14, 1581–1593. [Google Scholar] [CrossRef]
- Cuevas, E.; Rosas-Hernandez, H.; Burks, S.M.; Ramirez-Lee, M.A.; Guzman, A.; Imam, S.Z.; Ali, S.F.; Sarkar, S. Amyloid Beta 25-35 induces blood-brain barrier disruption in vitro. Metab. Brain Dis. 2019, 34, 1365–1374. [Google Scholar] [CrossRef]
- Rensink, A.A.; Verbeek, M.M.; Otte-Holler, I.; ten Donkelaar, H.T.; de Waal, R.M.; Kremer, B. Inhibition of amyloid-beta-induced cell death in human brain pericytes in vitro. Brain Res. 2002, 952, 111–121. [Google Scholar] [CrossRef]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Ripley, J.L.; Sultana, R.; Zhang, Z.; Niehoff, M.L.; Platt, T.L.; Murphy, M.P.; Morley, J.E.; Kumar, V.; Butterfield, D.A. Antisense oligonucelotide against GSK-3 beta in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer’s disease. Free Radic. Biol. Med. 2013, 67C, 387–395. [Google Scholar]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Robinson, S.M.; Verma, S.; Morley, J.E. Efflux of human and mouse amyloid · proteins 1-40 and 1-42 from brain: Impairment in a mouse model of Alzheimer’s disease. Neuroscience 2003, 121, 487–492. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Sita, G.; Hrelia, P.; Tarozzi, A.; Morroni, F. P-glycoprotein (ABCB1) and Oxidative Stress: Focus on Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 7905486. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.B.; Sultana, R.; Aluise, C.D.; Erickson, M.A.; Price, T.O.; Bu, G.; Banks, W.A.; Butterfield, D.A. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer’s disease: Implications for Abeta accumulation in AD brain. Free Radic. Biol. Med. 2010, 49, 1798–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, M.A.; Hansen, K.; Banks, W.A. Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood-brain barrier: Protection by the antioxidant N-acetylcysteine. Brain Behav. Immun. 2012, 26, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2017, 16, 17–26. [Google Scholar] [CrossRef]
- Van Skike, C.E.; Jahrling, J.B.; Olson, A.B.; Sayre, N.L.; Hussong, S.A.; Ungvari, Z.; Lechleiter, J.D.; Galvan, V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H693–H703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Banks, W.A.; Rhea, E.M. Effects of Rapamycin on Insulin Brain Endothelial Cell Binding and Blood-Brain Barrier Transport. Med. Sci. 2021, 9, 56. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood-brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2018, 243, 159–165. [Google Scholar] [CrossRef]
- Vamanu, E.; Rai, S.N. The Link between Obesity, Microbiota Dysbiosis, and Neurodegenerative Pathogenesis. Diseases 2021, 9, 45. [Google Scholar] [CrossRef]
- Velasquez-Jimenez, D.; Corella-Salazar, D.A.; Zuniga-Martinez, B.S.; Dominguez-Avila, J.A.; Montiel-Herrera, M.; Salazar-Lopez, N.J.; Rodrigo-Garcia, J.; Villegas-Ochoa, M.A.; Gonzalez-Aguilar, G.A. Phenolic compounds that cross the blood-brain barrier exert positive health effects as central nervous system antioxidants. Food Funct. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Benfeito, S.; Fernandes, C.; Borges, F. Chapter 9-Antioxidant therapy, oxidative stress, and blood-brain barrier: The road of dietary antioxidants. In Oxidative Stress and Dietary Antioxidants in Neurological Diseases; Martin, C.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 125–141. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.C.; Learman, C.R.; Baker, G.B.; Weaver, C.L.; Chung, P.S.; Kim, H.G.; Song, M.S. Regulation of Diabetes: A Therapeutic Strategy for Alzheimer’s Disease? J. Korean Med. Sci. 2019, 34, e297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banks, W.A.; Rhea, E.M. The Blood–Brain Barrier, Oxidative Stress, and Insulin Resistance. Antioxidants 2021, 10, 1695. https://doi.org/10.3390/antiox10111695
Banks WA, Rhea EM. The Blood–Brain Barrier, Oxidative Stress, and Insulin Resistance. Antioxidants. 2021; 10(11):1695. https://doi.org/10.3390/antiox10111695
Chicago/Turabian StyleBanks, William A., and Elizabeth M. Rhea. 2021. "The Blood–Brain Barrier, Oxidative Stress, and Insulin Resistance" Antioxidants 10, no. 11: 1695. https://doi.org/10.3390/antiox10111695
APA StyleBanks, W. A., & Rhea, E. M. (2021). The Blood–Brain Barrier, Oxidative Stress, and Insulin Resistance. Antioxidants, 10(11), 1695. https://doi.org/10.3390/antiox10111695