
Redox Dysregulation in Aging and COPD: Role of NOX Enzymes and Implications for Antioxidant Strategies


Abstract
1. Introduction
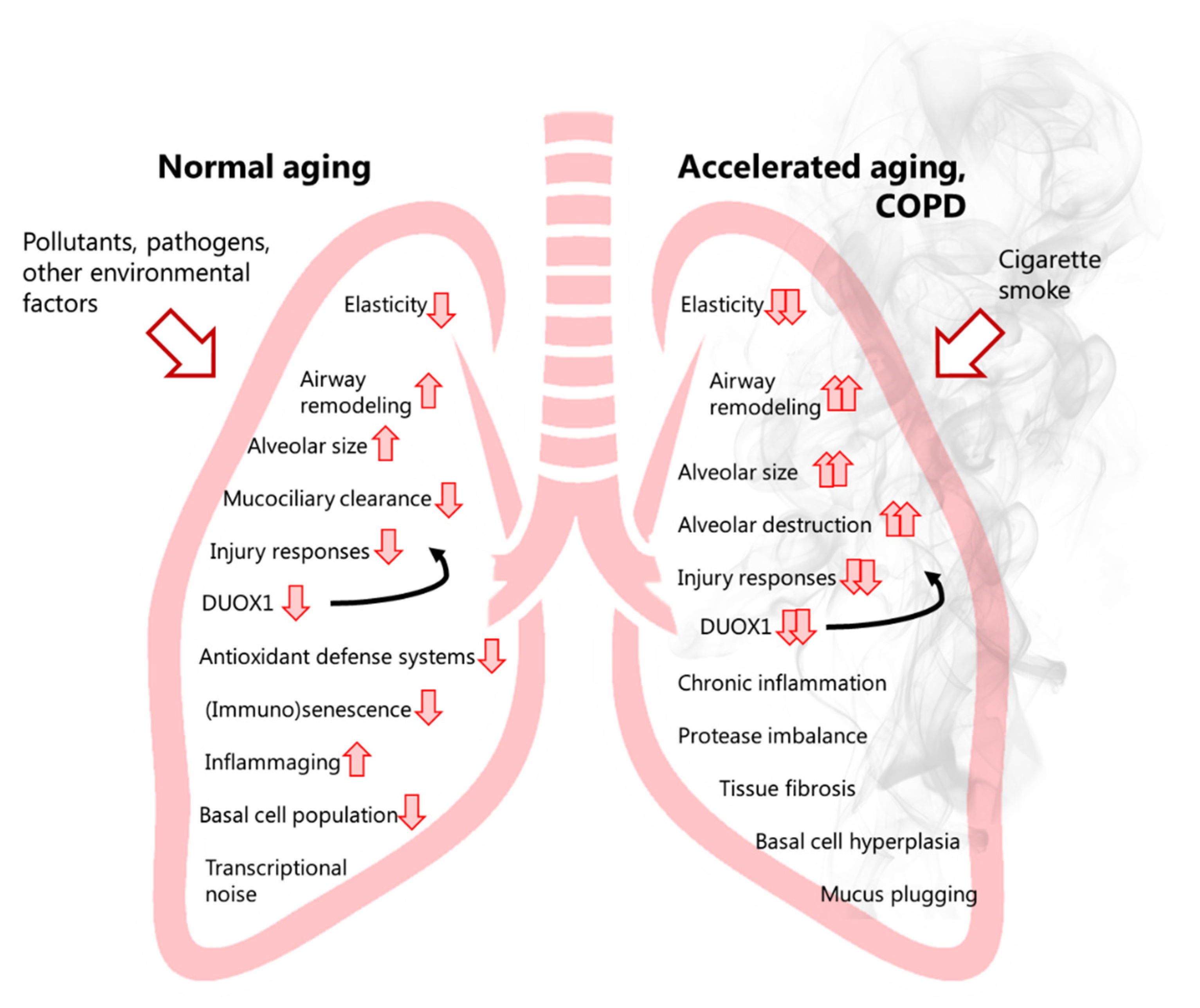
2. The Ageing Lung
3. Oxidative Stress in Ageing: Revisiting the Free Radical Theory (FRT) of Ageing
4. COPD a Disease of Accelerated Ageing?
5. Oxidative Stress in COPD: The Present Evidence
6. NADPH Oxidases (NOX) in Lung Physiology and Pathology
7. NOX Enzymes and Their Relation to Normal Ageing
8. NOX Enzymes in COPD Pathology
9. Therapeutic Targeting of Oxidative Stress in COPD: Current Status and Pitfalls
9.1. Supplementary “Antioxidants”
9.2. Small Molecule Activators of Endogenous Anti-Oxidant Responses
9.3. Inhibitors of ROS Production
10. Conclusions, and Future Perspectives
11. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | Antagonistic pleiotropy |
| ATP | Adenosine triphosphate |
| BLI | Blistered cuticle |
| CGD | Chronic granulomatous disease |
| COPD | Chronic obstructive pulmonary disease |
| CS | Cigarette smoke |
| CT | Computerized tomography |
| CysSSH | Cysteine persulfide |
| DAMP | Danger-associated molecular pattern |
| DNA | Deoxyribonucleic acid |
| DUOX | Dual oxidase |
| ECM | Extracellular matrix |
| EcSOD | Extracellular superoxide dismutase |
| EELV | End-expiratory lung volume |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ELF | Epithelial lining fluid |
| EMT | Epithelial mesenchymal transition |
| FAD/FADH2 | Flavin adenine dinucleotide |
| FEV1 | Forced expiratory volume in 1 s |
| FRC | Functional residual capacity |
| FRT | Free radical theory |
| FVC | Forced vital capacity |
| GOLD | Global initiative for chronic obstructive lung disease |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| GSSH | Glutathione persulfide |
| GSSSH | Glutathione trisulfide |
| GTEx | Genotype-tissue Expression |
| G6PD | Glucose-6-phosphate dehydrogenase |
| H2O2 | Hydrogen peroxide |
| H2S | Hydrogen sulfide |
| IL | Interleukin |
| ILC | Innate lymphoid cell |
| IPF | Idiopathic pulmonary fibrosis |
| JNK | C-jun N-terminal kinase |
| Mclk1 | Mitochondrial 5-demethoxyubiquinone hydroxylase |
| MDA | Malondialdehyde |
| MMP | Matrix metalloproteinase |
| MnSOD | Manganese superoxide dismutase |
| MPO | Myeloperoxidase |
| mtDNA | Mitochondrial deoxyribonucleic acid |
| mTOR | Mechanistic target of rapamycin |
| NAC | N-acetyl cysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NADH/NAD+ | Nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor kappa B |
| NOX | NADPH oxidase |
| NOXO1 | NADPH oxidase organizer 1 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| Nuo-6 | NADH dehydrogenase ubiquinone flavoprotein 1 |
| O2•− | Superoxide |
| OH• | Hydroxyl radical |
| OSCN− | Hypothiocyanite |
| RBC | Red blood cell |
| ROS | Reactive oxygen species |
| SASP | Senescence-associated secretory phenotype |
| SERPIN | Serine protease inhibitor |
| SOD | Superoxide dismutase |
| SIRT | Sirtuin |
| TGFβ | Transforming growth factor β |
| Th1 | T-helper type 1 |
| Th2 | T-helper type 2 |
| Th17 | T-helper type 17 |
| TLR | Toll-like receptor |
| Treg | Regulatory T-cell |
| Trx | Thioredoxin |
| TSP1 | Thrombospondin 1 |
References
- Dzau, V.J.; Inouye, S.K.; Rowe, J.W.; Finkelman, E.; Yamada, T. Enabling healthful aging for all—The national academy of medicine grand challenge in healthy longevity. N. Engl. J. Med. 2019, 381, 1699–1701. [Google Scholar] [CrossRef] [PubMed]
- Mangel, M. Complex adaptive systems, aging and longevity. J. Theor. Biol. 2001, 213, 559–571. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Konigshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Freeman, B.A.; Crapo, J.D. Biology of disease: Free radicals and tissue injury. Lab. Investig. 1982, 47, 412–426. [Google Scholar]
- Halliwell, B. The wanderings of a free radical. Free Radic. Biol. Med. 2009, 46, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.; Mercuri, A.; Wu, S.; Salar-Behzadi, S. Measurements of deposition, lung surface area and lung fluid for simulation of inhaled compounds. Front. Pharmacol. 2016, 7, 181. [Google Scholar] [CrossRef]
- Budinger, G.R.S.; Kohanski, R.A.; Gan, W.; Kobor, M.S.; Amaral, L.A.; Armanios, M.; Kelsey, K.T.; Pardo, A.; Tuder, R.; Macian, F.; et al. The intersection of aging biology and the pathobiology of lung diseases: A joint nhlbi/nia workshop. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1492–1500. [Google Scholar] [CrossRef]
- Ito, K.; Barnes, P.J. COPD as a disease of accelerated lung aging. Chest 2009, 135, 173–180. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Quaderi, S.A.; Hurst, J.R. The unmet global burden of COPD. Glob. Health Epidemiol. Genom. 2018, 3, e4. [Google Scholar] [CrossRef] [PubMed]
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444. [Google Scholar] [CrossRef]
- Bowdish, D.M.E. The aging lung: Is lung health good health for older adults? Chest 2019, 155, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.; Peto, R. The natural history of chronic airflow obstruction. BMJ 1977, 1, 1645–1648. [Google Scholar] [CrossRef]
- Agusti, A.; Faner, R. Lung function trajectories in health and disease. Lancet Respir. Med. 2019, 7, 358–364. [Google Scholar] [CrossRef]
- Agusti, A.; Hogg, J.C. Update on the pathogenesis of chronic obstructive pulmonary disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef]
- Brandenberger, C.; Muhlfeld, C. Mechanisms of lung aging. Cell Tissue Res. 2017, 367, 469–480. [Google Scholar] [CrossRef]
- Cho, S.J.; Stout-Delgado, H.W. Aging and lung disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef] [PubMed]
- Wansleeben, C.; Bowie, E.; Hotten, D.F.; Yu, Y.R.; Hogan, B.L. Age-related changes in the cellular composition and epithelial organization of the mouse trachea. PLoS ONE 2014, 9, e93496. [Google Scholar]
- Watson, J.K.; Sanders, P.; Dunmore, R.; Rosignoli, G.; Jule, Y.; Rawlins, E.L.; Mustelin, T.; May, R.; Clarke, D.; Finch, D.K. Distal lung epithelial progenitor cell function declines with age. Sci Rep. 2020, 10, 10490. [Google Scholar] [CrossRef] [PubMed]
- De Keizer, P.L. The fountain of youth by targeting senescent cells? Trends Mol. Med. 2017, 23, 6–17. [Google Scholar] [CrossRef]
- Gibbs, K.W.; Chuang Key, C.C.; Belfield, L.; Krall, J.; Purcell, L.; Liu, C.; Files, D.C. Aging influences the metabolic and inflammatory phenotype in an experimental mouse model of acute lung injury. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2021, 76, 770–777. [Google Scholar] [CrossRef]
- Schiffers, C.; Lundblad, L.K.A.; Hristova, M.; Habibovic, A.; Dustin, C.M.; Daphtary, N.; Aliyeva, M.; Seward, D.J.; Janssen-Heininger, Y.M.; Wouters, E.F.; et al. Downregulation of DUOX1 function contributes to aging-related impairment of innate airway injury responses and accelerated senile emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L144–L158. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Farshbafnadi, M.; Kamali Zonouzi, S.; Sabahi, M.; Dolatshahi, M.; Aarabi, M.H. Aging & COVID-19 susceptibility, disease severity, and clinical outcomes: The role of entangled risk factors. Exp. Gerontol. 2021, 154, 111507. [Google Scholar]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, immunity, and COVID-19: How age influences the host immune response to coronavirus infections? Front. Physiol. 2020, 11, 571416. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Panda, A.; Arjona, A.; Sapey, E.; Bai, F.; Fikrig, E.; Montgomery, R.R.; Lord, J.M.; Shaw, A.C. Human innate immunosenescence: Causes and consequences for immunity in old age. Trends Immunol. 2009, 30, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Capell, B.C. The senescence-associated secretory phenotype: Critical effector in skin cancer and aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef]
- Wang, L.; Green, F.H.; Smiley-Jewell, S.M.; Pinkerton, K.E. Susceptibility of the aging lung to environmental injury. Semin. Respir. Crit. Care Med. 2010, 31, 539–553. [Google Scholar] [CrossRef]
- Bustos, M.L.; Huleihel, L.; Kapetanaki, M.G.; Lino-Cardenas, C.L.; Mroz, L.; Ellis, B.M.; McVerry, B.J.; Richards, T.J.; Kaminski, N.; Cerdenes, N.; et al. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am. J. Respir. Crit. Care. Med. 2014, 189, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Katial, R.; Zheng, W. Allergy and immunology of the aging lung. Clin. Chest Med. 2007, 28, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Verbeken, E.K.; Cauberghs, M.; Mertens, I.; Clement, J.; Lauweryns, J.M.; Van de Woestijne, K.P. The senile lung. Comparison with normal and emphysematous lungs. 1. Structural aspects. Chest 1992, 101, 793–799. [Google Scholar] [CrossRef]
- Verbeken, E.K.; Cauberghs, M.; Mertens, I.; Clement, J.; Lauweryns, J.M.; Van de Woestijne, K.P. The senile lung. Comparison with normal and emphysematous lungs. 2. Functional aspects. Chest 1992, 101, 800–809. [Google Scholar] [CrossRef]
- Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and detrimental effects of reactive oxygen species on lifespan: A comprehensive review of comparative and experimental studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef]
- Sohal, R.S.; Sohal, B.H. Hydrogen peroxide release by mitochondria increases during aging. Mech. Ageing Dev. 1991, 57, 187–202. [Google Scholar] [CrossRef]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Lehner, M.; Garcia-de-la Asuncion, J.; Viña, J. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 1999, 299, 267–276. [Google Scholar]
- Lambert, A.J.; Boysen, H.M.; Buckingham, J.A.; Yang, T.; Podlutsky, A.; Austad, S.N.; Kunz, T.H.; Buffenstein, R.; Brand, M.D. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell 2007, 6, 607–618. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Redox theory of aging: Implications for health and disease. Clin. Sci. 2017, 131, 1669–1688. [Google Scholar] [CrossRef] [PubMed]
- Saldmann, F.; Viltard, M.; Leroy, C.; Friedlander, G. The naked mole rat: A unique example of positive oxidative stress. Oxid. Med. Cell. Longev. 2019, 2019, 4502819. [Google Scholar] [CrossRef]
- Rodriguez, K.A.; Wywial, E.; Perez, V.I.; Lambert, A.J.; Edrey, Y.H.; Lewis, K.N.; Grimes, K.; Lindsey, M.L.; Brand, M.D.; Buffenstein, R. Walking the oxidative stress tightrope: A perspective from the naked mole-rat, the longest-living rodent. Curr. Pharm. Des. 2011, 17, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Smith, M.; Buffenstein, R. Naked Mole-Rat mortality rates defy gompertzian laws by not increasing with age. Elife 2018, 7, e31157. [Google Scholar] [CrossRef]
- Hamilton, M.L.; Van Remmen, H.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Gianni, P.; Jan, K.J.; Douglas, M.J.; Stuart, P.M.; Tarnopolsky, M.A. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp. Gerontol. 2004, 39, 1391–1400. [Google Scholar] [CrossRef]
- Lee, H.C.; Lim, M.L.; Lu, C.Y.; Liu, V.W.; Fahn, H.J.; Zhang, C.; Nagley, P.; Wei, Y.H. Concurrent increase of oxidative DNA damage and lipid peroxidation together with mitochondrial DNA mutation in human lung tissues during aging—Smoking enhances oxidative stress on the aged tissues. Arch. Biochem. Biophys. 1999, 362, 309–316. [Google Scholar] [CrossRef]
- Wozniak, A.; Drewa, G.; Wozniak, B.; Schachtschabel, D.O. Activity of antioxidant enzymes and concentration of lipid peroxidation products in selected tissues of mice of different ages, both healthy and melanoma-bearing. Z. Für Gerontol. Und Geriatr. 2004, 37, 184–189. [Google Scholar] [CrossRef]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Elko, E.A.; Mahoney, J.M.; Vacek, P.; Van der Vliet, A.; Anathy, V.; Van der Velden, J.; Janssen-Heininger, Y.M.; Seward, D.J. Age-dependent dysregulation of redox genes may contribute to fibrotic pulmonary disease susceptibility. Free Radic. Biol. Med. 2019, 141, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Xiong, C.; Lucero, J.; Behrens, M.M.; Dugan, L.L.; Quick, K.L. Gender differences in free radical homeostasis during aging: Shorter-lived female C57BL6 mice have increased oxidative stress. Aging Cell 2006, 5, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88 Pt B, 314–336. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of antioxidants supplementation on aging and longevity. Biomed. Res. Int. 2014, 2014, 404680. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Scheibye-Knudsen, M.; Longo, D.L.; De Cabo, R. Animal models of aging research: Implications for human aging and age-related diseases. Annu. Rev. Anim. Biosci. 2015, 3, 283–303. [Google Scholar] [CrossRef]
- Perez, V.I.; Bokov, A.; Van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 2009, 1790, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective persulfide detection reveals evolutionarily conserved antiaging effects of s-sulfhydration. Cell Metab. 2019, 30, 1152–1170. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.A.; Forman, H.J. Does Bach1 & c-Myc dependent redox dysregulation of Nrf2 & adaptive homeostasis decrease cancer risk in ageing? Free Radic. Biol. Med. 2019, 134, 708–714. [Google Scholar]
- Zhou, L.; Zhang, H.; Davies, K.J.A.; Forman, H.J. Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells. Redox Biol. 2018, 14, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.L.; Schmitt, F.A.; Mendiondo, M.S.; Marcum, J.L.; Kryscio, R.J. Vitamin E and all-cause mortality: A meta-analysis. Curr. Aging Sci. 2011, 4, 158–170. [Google Scholar] [CrossRef]
- Flores, L.C.; Roman, M.G.; Cunningham, G.M.; Cheng, C.; Dube, S.; Allen, C.; Van Remmen, H.; Hubbard, G.B.; Saunders, T.L.; Ikeno, Y. Continuous overexpression of thioredoxin 1 enhances cancer development and does not extend maximum lifespan in male C57BL/6 mice. Pathobiol. Aging Age Relat. Dis. 2018, 8, 1533754. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.M.; Flores, L.C.; Roman, M.G.; Cheng, C.; Dube, S.; Allen, C.; Valentine, J.M.; Hubbard, G.B.; Bai, Y.; Saunders, T.L.; et al. Thioredoxin overexpression in both the cytosol and mitochondria accelerates age-related disease and shortens lifespan in male C57BL/6 mice. Geroscience 2018, 40, 453–468. [Google Scholar] [CrossRef]
- Kirkwood, T.B. Evolution of ageing. Nature 1977, 270, 301–304. [Google Scholar] [CrossRef]
- Austad, S.N.; Hoffman, J.M. Is antagonistic pleiotropy ubiquitous in aging biology? Evol. Med. Public Health 2018, 2018, 287–294. [Google Scholar] [CrossRef]
- Williams, G.C. Pleiotropy, natural selection, and the evolution of senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Burney, P.G.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15076. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- McDonough, J.E.; Yuan, R.; Suzuki, M.; Seyednejad, N.; Elliott, W.M.; Sanchez, P.G.; Wright, A.C.; Gefter, W.B.; Litzky, L.; Coxson, H.O.; et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N. Engl. J. Med. 2011, 365, 1567–1575. [Google Scholar] [CrossRef]
- Janssen, R.; Wouters, E.F.M. Loss of alveolar attachments as a pathomechanistic link between small airway disease and emphysema. Am. J. Respir. Crit. Care. Med. 2020, 201, 878–879. [Google Scholar] [CrossRef]
- Kirby, M.; Tanabe, N.; Tan, W.C.; Zhou, G.; Obeidat, M.; Hague, C.J.; Leipsic, J.; Bourbeau, J.; Sin, D.D.; Hogg, J.C.; et al. Total airway count on computed tomography and the risk of chronic obstructive pulmonary disease progression. Findings from a population-based study. Am. J. Respir. Crit. Care. Med. 2018, 197, 56–65. [Google Scholar] [CrossRef]
- Mitzner, W. Emphysema—A disease of small airways or lung parenchyma? N. Engl. J. Med. 2011, 365, 1637–1639. [Google Scholar] [CrossRef]
- Berg, K.; Wright, J.L. The pathology of chronic obstructive pulmonary disease: Progress in the 20th and 21st centuries. Arch. Pathol. Lab. Med. 2016, 140, 1423–1428. [Google Scholar] [CrossRef]
- Hall, R.; Hall, I.P.; Sayers, I. Genetic risk factors for the development of pulmonary disease identified by genome-wide association. Respirology 2019, 24, 204–214. [Google Scholar] [CrossRef]
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431. [Google Scholar] [CrossRef]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, C.A.; De Vries, M.; Costa, R.; Woldhuis, R.R.; Konigshoff, M.; Timens, W. Lung ageing and COPD: Is there a role for ageing in abnormal tissue repair? Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2017, 26, 170073. [Google Scholar] [CrossRef] [PubMed]
- Morla, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agusti, A.G. Telomere shortening in smokers with and without COPD. Eur. Respir. J. 2006, 27, 525–528. [Google Scholar] [CrossRef]
- Rutten, E.P.; Gopal, P.; Wouters, E.F.; Franssen, F.M.; Hageman, G.J.; Vanfleteren, L.E.; Spruit, M.A.; Reynaert, N.L. Various mechanistic pathways representing the aging process are altered in COPD. Chest 2016, 149, 53–61. [Google Scholar] [CrossRef]
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D.; Prakash, Y.S., Jr. Cellular senescence in the lung across the age spectrum. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L826–L842. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Starke, R.D.; Hu, Y.; Mercado, N.; Margariti, A.; Gorgoulis, V.G.; Randi, A.M.; Barnes, P.J. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells 2013, 31, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; Heijink, I.H.; Van den Berge, M.; Timens, W.; Oliver, B.G.G.; De Vries, M.; Brandsma, C.A. COPD-derived fibroblasts secrete higher levels of senescence-associated secretory phenotype proteins. Thorax 2021, 76, 508–511. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Crystal, R.G. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. S5), S252–S258. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; McCray, P.B.; Bals, R., Jr. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Puchelle, E.; Zahm, J.M.; Tournier, J.M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Wijk, S.C.; Prabhala, P.; Michalikova, B.; Sommarin, M.; Doyle, A.; Lang, S.; Kanzenbach, K.; Tufvesson, E.; Lindstedt, S.; Leigh, N.D.; et al. Human primary airway basal cells display a continuum of molecular phases from health to disease in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2021, 65, 103–113. [Google Scholar] [CrossRef]
- Yang, J.; Zuo, W.L.; Fukui, T.; Chao, I.; Gomi, K.; Lee, B.; Staudt, M.R.; Kaner, R.J.; Strulovici-Barel, Y.; Salit, J.; et al. Smoking-dependent distal-to-proximal repatterning of the adult human small airway epithelium. Am. J. Respir. Crit. Care Med. 2017, 196, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, E.; Turato, G.; Tine, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef]
- Doyle, I.; Ratcliffe, M.; Walding, A.; Vanden Bon, E.; Dymond, M.; Tomlinson, W.; Tilley, D.; Shelton, P.; Dougall, I. Differential gene expression analysis in human monocyte-derived macrophages: Impact of cigarette smoke on host defence. Mol. Immunol. 2010, 47, 1058–1065. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O’Connor, T.P.; Crystal, R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: Implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009, 183, 2867–2883. [Google Scholar] [CrossRef]
- Hristova, M.; Spiess, P.C.; Kasahara, D.I.; Randall, M.J.; Deng, B.; Van der Vliet, A. The tobacco smoke component, acrolein, suppresses innate macrophage responses by direct alkylation of c-Jun N-terminal kinase. Am. J. Respir. Cell Mol. Biol. 2012, 46, 23–33. [Google Scholar] [CrossRef]
- Singh, D.; Agusti, A.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Criner, G.J.; Frith, P.; Halpin, D.M.; Han, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: The GOLD science committee report 2019. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Barnes, P.J.; Vestbo, J.; Calverley, P.M. The pressing need to redefine “COPD”. Chronic Obstr. Pulm. Dis. 2019, 6, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.E.; Regan, E.A.; Anzueto, A.; Austin, E.; Austin, J.H.M.; Beaty, T.H.; Benos, P.V.; Benway, C.J.; Bhatt, S.P.; Bleecker, E.R.; et al. COPDGene((R)) 2019: Redefining the diagnosis of chronic obstructive pulmonary disease. Chronic Obstr. Pulm. Dis. 2019, 6, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.; Crystal, R.G. Oxidants, antioxidants and the pathogenesis of emphysema. Eur. J. Respir. Dis. Suppl. 1985, 139, 7–17. [Google Scholar]
- Laurell, C.B.; Eriksson, S. The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. 1963. COPD 2013, 10 (Suppl. S1), 3–8. [Google Scholar]
- Salvi, S.; Barnes, P.J. Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest 2010, 138, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Van der Vaart, H.; Postma, D.S.; Timens, W.; Ten Hacken, N.H. Acute effects of cigarette smoke on inflammation and oxidative stress: A review. Thorax 2004, 59, 713–721. [Google Scholar] [CrossRef]
- Yanbaeva, D.G.; Dentener, M.A.; Creutzberg, E.C.; Wesseling, G.; Wouters, E.F. Systemic effects of smoking. Chest 2007, 131, 1557–1566. [Google Scholar] [CrossRef]
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Anderson, R.K.; Correia-Melo, C.; Jurk, D.; Hewitt, G.; Marques, F.M.; Green, N.J.; Moisey, E.; Birrell, M.A.; Belvisi, M.G.; et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1124–L1137. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Milevoj Kopcinovic, L.; Domijan, A.M.; Posavac, K.; Cepelak, I.; Zanic Grubisic, T.; Rumora, L. Systemic redox imbalance in stable chronic obstructive pulmonary disease. Biomarkers 2016, 21, 692–698. [Google Scholar] [CrossRef]
- Schaberg, T.; Klein, U.; Rau, M.; Eller, J.; Lode, H. Subpopulations of alveolar macrophages in smokers and nonsmokers: Relation to the expression of CD11/CD18 molecules and superoxide anion production. Am. J. Respir. Crit. Care Med. 1995, 151, 1551–1558. [Google Scholar] [CrossRef]
- Van der Vliet, A.; Janssen-Heininger, Y.M.W.; Anathy, V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Asp. Med. 2018, 63, 59–69. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef]
- Harju, T.; Kaarteenaho-Wiik, R.; Sirvio, R.; Paakko, P.; Crapo, J.D.; Oury, T.D.; Soini, Y.; Kinnula, V.L. Manganese superoxide dismutase is increased in the airways of smokers’ lungs. Eur. Respir. J. 2004, 24, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Regan, E.A.; Mazur, W.; Meoni, E.; Toljamo, T.; Millar, J.; Vuopala, K.; Bowler, R.P.; Rahman, I.; Nicks, M.E.; Crapo, J.D.; et al. Smoking and COPD increase sputum levels of extracellular superoxide dismutase. Free Radic. Biol. Med. 2011, 51, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, G.; Pietraforte, D.; Matarrese, P.; Gambardella, L.; Metere, A.; Paone, G.; Bianchi, E.L.; Straface, E. The red blood cell as a biosensor for monitoring oxidative imbalance in chronic obstructive pulmonary disease: An ex vivo and in vitro study. Antioxid. Redox Signal. 2006, 8, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Straface, E.; Matarrese, P.; Gambardella, L.; Forte, S.; Carlone, S.; Libianchi, E.; Schmid, G.; Malorni, W. N-Acetylcysteine counteracts erythrocyte alterations occurring in chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2000, 279, 552–556. [Google Scholar] [CrossRef]
- Remels, A.H.; Gosker, H.R.; Langen, R.C.; Schols, A.M. The mechanisms of cachexia underlying muscle dysfunction in COPD. J. Appl. Physiol. 2013, 114, 1253–1262. [Google Scholar] [CrossRef]
- Abrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid. Med. Cell. Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef]
- Rahman, I.; Kinnula, V.L. Strategies to decrease ongoing oxidant burden in chronic obstructive pulmonary disease. Expert Rev. Clin. Pharmacol. 2012, 5, 293–309. [Google Scholar] [CrossRef]
- Randall, M.J.; Hristova, M.; Van der Vliet, A. Protein alkylation by the alpha, beta-unsaturated aldehyde acrolein. A reversible mechanism of electrophile signaling? FEBS Lett. 2013, 587, 3808–3814. [Google Scholar] [CrossRef] [PubMed]
- Moretto, N.; Volpi, G.; Pastore, F.; Facchinetti, F. Acrolein effects in pulmonary cells: Relevance to chronic obstructive pulmonary disease. Ann. N. Y. Acad. Sci. 2012, 1259, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Finkelstein, E.I.; Wong, P.S.; Phung, A.; Cross, C.E.; Van der Vliet, A. Identification of glutathione modifications by cigarette smoke. Free Radic. Biol. Med. 2002, 33, 1490–1498. [Google Scholar] [CrossRef]
- Van der Toorn, M.; Smit-de Vries, M.P.; Slebos, D.J.; De Bruin, H.G.; Abello, N.; Van Oosterhout, A.J.; Bischoff, R.; Kauffman, H.F. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1156–L1162. [Google Scholar] [CrossRef] [PubMed]
- Willemse, B.W.; ten Hacken, N.H.; Rutgers, B.; Lesman-Leegte, I.G.; Postma, D.S.; Timens, W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur. Respir. J. 2005, 26, 835–845. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.E.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Z.; Zhang, P.; Qu, J.; Zheng, C.; Mo, X.; Zhou, W.; Xu, L.; Yao, H.; Gao, J. Nrf2 attenuates inflammatory response in COPD/emphysema: Crosstalk with Wnt3a/beta-catenin and AMPK pathways. J. Cell. Mol. Med. 2018, 22, 3514–3525. [Google Scholar] [CrossRef]
- Morrison, D.; Rahman, I.; Lannan, S.; MacNee, W. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am. J. Respir. Crit. Care Med. 1999, 159, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Turgut, T.; Ilhan, N.; Deveci, F.; Akpolat, N.; Erden, E.S.; Muz, M.H. Glutathione and nitrite levels in induced sputum at COPD patients and healthy smokers. J. Thorac. Dis. 2014, 6, 765–771. [Google Scholar] [PubMed]
- Beeh, K.M.; Beier, J.; Koppenhoefer, N.; Buhl, R. Increased glutathione disulfide and nitrosothiols in sputum supernatant of patients with stable COPD. Chest 2004, 126, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.G.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef]
- Numakura, T.; Sugiura, H.; Akaike, T.; Ida, T.; Fujii, S.; Koarai, A.; Yamada, M.; Onodera, K.; Hashimoto, Y.; Tanaka, R.; et al. Production of reactive persulfide species in chronic obstructive pulmonary disease. Thorax 2017, 72, 1074–1083. [Google Scholar] [CrossRef]
- De Deken, X.; Corvilain, B.; Dumont, J.E.; Miot, F. Roles of DUOX-mediated hydrogen peroxide in metabolism, host defense, and signaling. Antioxid. Redox Signal. 2014, 20, 2776–2793. [Google Scholar] [CrossRef]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; Van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Protein oxidation. Ann. N. Y. Acad. Sci. 2000, 899, 191–208. [Google Scholar] [CrossRef]
- Sirokmany, G.; Geiszt, M. The relationship of NADPH Oxidases and Heme Peroxidases: Fallin’ in and out. Front. Immunol. 2019, 10, 394. [Google Scholar] [CrossRef]
- Lee, I.T.; Yang, C.M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef]
- Nauseef, W.M. How human neutrophils kill and degrade microbes: An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490. [Google Scholar] [CrossRef]
- Roos, D. Chronic granulomatous disease. Methods Mol. Biol. 2019, 1982, 531–542. [Google Scholar]
- Fu, P.; Mohan, V.; Mansoor, S.; Tiruppathi, C.; Sadikot, R.T.; Natarajan, V. Role of nicotinamide adenine dinucleotide phosphate-reduced oxidase proteins in Pseudomonas aeruginosa-induced lung inflammation and permeability. Am. J. Respir. Cell Mol. Biol. 2013, 48, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove, R.J.; Edwards, L.; Rae, A.J.; Hussell, T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006, 36, 1364–1373. [Google Scholar] [CrossRef]
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-kappaB in airway epithelial cells. J. Immunol. 2008, 180, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Metrailler-Ruchonnet, I.; Schappi, M.; Donati, Y.; Matthay, M.A.; Krause, K.H.; Barazzone Argiroffo, C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care. Med. 2009, 180, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wang, Y.; Wu, X.; Peshavariya, H.M.; Dusting, G.J.; Zhang, M.; Jiang, F. Nox4 and redox signaling mediate TGF-beta-induced endothelial cell apoptosis and phenotypic switch. Cell. Death Dis. 2014, 5, e1010. [Google Scholar] [CrossRef]
- He, C.; Larson-Casey, J.L.; Davis, D.; Hanumanthu, V.S.; Longhini, A.L.F.; Thannickal, V.J.; Gu, L.; Carter, A.B. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Veith, C.; Hristova, M.; Danyal, K.; Habibovic, A.; Dustin, C.M.; McDonough, J.E.; Vanaudenaerde, B.M.; Kreuter, M.; Schneider, M.A.; Kahn, N.; et al. Profibrotic epithelial TGF-beta1 signaling involves NOX4-mitochondria cross talk and redox-mediated activation of the tyrosine kinase FYN. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L356–L367. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef]
- Li, Z.M.; Xu, S.Y.; Feng, Y.Z.; Cheng, Y.R.; Xiong, J.B.; Zhou, Y.; Guan, C.X. The role of NOX4 in pulmonary diseases. J. Cell. Physiol. 2021, 236, 1628–1637. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003, 17, 1502–1504. [Google Scholar] [CrossRef]
- Van der Vliet, A.; Danyal, K.; Heppner, D.E. Dual oxidase: A novel therapeutic target in allergic disease. Br. J. Pharmacol. 2018, 175, 1401–1418. [Google Scholar] [CrossRef]
- Sarr, D.; Gingerich, A.D.; Asthiwi, N.M.; Almutairi, F.; Sautto, G.A.; Ecker, J.; Nagy, T.; Kilgore, M.B.; Chandler, J.D.; Ross, T.M.; et al. Dual oxidase 1 promotes antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Schiffers, C.; Hristova, M.; Habibovic, A.; Dustin, C.M.; Danyal, K.; Reynaert, N.L.; Wouters, E.F.; Van der Vliet, A. The transient receptor potential channel vanilloid 1 is critical in innate airway epithelial responses to protease allergens. Am. J. Respir. Cell Mol. Biol. 2020, 63, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Hristova, M.; Habibovic, A.; Veith, C.; Janssen-Heininger, Y.M.; Dixon, A.E.; Geiszt, M.; Van der Vliet, A. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J. Allergy Clin. Immunol. 2016, 137, 1545–1556. [Google Scholar] [CrossRef]
- Wesley, U.V.; Bove, P.F.; Hristova, M.; McCarthy, S.; Van der Vliet, A. Airway epithelial cell migration and wound repair by ATP-mediated activation of dual oxidase 1. J. Biol. Chem. 2007, 282, 3213–3220. [Google Scholar] [CrossRef]
- Van der Vliet, A.; Janssen-Heininger, Y.M. Hydrogen peroxide as a damage signal in tissue injury and inflammation: Murderer, mediator, or messenger? J. Cell. Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef]
- Joo, J.H.; Ryu, J.H.; Kim, C.H.; Kim, H.J.; Suh, M.S.; Kim, J.O.; Chung, S.Y.; Lee, S.N.; Kim, H.M.; Bae, Y.S.; et al. Dual oxidase 2 is essential for the toll-like receptor 5-mediated inflammatory response in airway mucosa. Antioxid. Redox Signal. 2012, 16, 57–70. [Google Scholar] [CrossRef]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; De Deken, X.; Brochiero, E.; Miot, F.; Grandvaux, N. IFNbeta/TNFalpha synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell Res. 2013, 23, 673–690. [Google Scholar] [CrossRef]
- Sahoo, S.; Meijles, D.N.; Pagano, P.J. NADPH oxidases: Key modulators in aging and age-related cardiovascular diseases? Clin. Sci. 2016, 130, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef]
- McCrann, D.J.; Yang, D.; Chen, H.; Carroll, S.; Ravid, K. Upregulation of Nox4 in the aging vasculature and its association with smooth muscle cell polyploidy. Cell Cycle 2009, 8, 902–908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rezende, F.; Schurmann, C.; Schutz, S.; Harenkamp, S.; Herrmann, E.; Seimetz, M.; Weißmann, N.; Schröder, K. Knock out of the NADPH oxidase Nox4 has no impact on life span in mice. Redox Biol. 2017, 11, 312–314. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]
- Ghatak, S.; Hascall, V.C.; Markwald, R.R.; Feghali-Bostwick, C.; Artlett, C.M.; Gooz, M.; Bogatkevich, G.S.; Atanelishvili, I.; Silver, R.M.; Wood, J.; et al. Transforming growth factor beta1 (TGFbeta1)-induced CD44V6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis. J. Biol. Chem. 2017, 292, 10490–10519. [Google Scholar] [CrossRef]
- Palumbo, S.; Shin, Y.J.; Ahmad, K.; Desai, A.A.; Quijada, H.; Mohamed, M.; Knox, A.; Sammani, S.; Colson, B.A.; Wang, T.; et al. Dysregulated Nox4 ubiquitination contributes to redox imbalance and age-related severity of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L297–L308. [Google Scholar] [CrossRef]
- Wen, Z.; Shimojima, Y.; Shirai, T.; Li, Y.; Ju, J.; Yang, Z.; Tian, L.; Goronzy, J.J.; Weyand, C.M. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J. Clin. Investig. 2016, 126, 1953–1967. [Google Scholar] [CrossRef]
- Lee, K.; Won, H.Y.; Bae, M.A.; Hong, J.H.; Hwang, E.S. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9548–9553. [Google Scholar] [CrossRef] [PubMed]
- Meijles, D.N.; Sahoo, S.; Al Ghouleh, I.; Amaral, J.H.; Bienes-Martinez, R.; Knupp, H.E.; Attaran, S.; Sembrat, J.C.; Nouraie, S.M.; Rojas, M.M.; et al. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kracun, D.; Dustin, C.M.; El Massry, M.; Yuan, S.; Goossen, C.J.; DeVallance, E.R.; Sahoo, S.; Hilaire, C.S.; Gurkar, A.U.; et al. Forestalling age-impaired angiogenesis and blood flow by targeting NOX: Interplay of NOX1, IL-6, and SASP in propagating cell senescence. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Boe, D.M.; Boule, L.A.; Kovacs, E.J. Innate immune responses in the ageing lung. Clin. Exp. Immunol. 2017, 187, 16–25. [Google Scholar] [CrossRef]
- Wu, Y.; Goplen, N.P.; Sun, J. Aging and respiratory viral infection: From acute morbidity to chronic sequelae. Cell Biosci. 2021, 11, 112. [Google Scholar] [CrossRef]
- Khanna, K.; Raymond, W.; Charbit, A.R.; Jin, J.; Gitlin, I.; Tang, M.; Sperber, H.S.; Franz, S.; Pillai, S.; Simmons, G.; et al. Binding of SARS-CoV-2 spike protein to ACE2 is disabled by thiol-based drugs; evidence from in vitro SARS-CoV-2 infection studies. Biorxiv 2020. [Google Scholar] [CrossRef]
- Edens, W.A.; Sharling, L.; Cheng, G.; Shapira, R.; Kinkade, J.M.; Lee, T.; Edens, H.A.; Tang, X.; Sullards, C.; Flaherty, D.B.; et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 2001, 154, 879–891. [Google Scholar] [CrossRef]
- Sasakura, H.; Moribe, H.; Nakano, M.; Ikemoto, K.; Takeuchi, K.; Mori, I. Lifespan extension by peroxidase and dual oxidase-mediated ROS signaling through pyrroloquinoline quinone in C. elegans. J. Cell Sci. 2017, 130, 2631–2643. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Hourihan, J.M.; Bland, M.S.; Obieglo, C.; Katic, I.; Moronetti Mazzeo, L.E.; Alcedo, J.; Blackwell, T.K.; Hynes, N.E. NADPH oxidase-mediated redox signaling promotes oxidative stress resistance and longevity through memo-1 in C. elegans. Elife 2017, 6, e19493. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernandez-Cardenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoeven, R.; Cruz, M.R.; Chavez, V.; Garsin, D.A. Localization of the Dual Oxidase BLI-3 and Characterization of Its NADPH Oxidase Domain during Infection of Caenorhabditis elegans. PLoS ONE 2015, 10, e0124091. [Google Scholar] [CrossRef]
- Schader, T.; Reschke, C.; Spaeth, M.; Wienstroer, S.; Wong, S.; Schroder, K. NoxO1 Knockout Promotes Longevity in Mice. Antioxidants 2020, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, P.C. Cytoplasmic and mitochondrial NADPH-coupled redox systems in the regulation of aging. Nutrients 2019, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Pagliarani, A. Nicotinamide nucleotide transhydrogenase as a sensor of mitochondrial biology. Trends Cell Biol. 2020, 30, 1–3. [Google Scholar] [CrossRef]
- Murphy, M.P. Redox Modulation by reversal of the mitochondrial nicotinamide nucleotide transhydrogenase. Cell Metab. 2015, 22, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.C.; Krause, K.H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2015, 235, 65–78. [Google Scholar] [CrossRef]
- Tollefson, A.K.; Oberley-Deegan, R.E.; Butterfield, K.T.; Nicks, M.E.; Weaver, M.R.; Remigio, L.K.; Decsesznak, J.; Chu, H.W.; Bratton, D.L.; Riches, D.W.; et al. Endogenous enzymes (NOX and ECSOD) regulate smoke-induced oxidative stress. Free Radic. Biol. Med. 2010, 49, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Edirisinghe, I.; Yang, S.R.; Rajendrasozhan, S.; Kode, A.; Caito, S.; Adenuga, D.; Rahman, I. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am. J. Pathol. 2008, 172, 1222–1237. [Google Scholar] [CrossRef] [PubMed]
- Hollins, F.; Sutcliffe, A.; Gomez, E.; Berair, R.; Russell, R.; Szyndralewiez, C.; Saunders, R.; Brightling, C. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir. Res. 2016, 17, 84. [Google Scholar] [CrossRef]
- Liu, X.; Hao, B.; Ma, A.; He, J.; Liu, X.; Chen, J. The expression of NOX4 in smooth muscles of small airway correlates with the disease severity of COPD. Biomed. Res. Int. 2016, 2016, 2891810. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Fan, Y.; Cui, J.; Hao, B.; Zhu, L.; Sun, X.; He, J.; Yang, J.; Dong, J.; Wang, Y.; et al. NOX4 expression and distal arteriolar remodeling correlate with pulmonary hypertension in COPD. BMC Pulm. Med. 2018, 18, 111. [Google Scholar] [CrossRef]
- Hernandez-Saavedra, D.; Sanders, L.; Perez, M.J.; Kosmider, B.; Smith, L.P.; Mitchell, J.D.; Yoshida, T.; Tuder, R.M. RTP801 amplifies nicotinamide adenine dinucleotide phosphate oxidase-4-dependent oxidative stress induced by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2017, 56, 62–73. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059. [Google Scholar] [CrossRef]
- Nagai, K.; Betsuyaku, T.; Suzuki, M.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid. Redox Signal. 2008, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Pierrou, S.; Broberg, P.; O’Donnell, R.A.; Pawlowski, K.; Virtala, R.; Lindqvist, E.; Richter, A.; Wilson, S.J.; Angco, G.; Moller, S.; et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Schiffers, C.; Van de Wetering, C.; Bauer, R.A.; Habibovic, A.; Hristova, M.; Dustin, C.M.; Lambrichts, S.; Vacek, P.M.; Wouters, E.F.; Reynaert, N.L.; et al. Downregulation of epithelial DUOX1 in chronic obstructive pulmonary disease. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, H.; Dixon, J.; Traphagen, N.; Wyatt, T.A.; Kharbanda, K.; Chadwick, S.S.; Kolliputi, N.; Allen-Gipson, D.S. Cigarette smoke impairs A2A adenosine receptor mediated wound repair through up-regulation of Duox-1 expression. Sci. Rep. 2017, 7, 44405. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, R.; Farne, H.; Ritchie, A.; Luke, E.; Johnston, S.L.; Mallia, P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther. Adv. Respir. Dis. 2016, 10, 158–174. [Google Scholar] [CrossRef]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta 2012, 1822, 714–728. [Google Scholar] [CrossRef]
- Vezina, F.A.; Cantin, A.M. Antioxidants and chronic obstructive pulmonary disease. Chronic. Obstr. Pulm. Dis. 2018, 5, 277–288. [Google Scholar] [CrossRef]
- Chocry, M.; Leloup, L. The NADPH oxidase family and its inhibitors. Antioxid. Redox Signal. 2020, 33, 332–353. [Google Scholar] [CrossRef]
- Kang, C. Senolytics and senostatics: A two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cells 2019, 42, 821–827. [Google Scholar] [PubMed]
- Zhang, M.; Swarts, S.G.; Yin, L.; Liu, C.; Tian, Y.; Cao, Y.; Swarts, M.; Yang, S.; Zhang, S.B.; Zhang, K.; et al. Antioxidant properties of quercetin. Adv. Exp. Med. Biol. 2011, 701, 283–289. [Google Scholar] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Navarro-Nunez, L.; Lozano, M.L.; Martinez, C.; Vicente, V.; Rivera, J. Effect of quercetin on platelet spreading on collagen and fibrinogen and on multiple platelet kinases. Fitoterapia 2010, 81, 75–80. [Google Scholar] [CrossRef]
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Rogliani, P.; Matera, M.G.; Page, C.; Puxeddu, E.; Cazzola, M.; Calzetta, L. Efficacy and safety profile of mucolytic/antioxidant agents in chronic obstructive pulmonary disease: A comparative analysis across erdosteine, carbocysteine, and N-acetylcysteine. Respir. Res. 2019, 20, 104. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Antioxidant pharmacological therapies for COPD. Curr. Opin. Pharmacol. 2012, 12, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.P.; Kang, J.; Huang, S.G.; Chen, P.; Yao, W.Z.; Yang, L.; Bai, C.X.; Wang, C.Z.; Wang, C.; Chen, B.Y.; et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study): A randomised placebo-controlled study. Lancet 2008, 371, 2013–2018. [Google Scholar] [CrossRef]
- Tatsumi, K.; Fukuchi, Y.; Group, P.S. Carbocisteine improves quality of life in patients with chronic obstructive pulmonary disease. J. Am. Geriatr. Soc. 2007, 55, 1884–1886. [Google Scholar] [CrossRef]
- Poole, P.J. Role of mucolytics in the management of COPD. Int. J. Chron Obstruct Pulmon Dis. 2006, 1, 123–128. [Google Scholar] [CrossRef][Green Version]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef]
- Hanson, C.; Rutten, E.P.; Wouters, E.F.; Rennard, S. Influence of diet and obesity on COPD development and outcomes. Int. J. Chron. Obstruct. Pulmon. Dis. 2014, 9, 723–733. [Google Scholar] [CrossRef]
- Scoditti, E.; Massaro, M.; Garbarino, S.; Toraldo, D.M. Role of diet in chronic obstructive pulmonary disease prevention and treatment. Nutrients 2019, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Tsiligianni, I.G.; Van der Molen, T. A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir. Res. 2010, 11, 171. [Google Scholar] [CrossRef]
- Gouzi, F.; Maury, J.; Heraud, N.; Molinari, N.; Bertet, H.; Ayoub, B.; Blaquière, M.; Bughin, F.; De Rigal, P.; Poulain, M.; et al. Additional effects of nutritional antioxidant supplementation on peripheral muscle during pulmonary rehabilitation in copd patients: A randomized controlled trial. Oxid. Med. Cell Longev. 2019, 2019, 5496346. [Google Scholar] [CrossRef] [PubMed]
- Alpha-Tocopherol BCCPSG. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, J.; Wang, J.; Li, S.; Fukunaga, A.; Yodoi, J.; Tian, H. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct. Target. Ther. 2020, 5, 248. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Thimmulappa, R.K.; Harvey, C.J. Experimental therapeutics of Nrf2 as a target for prevention of bacterial exacerbations in COPD. Proc. Am. Thorac. Soc. 2012, 9, 47–51. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD(+) metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Rahman, I.; Kinnula, V.L.; Gorbunova, V.; Yao, H. SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev. Med. 2012, 54, S20–S28. [Google Scholar] [CrossRef]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Radenkovic, D.; Reason Verdin, E. Clinical evidence for targeting NAD therapeutically. Pharmaceuticals 2020, 13, 247. [Google Scholar] [CrossRef]
- Partridge, L.; Fuentealba, M.; Kennedy, B.K. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 2020, 19, 513–532. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Bernardo, I.; Mastronardo, C.; Mou, K.; De Luca, S.N.; Seow, H.J.; Dobric, A.; Brassington, K.; Selemidis, S.; Bozinovski, S.; et al. Apocynin prevents cigarette smoking-induced loss of skeletal muscle mass and function in mice by preserving proteostatic signalling. Br. J. Pharmacol. 2021, 178, 3049–3066. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Thannickal, V.J. NADPH oxidase inhibition in fibrotic pathologies. Antioxid. Redox Signal. 2020, 33, 455–479. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant supplements and mortality. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Osar, Z.; Samanci, T.; Demirel, G.Y.; Damci, T.; Ilkova, H. Nicotinamide effects oxidative burst activity of neutrophils in patients with poorly controlled type 2 diabetes mellitus. Exp. Diabesity Res. 2004, 5, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Rabani, R.; Cossette, C.; Graham, F.; Powell, W.S. Protein kinase C activates NAD kinase in human neutrophils. Free Radic. Biol. Med. 2020, 161, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, P.M.; Bansal, N.; Kerrigan, J.E.; Abali, E.E.; Scotto, K.W.; Bertino, J.R. NAD+ kinase as a therapeutic target in cancer. Clin. Cancer Res. 2016, 22, 5189–5195. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Cai, P.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Inhibition of glucose-6-phosphate dehydrogenase reverses cisplatin resistance in lung cancer cells via the redox system. Front. Pharmacol. 2018, 9, 43. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| The Free Radical Theory (FRT) of Aging [8] | |
|---|---|
| Supporting evidence | Conflicting evidence |
|
|
| Normalized Expression | Lung Cell Expression [157] | ROS Production | |
|---|---|---|---|
| NOX1 | 0.2 | Endothelium Immune cells | O2•− |
| NOX2/CYBB | 49.4 | Endothelium Immune cells Alveolar and airway epithelium | O2•− |
| NOX3/gp91phox | 0.1 | Endothelium N.A. | O2•− |
| NOX4 | 1.2 | Smooth muscle and endothelium Myofibroblasts Immune cells Alveolar and airway epithelium | O2•− H2O2 |
| NOX5 | 0.6 | N.A. | O2•− |
| DUOX1 | 48.7 | Alveolar and airway epithelium | H2O2 |
| DUOX2 | 1.2 | Alveolar and airway epithelium | H2O2 |
| Compounds | Examples | Pitfalls | Findings |
|---|---|---|---|
| Thiol compounds with mucolytic properties | N-acetyl-cysteine (NAC) Erdosteine Carbocysteine Fudosteine | Most likely function as mucolytics, rather than ROS scavengers | Clinical studies indicated some efficacy (reduced exacerbations, mortality risk, hospitalization); others did not (no improvement in lung function) [92,222,229,230,231,232] |
| Mimetics of glutathione peroxidase superoxide dismutase | Ebselen (Manganese) Metaloporphyrins | These ‘anti-oxidants’ do not scavenge all ROS | Clinical efficacy in COPD patients unknown; Performed well in phase I safety studies; Currently being developed for COPD patients [234] |
| Dietary agents, polyphenols | Ascorbate α-tocopherol Carotenoids Flavonoids Zinc gluconate Selenomethionine | Not necessarily effective against all ROS; Some flavonoids (e.g., quercetin) may function as senolytics, potentially by tyrosine kinase inhibition; Flavonoid oxidation to (semi) quinones may generate electrophiles that activate Nrf2 | Mixed findings; No improvement in lung function, clinical features of COPD [237]; Improved muscle strength [238]; Adverse health effects: increased incidence of lung cancer [239] |
| Nrf2 activators | CDDO-Im Sulforaphane | Do not directly target ROS | Alleviates inflammation; improves innate antibacterial defenses; restores corticosteroid-induced responses [244,245] |
| Potential Strategies | Examples | Potential Pitfalls | Findings |
| Mitochondria-targeted anti-oxidant compounds | MitoQ MitoTEMPO | May interfere with mtROS signaling | No clinical knowledge in COPD yet |
| Activators of endogenous anti-oxidant responses | Nicotinamide adenine dinucleotide (NAD) precursors | May also enhance NOX function | No clinical knowledge in COPD yet |
| Pharmacological inhibitors of of NADPH oxidase enzymes | Apocynin Setanaxib | Inhibition of DUOX1 function | No clinical knowledge in COPD yet; Setanaxib currently in clinical trial for IPF [223] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiffers, C.; Reynaert, N.L.; Wouters, E.F.M.; van der Vliet, A. Redox Dysregulation in Aging and COPD: Role of NOX Enzymes and Implications for Antioxidant Strategies. Antioxidants 2021, 10, 1799. https://doi.org/10.3390/antiox10111799
Schiffers C, Reynaert NL, Wouters EFM, van der Vliet A. Redox Dysregulation in Aging and COPD: Role of NOX Enzymes and Implications for Antioxidant Strategies. Antioxidants. 2021; 10(11):1799. https://doi.org/10.3390/antiox10111799
Chicago/Turabian StyleSchiffers, Caspar, Niki L. Reynaert, Emiel F. M. Wouters, and Albert van der Vliet. 2021. "Redox Dysregulation in Aging and COPD: Role of NOX Enzymes and Implications for Antioxidant Strategies" Antioxidants 10, no. 11: 1799. https://doi.org/10.3390/antiox10111799
APA StyleSchiffers, C., Reynaert, N. L., Wouters, E. F. M., & van der Vliet, A. (2021). Redox Dysregulation in Aging and COPD: Role of NOX Enzymes and Implications for Antioxidant Strategies. Antioxidants, 10(11), 1799. https://doi.org/10.3390/antiox10111799