
Tumour Microenvironment Stress Promotes the Development of Drug Resistance

 , and
, and 
Abstract
1. Introduction
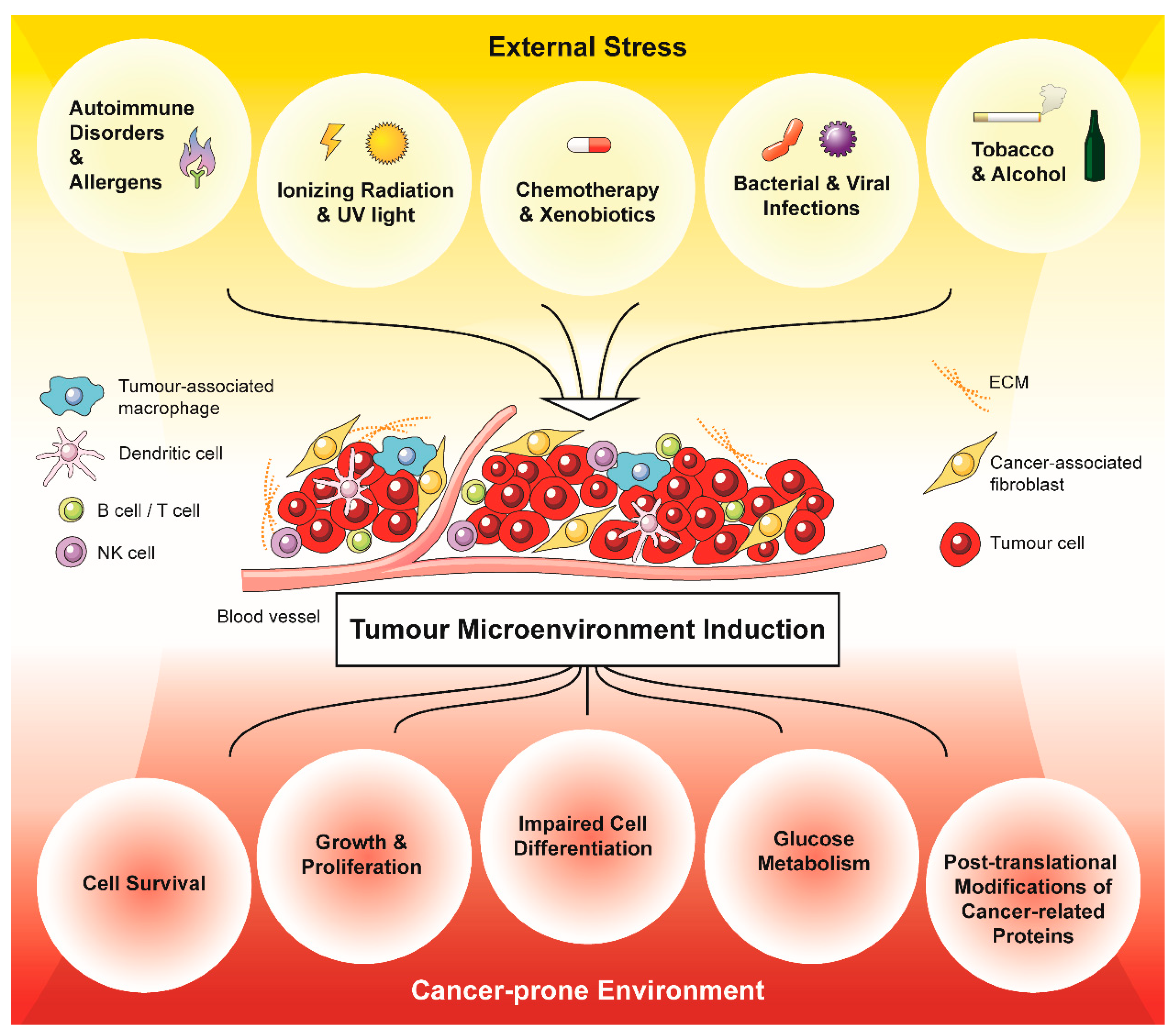
2. External Stress Mediates the Development of a Cancer-Prone Microenvironment
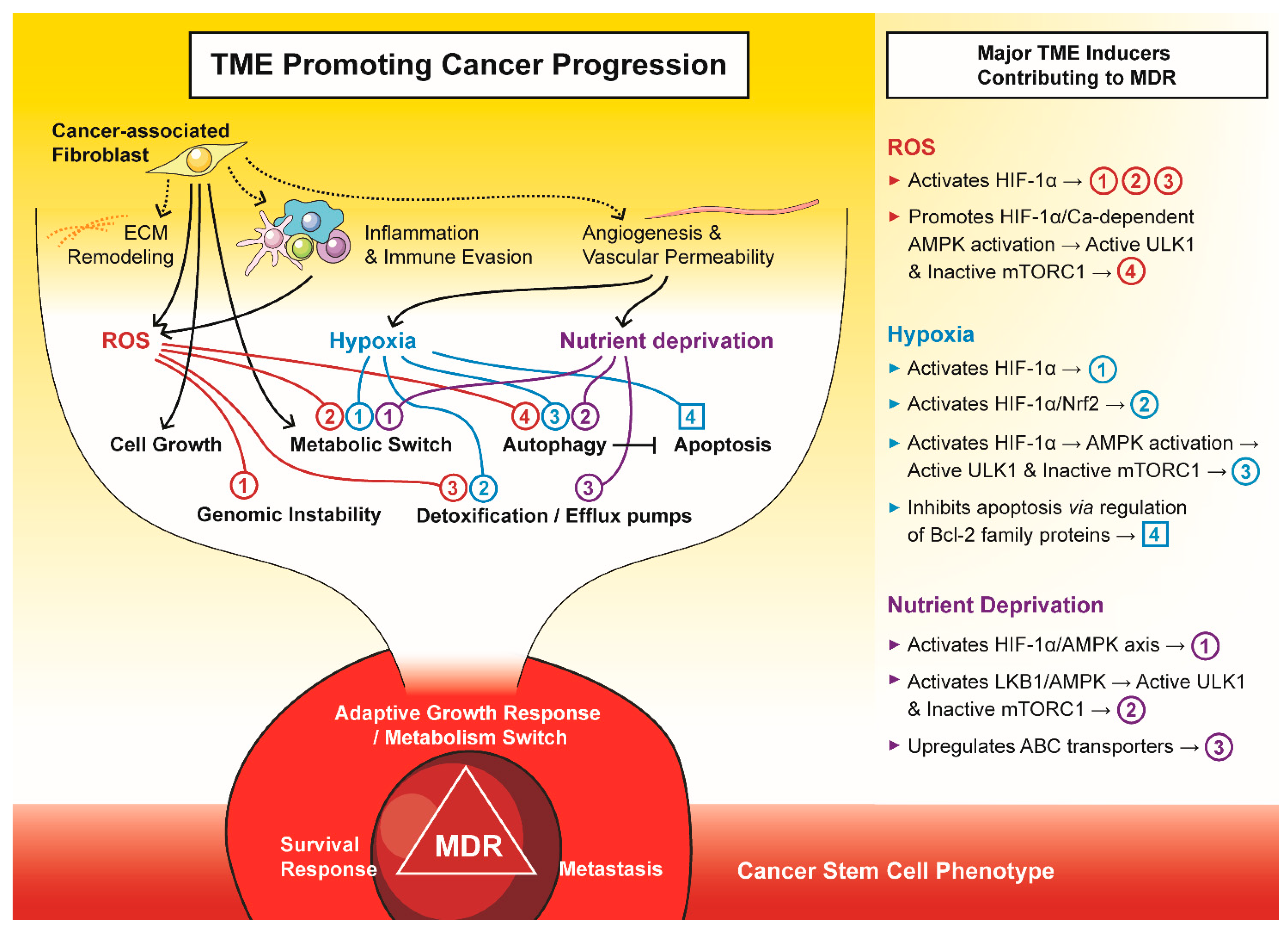
3. Microenvironmental Stress and the Development of Drug Resistance
3.1. Metabolic Reprogramming, the ROS/HIF-Axis and the Development of Multi-Drug Resistance
3.2. Stromal Cells and the TME
3.3. The TME Modulates Autophagy and Apoptosis to Enhance Cancer Cell Survival
3.4. TME Induces a Cancer Stem Cell (CSC) Phenotype
4. Clinical Use of Agents Targeting the Stress Factors within the TME
4.1. Targeting the ROS/HIF Axis
4.2. Stroma-Targeting Therapies

{kind=link}
{kind=link}
{kind=link}
| Stromal Targets | Compounds Involved in Cancer Clinical Trials |
|---|---|
| ECM | |
| collagen type I | nanoparticle albumin-bound paclitaxel [287], halofuginone [285] |
| hyaluronic acid | PEGPH20 [282] |
| integrins | cilengitide [281] |
| lysyl oxidase | all-trans retinoic acid (ATRA) [280], calcipotriol [284] |
| matrix metalloproteinases | marimastat [286] |
| Stroma-specific proteins | |
| CYP3A4 | clarithromycin, itraconazole [159] |
| FAP | ATRA [289], sibrotuzumab [288], RO6874813 [290] |
| Cancer cell-stroma signalling | |
| CXCR4 | plerixafor [296] |
| FAK | defactinib [291] |
| FGFR | AZD4547 [293], dovitinib [294] |
| TGFβ | fresolimumab, galunisertib [295] |
| VEGF | aflibercept, bevacizumab [306], PTK787 [297] |
| VEGFR | pazopanib, sorafenib, sunitinib, vandetanib [292] |
| Inflammation inhibition | |
| pro-inflammatory immune cells | gemcitabine [301], sunitinib [300] |
| mediators of inflammation | celecoxib [307], dexamethasone [304], metformin [302], NSAIDs [305] |
4.3. Clinical Use of Autophagy and Apoptosis-Targeted Therapies
4.4. Clinical Potential of Targeting the CSC–TME Feedback Loop
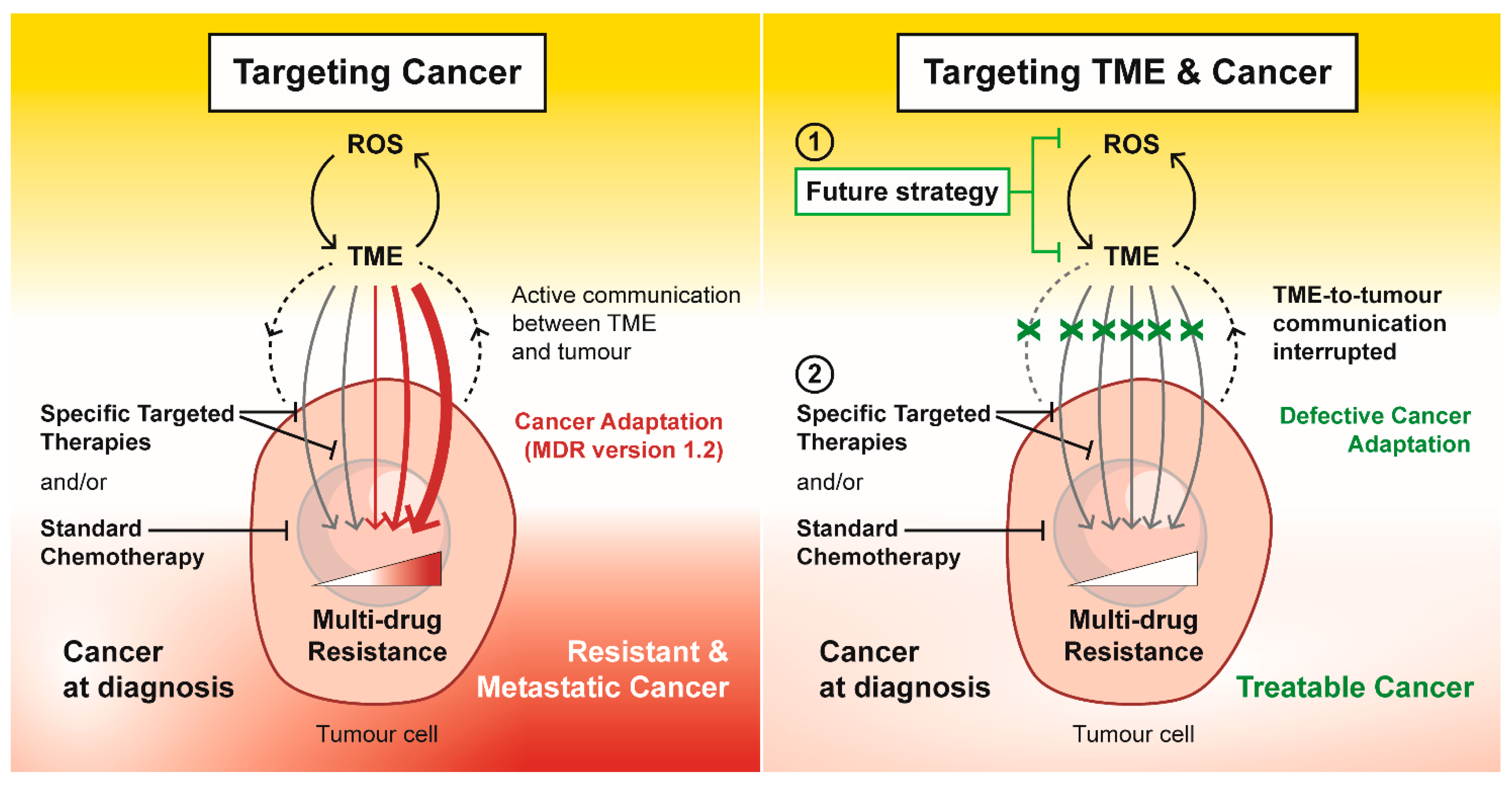
5. Conclusions and New Directions for Anti-Cancer Strategies
Author Contributions
Funding
Conflicts of Interest
References
- Brown, L.F.; Guidi, A.J.; Schnitt, S.J.; van de Water, L.; Iruela-Arispe, M.L.; Yeo, T.K.; Tognazzi, K.; Dvorak, H.F. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin. Cancer Res. 1999, 5, 1041–1056. [Google Scholar]
- Swann, J.B.; Vesely, M.; Silva, A.; Sharkey, J.; Akira, S.; Schreiber, R.D.; Smyth, M.J. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 652–656. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13–secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2017, 116, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Ma, Q.; Cavallin, L.E.; Yan, B.; Zhu, S.; Duran, E.M.; Wang, H.; Hale, L.P.; Dong, C.; Cesarman, E.; Mesri, E.A.; et al. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi’s sarcoma. Proc. Natl. Acad. Sci. USA 2009, 106, 8683–8688. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kikani, C.K.; Verona, E.V.; Ryu, J.; Shen, Y.; Ye, Q.; Zheng, L.; Qian, Z.; Sakaue, H.; Nakamura, K.; Du, J.; et al. Proliferative and Antiapoptotic Signaling Stimulated by Nuclear-Localized PDK1 Results in Oncogenesis. Sci. Signal. 2012, 5, ra80. [Google Scholar] [CrossRef]
- Lluis, J.M.; Buricchi, F.; Chiarugi, P.; Morales, A.; Fernández-Checa, J.C. Dual Role of Mitochondrial Reactive Oxygen Species in Hypoxia Signaling: Activation of Nuclear Factor-κB via c-SRC− and Oxidant-Dependent Cell Death. Cancer Res. 2007, 67, 7368–7377. [Google Scholar] [CrossRef]
- Huang, S.; Pettaway, C.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-κB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef] [PubMed]
- Khromova, N.; Kopnin, P.; Stepanova, E.; Agapova, L.; Kopnin, B. p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett. 2009, 276, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in Enterocytes Generates an Inflammatory Microenvironment Enabling Invasion and Lymph Node Metastasis of Carcinogen-Induced Colorectal Tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, N.; Richardson, D.; Jansson, P.J. Glucose modulation induces reactive oxygen species and increases P-glycoprotein-mediated multidrug resistance to chemotherapeutics. Br. J. Pharmacol. 2015, 172, 2557–2572. [Google Scholar] [CrossRef]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.; Pugh, C.; Ratcliffe, P.; Maxwell, P. Hypoxia Inducible Factor-α Binding and Ubiquitylation by the von Hippel-Lindau Tumor Suppressor Protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Xu, W.; Wang, Z.; Qi, X.; Wang, Y.; Ni, Y.; Shen, H.; Hu, Q.; Han, W. Crosstalk between the HIF-1 and Toll-like receptor/nuclear factor-κB pathways in the oral squamous cell carcinoma microenvironment. Oncotarget 2016, 7, 37773–37789. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-Inducible Factor 1α Is Essential for Cell Cycle Arrest during Hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef]
- Lin, X.; Zheng, W.; Liu, J.; Zhang, Y.; Qin, H.; Wu, H.; Xue, B.; Lu, Y.; Shen, P. Oxidative Stress in Malignant Melanoma Enhances Tumor Necrosis Factor-α Secretion of Tumor-Associated Macrophages That Promote Cancer Cell Invasion. Antioxid. Redox Signal. 2013, 19, 1337–1355. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial–mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Jian, S.L.; Chen, W.W.; Su, Y.C.; Su, Y.W.; Chuang, T.H.; Hsu, S.C.; Huang, L.R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of Peroxiredoxin I by Phosphorylation Allows Localized H2O2 Accumulation for Cell Signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.-T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 Regulates the Angiogenic Phenotype via PTEN Oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Clydesdale, G.J.; Dandie, G.W.; Muller, H.K. Ultraviolet light induced injury: Immunological and inflammatory effects. Immunol. Cell Biol. 2001, 79, 547–568. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Henkler, F.; Brinkmann, J.; Luch, A. The Role of Oxidative Stress in Carcinogenesis Induced by Metals and Xenobiotics. Cancers 2010, 2, 376–396. [Google Scholar] [CrossRef]
- Conklin, K.A. Chemotherapy-Associated Oxidative Stress: Impact on Chemotherapeutic Effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress in Infection and Consequent Disease. Oxid. Med. Cell. Longev. 2017, 2017, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Gani, A.R.; Ansarullah; Ramachandran, A.V.; Dalai, S.; Begum, R. Vitiligo: Interplay between oxidative stress and immune system. Exp. Dermatol. 2013, 22, 245–250. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Hirshberg, J.; Lyle, D.; Freij, J.B.; Caturegli, P. Reactive oxygen species in organ-specific autoimmunity. Autoimmun. Highlights 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Li, Y.; Zhong, W.; Gao, P.; Hu, C. Recent developments in the role of reactive oxygen species in allergic asthma. J. Thorac. Dis. 2017, 9, E32–E43. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Alcohol, Oxidative Stress, and Free Radical Damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Schetter, A.J.; Heegaard, N.H.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2009, 31, 37–49. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Salzano, S.; Checconi, P.; Hanschmann, E.-M.; Lillig, C.H.; Bowler, L.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef]
- Kennel, K.B.; Greten, F.R. Immune cell—Produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef]
- Kamata, T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009, 100, 1382–1388. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Wu, Q.; Chen, Y.; Deng, Y.; Yang, Z.; Zhang, L.; Liu, B. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol. 2019, 22, 101116. [Google Scholar] [CrossRef]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. J. Immun. 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.-J.; Wernicke, D.; Alder, H.; Costinean, S.; Volinia, S.; Croce, C.M. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4908–4913. [Google Scholar] [CrossRef]
- Meena, A.S.; Sharma, A.; Kumari, R.; Muhammad, N.; Singh, S.V.; Bhat, M.K. Inherent and Acquired Resistance to Paclitaxel in Hepatocellular Carcinoma: Molecular Events Involved. PLoS ONE 2013, 8, e61524. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microen-vironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Runa, F.; Hamalian, S.; Meade, K.; Shisgal, P.; Gray, P.C.; Kelber, J.A. Tumor Microenvironment Heterogeneity: Challenges and Opportunities. Curr. Mol. Biol. Rep. 2017, 3, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updates 2012, 15, 39–49. [Google Scholar] [CrossRef]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell Adhesion Mediated Drug Resistance (CAM-DR): Role of Integrins and Resistance to Apoptosis in Human Myeloma Cell Lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Marleau, A.M.; Chen, C.-S.; Joyce, J.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.; Allen, T.; Higashi, R.M.; Ferraris, D.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Höckel, M.; Schlenger, K.; Knoop, C.; Vaupel, P. Oxygenation of carcinomas of the uterine cervix: Evaluation by computerized O2 tension measurements. Cancer Res. 1991, 51, 6098–6102. [Google Scholar]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and Characterization of Tumor Hypoxia Using pO2 Histography. Antioxid. Redox Signal 2007, 9, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.-T.; Chen, E.; Salim, A.; Cao, H.; Kong, C.S.; Whyte, R.; Donington, J.; Cannon, W.; Wakelee, H.; Tibshirani, R.; et al. An Evaluation of Tumor Oxygenation and Gene Expression in Patients with Early Stage Non–Small Cell Lung Cancers. Clin. Cancer Res. 2006, 12, 1507–1514. [Google Scholar] [CrossRef]
- Hielscher, A.; Gerecht, S. Hypoxia and free radicals: Role in tumor progression and the use of engineering-based platforms to address these relationships. Free Radic. Biol. Med. 2015, 79, 281–291. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A Conserved Family of Prolyl-4-Hydroxylases That Modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noel, A.; Pouysségur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Lluis, J.M.; Morales, A.; Blasco, C.; Colell, A.; Mari, M.; Garcia-Ruiz, C.; Fernández-Checa, J.C. Critical Role of Mitochondrial Glutathione in the Survival of Hepatocytes during Hypoxia. J. Biol. Chem. 2005, 280, 3224–3232. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kanai, H.; Uchiyama, T.; Iso, T.; Ohyama, Y.; Sakamoto, H.; Tamura, J.; Nagai, R.; Kurabayashi, M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc. Res. 2005, 67, 714–722. [Google Scholar] [CrossRef]
- Xu, W.; Chi, L.; Row, B.; Xu, R.; Ke, Y.; Xu, B.; Luo, C.; Kheirandish, L.; Gozal, L.; Liu, R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. J. Neurosci. 2004, 126, 313–323. [Google Scholar] [CrossRef]
- Peng, Y.-J.; Yuan, G.; Ramakrishnan, D.; Sharma, S.; Bosch-Marce, M.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J. Physiol. 2006, 577, 705–716. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Chen, J.-Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1826, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The Plasma Membrane Lactate Transporter MCT4, but Not MCT1, Is Up-regulated by Hypoxia through a HIF-1α-dependent Mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.; Granter, S.R.; Widlund, H.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Hu, K.; Babapoor-Farrokhran, S.; Rodrigues, M.; Deshpande, M.; Puchner, B.; Kashiwabuchi, F.; Hassan, S.J.; Asnaghi, L.; Handa, J.T.; Merbs, S.; et al. Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget 2016, 7, 7816–7828. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Baba, M.; Inoue, M.; Itoh, K.; Nishizawa, Y. Blocking CD147 induces cell death in cancer cells through impairment of glycolytic energy metabolism. Biochem. Biophys. Res. Commun. 2008, 374, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Beloueche-Babari, M.; Wantuch, S.; Galobart, T.C.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.-L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 Inhibitor AZD3965 Increases Mitochondrial Metabolism, Facilitating Combination Therapy and Noninvasive Magnetic Resonance Spectroscopy. Cancer Res. 2017, 77, 5913–5924. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Sanità, P.; Capulli, M.; Teti, A.; Galatioto, G.P.; Vicentini, C.; Chiarugi, P.; Bologna, M.; Angelucci, A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 2014, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Whitaker-Menezes, D.; Dasgupta, A.; Philp, N.J.; Lin, Z.; Gandara, R.; Sneddon, S.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Using the “reverse Warburg effect” to identify high-risk breast cancer patients. Stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 2012, 11, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef]
- Tavares-Valente, D.; Baltazar, F.; Moreira, R.; Queirós, O. Cancer cell bioenergetics and pH regulation influence breast cancer cell resistance to paclitaxel and doxorubicin. J. Bioenerg. Biomembr. 2013, 45, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- El Hassouni, B.; Granchi, C.; Vallés-Martí, A.; Supadmanaba, I.G.P.; Bononi, G.; Tuccinardi, T.; Funel, N.; Jimenez, C.R.; Peters, G.J.; Giovannetti, E.; et al. The dichotomous role of the glycolytic metabolism pathway in cancer metastasis: Interplay with the complex tumor microenvironment and novel therapeutic strategies. Semin. Cancer Biol. 2020, 60, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, M.; Xu, X.; Xuan, L.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334–342. [Google Scholar] [CrossRef]
- Sowa, T.; Menju, T.; Chen-Yoshikawa, T.F.; Takahashi, K.; Nishikawa, S.; Nakanishi, T.; Shikuma, K.; Motoyama, H.; Hijiya, K.; Aoyama, A.; et al. Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med. 2016, 6, 288–297. [Google Scholar] [CrossRef]
- Calvani, M.; Comito, G.; Giannoni, E.; Chiarugi, P. Time-Dependent Stabilization of Hypoxia Inducible Factor-1α by Different Intracellular Sources of Reactive Oxygen Species. PLoS ONE 2012, 7, e38388. [Google Scholar] [CrossRef]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, D.H.; Landon, C.D.; Nien, C.Y.-C.; Hanna, G.; Rich, J.N.; Provenzale, J.M.; et al. Tumor Cells Upregulate Normoxic HIF-1α in Response to Doxorubicin. Cancer Res. 2013, 73, 6230–6242. [Google Scholar] [CrossRef]
- Zolotoff, C.; Voirin, A.-C.; Puech, C.; Roche, F.; Perek, N. Intermittent Hypoxia and Its Impact on Nrf2/HIF-1α Expression and ABC Transporters: An in Vitro Human Blood-Brain Barrier Model Study. Cell. Physiol. Biochem. 2020, 54, 1231–1248. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. OncoTargets Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Robles, R.J.; Mukherjee, S.; Zhang, H.; Feldbrügge, L.; Csizmadia, E.; Wu, Y.; Enjyoji, K.; Moss, A.C.; Otterbein, L.E.; et al. HIF-1α-induced xenobiotic transporters promote Th17 responses in Crohn’s disease. J. Autoimmun. 2018, 94, 122–133. [Google Scholar] [CrossRef]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 2016, 17, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Xu, J.; Pabbisetty, S.K.; Alonso, M.; Liu, D.; Lee, O.-H.; Gumin, J.; Bhat, K.P.; Colman, H.; Lang, F.F.; et al. Tie2-mediated multidrug resistance in malignant gliomas is associated with upregulation of ABC transporters. Oncogene 2009, 28, 2358–2363. [Google Scholar] [CrossRef]
- Seebacher, N.; Lane, D.; Richardson, D.; Jansson, P. Turning the gun on cancer: Utilizing lysosomal P-glycoprotein as a new strategy to overcome multi-drug resistance. Free Radic. Biol. Med. 2016, 96, 432–445. [Google Scholar] [CrossRef]
- Altan, N.; Chen, Y.; Schindler, M.; Simon, S.M. Defective Acidification in Human Breast Tumor Cells and Implications for Chemotherapy. J. Exp. Med. 1998, 187, 1583–1598. [Google Scholar] [CrossRef]
- Krchniakova, M.; Skoda, J.; Neradil, J.; Chlapek, P.; Veselska, R. Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. Int. J. Mol. Sci. 2020, 21, 3157. [Google Scholar] [CrossRef]
- Bruning, U.; Cerone, L.; Neufeld, Z.; Fitzpatrick, S.F.; Cheong, A.; Scholz, C.C.; Simpson, D.A.; Leonard, M.O.; Tambuwala, M.M.; Cummins, E.P.; et al. MicroRNA-155 Promotes Resolution of Hypoxia-Inducible Factor 1 Activity during Prolonged Hypoxia. Mol. Cell. Biol. 2011, 31, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Cascio, S.; D’Andrea, A.; Ferla, R.; Surmacz, E.; Gulotta, E.; Amodeo, V.; Bazan, V.; Gebbia, N.; Russo, A. miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J. Cell. Physiol. 2010, 224, 242–249. [Google Scholar] [CrossRef]
- Ding, G.; Huang, G.; Liu, H.-D.; Liang, H.-X.; Ni, Y.-F.; Ding, Z.-H.; Ni, G.-Y.; Hua, H.-W. MiR-199a suppresses the hypoxia-induced proliferation of non-small cell lung cancer cells through targeting HIF1α. Mol. Cell. Biochem. 2013, 384, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Subramanian, I.V.; Adhikari, N.; Zhang, X.; Joshi, H.P.; Basi, D.; Chandrashekhar, Y.; Hall, J.L.; Roy, S.; Zeng, Y.; et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Investig. 2010, 120, 4141–4154. [Google Scholar] [CrossRef]
- Castells, M.; Thibault, B.; Delord, J.-P.; Couderc, B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; Rachida, S.B.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter Via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hänze, J. HIF-1α signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 2010, 48, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Naranjo-Suarez, S.; Carlson, B.A.; Tsuji, P.A.; Yoo, M.-H.; Gladyshev, V.N.; Hatfield, D.L. HIF-Independent Regulation of Thioredoxin Reductase 1 Contributes to the High Levels of Reactive Oxygen Species Induced by Hypoxia. PLoS ONE 2012, 7, e30470. [Google Scholar] [CrossRef]
- Jeddi, F.; Soozangar, N.; Sadeghi, M.R.; Somi, M.H.; Shirmohamadi, M.; Eftekhar-Sadat, A.-T.; Samadi, N. Nrf2 overexpression is associated with P-glycoprotein upregulation in gastric cancer. Biomed. Pharmacother. 2018, 97, 286–292. [Google Scholar] [CrossRef]
- Ji, L.; Li, H.; Gao, P.; Shang, G.; Zhang, D.D.; Zhang, N.; Jiang, T. Nrf2 Pathway Regulates Multidrug-Resistance-Associated Protein 1 in Small Cell Lung Cancer. PLoS ONE 2013, 8, e63404. [Google Scholar] [CrossRef]
- Bao, L.; Wu, J.; Dodson, M.; De La Vega, E.M.R.; Ning, Y.; Zhang, Z.; Yao, M.; Zhang, N.D.; Xu, C.; Yi, X. ABCF2, an Nrf2 target gene, contributes to cisplatin resistance in ovarian cancer cells. Mol. Carcinog. 2017, 56, 1543–1553. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Höpken, U.E.; Rehm, A. Targeting the Tumor Microenvironment of Leukemia and Lymphoma. Trends Cancer 2019, 5, 351–364. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Martinez-Outschoorn, U.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.K.; Lin, Z.; Balliet, R.M.; Howell, A.; et al. Understanding the "lethal" drivers of tumor-stroma co-evolution: Emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol. Ther. 2010, 10, 537–542. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Kanugula, S.; Sudhir, S.; Pereira, M.; Jain, S.; Aghi, M. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.; Santner, S.; Carolin, K.A.; Tait, L. Direct Involvement of Breast Tumor Fibroblasts in the Modulation of Tamoxifen Sensitivity. Am. J. Pathol. 2007, 170, 1546–1560. [Google Scholar] [CrossRef]
- Pontiggia, O.; Sampayo, R.; Raffo, D.; Motter, A.; Xu, R.; Bissell, M.J.; Joffé, E.B.D.K.; Simian, M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res. Treat. 2011, 133, 459–471. [Google Scholar] [CrossRef]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 1–22. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer Associated Fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef]
- Nagasaki, T.; Hara, M.R.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Lin, Z.; Pestell, R.G.; Howell, A.; Zimmers, T.; Casimiro, M.C.; Aquila, S.; Ando’, S.; Martinez-Outschoorn, U.; et al. Mitochondrial Fission Induces Glycolytic Reprogramming in Cancer-Associated Myofibroblasts, Driving Stromal Lactate Production, and Early Tumor Growth. Oncotarget 2012, 3, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Wu, X.; Li, W.; Su, P.; Cheng, H.; Xiang, L.; Gao, P.; Zhou, G. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/β-catenin pathway. Cancer Lett. 2012, 323, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Chau, W.K.; Ip, C.K.; Mak, A.S.C.; Lai, H.-C.; Wong, A.S.T. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin–ATP-binding cassette G2 signaling. Oncogene 2012, 32, 2767–2781. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Hemann, M.T. DNA Damage-Mediated Induction of a Chemoresistant Niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Goto, H.; Saijo, A.; Trung, V.T.; Aono, Y.; Ogino, H.; Kuramoto, T.; Tabata, S.; Uehara, H.; Izumi, K.; et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 2015, 6, 8792. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Yoshida, T.; Ishii, G.; Goto, K.; Neri, S.; Hashimoto, H.; Yoh, K.; Niho, S.; Umemura, S.; Matsumoto, S.; Ohmatsu, H.; et al. Podoplanin-Positive Cancer-Associated Fibroblasts in the Tumor Microenvironment Induce Primary Resistance to EGFR-TKIs in Lung Adenocarcinoma with EGFR Mutation. Clin. Cancer Res. 2015, 21, 642–651. [Google Scholar] [CrossRef]
- Mueller, K.L.; Madden, J.M.; Zoratti, G.L.; Kuperwasser, C.; List, K.; Boerner, J.L. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012, 14, R104. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments with High Integrin β1/FAK Signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Yui, Y.; Damiano, L.; Bainer, R.O.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M.; et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360–367. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Hazlehurst, L.; Damiano, J.S.; Buyuksal, I.; Pledger, W.J.; Dalton, W.S. Adhesion to fibronectin via β1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 2000, 19, 4319–4327. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; Pardo, E.M.D.J.; Kumar, S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef]
- Munson, J.M.; Bellamkonda, R.V.; Swartz, M.A. Interstitial Flow in a 3D Microenvironment Increases Glioma Invasion by a CXCR4-Dependent Mechanism. Cancer Res. 2013, 73, 1536–1546. [Google Scholar] [CrossRef]
- Alonso, S.; Su, M.; Jones, J.; Ganguly, S.; Kane, M.A.; Jones, R.J.; Ghiaur, G. Human bone marrow niche chemoprotection mediated by cytochrome p450 enzymes. Oncotarget 2015, 6, 14905–14912. [Google Scholar] [CrossRef]
- Alonso, S.; Hernandez, D.; Chang, Y.-T.; Gocke, C.D.; McCray, M.; Varadhan, R.; Matsui, W.H.; Jones, R.J.; Ghiaur, G. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. J. Clin. Investig. 2016, 126, 4460–4468. [Google Scholar] [CrossRef] [PubMed]
- Hirth, J.; Watkins, P.B.; Strawderman, M.; Schott, A.; Bruno, R.; Baker, L.H. The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clin. Cancer Res. 2000, 6, 1255–1258. [Google Scholar]
- Yeung, C.L.A.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Smolarczyk, R.; Czapla, J.; Jarosz-Biej, M.; Czerwinski, K.; Cichoń, T. Vascular disrupting agents in cancer therapy. Eur. J. Pharmacol. 2021, 891, 173692. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Wang, J.; Lv, X.-J.; Wang, Q.; Qiu, L.-X.; Lin, X.-Q.; Yu, L.-K.; Song, Y. Prognostic Value of Vascular Endothelial Growth Factor Expression in Patients with Lung Cancer: A Systematic Review with Meta-Analysis. J. Thorac. Oncol. 2009, 4, 1094–1103. [Google Scholar] [CrossRef]
- Guetz, G.D.; Uzzan, B.; Nicolas, P.; Cucherat, M.; Morere, J.-F.; Benamouzig, R.; Breau, J.-L.; Perret, G.-Y. Microvessel density and VEGF expression are prognostic factors in colorectal cancer. Meta-analysis of the literature. Br. J. Cancer 2006, 94, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Lobitz, S.; Daskalow, K.; Jöns, T.; Vieth, M.; Schlag, P.M.; Kemmner, W.; Wiedenmann, B.; Cramer, T.; Höcker, M. HIF-1α determines the metastatic potential of gastric cancer cells. Br. J. Cancer 2009, 100, 772–781. [Google Scholar] [CrossRef]
- Muz, B.; De La Puente, P.; Azab, F.; Luderer, M.; Azab, A.K. The Role of Hypoxia and Exploitation of the Hypoxic Environment in Hematologic Malignancies. Mol. Cancer Res. 2014, 12, 1347–1354. [Google Scholar] [CrossRef]
- Abramsson, A.; Lindblom, P.; Betsholtz, C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Investig. 2003, 112, 1142–1151. [Google Scholar] [CrossRef]
- Helmlinger, G.; Netti, P.; Lichtenbeld, H.C.; Melder, R.J.; Jain, R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997, 15, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 911–916. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.; Chauhan, V.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef]
- Martin, J.; Seano, G.; Jain, R.K. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef]
- Naito, H.; Wakabayashi, T.; Kidoya, H.; Muramatsu, F.; Takara, K.; Eino, D.; Yamane, K.; Iba, T.; Takakura, N. Endothelial Side Population Cells Contribute to Tumor Angiogenesis and Antiangiogenic Drug Resistance. Cancer Res. 2016, 76, 3200–3210. [Google Scholar] [CrossRef]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor Endothelial Cells Acquire Drug Resistance by MDR1 Up-Regulation via VEGF Signaling in Tumor Microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Akiyama, K.; Ohmura-Kakutani, H.; Torii, C.; Ohga, N.; Osawa, T.; Kikuchi, H.; Morimoto, H.; Morimoto, M.; et al. Tumor endothelial cells with high aldehyde dehydrogenase activity show drug resistance. Cancer Sci. 2017, 108, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Mountzios, G.; Pentheroudakis, G.; Carmeliet, P. Bevacizumab and micrometastases: Revisiting the preclinical and clinical rollercoaster. Pharmacol. Ther. 2014, 141, 117–124. [Google Scholar] [CrossRef]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-Mediated Down-Regulation of Bid and Bax in Tumors Occurs via Hypoxia-Inducible Factor 1-Dependent and -Independent Mechanisms and Contributes to Drug Resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef]
- Dancescu, M.; Rubio-Trujillo, M.; Biron, G.; Bron, D.; Delespesse, G.; Sarfati, M. Interleukin 4 protects chronic lymphocytic leukemic B cells from death by apoptosis and upregulates Bcl-2 expression. J. Exp. Med. 1992, 176, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Jewell, A.P.; Worman, C.P.; Lydyard, P.M.; Yong, K.L.; Giles, F.J.; Goldstone, A.H. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br. J. Haematol. 1994, 88, 268–274. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Menzel, T.; Lynen, S.; Wrazel, L.; Rosén, A.; Al-Katib, A.; Raveche, E.; Gabrilove, J. Basic fibroblast growth factor (bFGF) upregulates the expression of bcl-2 in B cell chronic lymphocytic leukemia cell lines resulting in delaying apoptosis. J. Leuk. 1997, 11, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes Impair Leukemia Treatment in Mice. Cancer Res. 2009, 69, 7867–7874. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Chan, E.Y.W.; Kir, S.; Tooze, S. siRNA Screening of the Kinome Identifies ULK1 as a Multidomain Modulator of Autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 1–10. [Google Scholar] [CrossRef]
- Sun, W.L.; Lan, D.; Gan, T.Q.; Cai, Z.W. Autophagy facilitates multidrug resistance development through inhibition of apoptosis in breast cancer cells. Neoplasma 2015, 62, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zeng, P.; Kang, R.; Yu, Y.; Yang, L.; Tang, D.; Cao, L. S100A8 Contributes to Drug Resistance by Promoting Autophagy in Leukemia Cells. PLoS ONE 2014, 9, e97242. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, J.; Shen, C.; Luo, Y.; Xia, L.; Xue, F.; Xia, Q. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem. Biophys. Res. Commun. 2012, 423, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem. Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Skoda, J.; Veselska, R. Cancer stem cells in sarcomas: Getting to the stemness core. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Borankova, K.; Jansson, P.; Huang, M.L.; Veselska, R.; Richardson, D.R. Pharmacological targeting of mitochondria in cancer stem cells: An ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019, 139, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, H.L.; Laterra, J. The cancer stem cell phenotype: You can’t win until you learn how to lose it. Mol. Cell. Oncol. 2015, 2, e989760. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, T.S.R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Hitomi, M.; Chumakova, A.P.; Silver, D.J.; Knudsen, A.M.; Pontius, W.D.; Murphy, S.; Anand, N.; Kristensen, B.W.; Lathia, J.D. Asymmetric cell division promotes therapeutic resistance in glioblastoma stem cells. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, Y.; Wang, X.; Zhao, H.; Ji, Z.; Cheng, C.; Li, L.; Fang, Y.; Xu, D.; Zhu, H.H.; et al. WNT/β-Catenin Directs Self-Renewal Symmetric Cell Division of hTERThigh Prostate Cancer Stem Cells. Cancer Res. 2017, 77, 2534–2547. [Google Scholar] [CrossRef]
- Lukaszewicz, A.I.; Nguyen, C.; Melendez, E.; Lin, D.P.; Teo, J.-L.; Lai, K.K.Y.; Huttner, W.B.; Shi, S.-H.; Kahn, M. The Mode of Stem Cell Division Is Dependent on the Differential Interaction of β-Catenin with the Kat3 Coactivators CBP or p300. Cancers 2019, 11, 962. [Google Scholar] [CrossRef]
- Cho, I.J.; Lui, P.; Obajdin, J.; Riccio, F.; Stroukov, W.; Willis, T.; Spagnoli, F.; Watt, F.M. Mechanisms, Hallmarks, and Implications of Stem Cell Quiescence. Stem Cell Rep. 2019, 12, 1190–1200. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wan, W.-W.; Xiong, S.-L.; Feng, H.; Wu, N. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 2017, 3, 16105. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Nelson, A.; Lamba, D.A.; Reh, T.A.; Ware, C.; Ruohola-Baker, H. Hypoxia induces re-entry of committed cells into pluripotency. Stem Cells 2013, 31, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86.e6. [Google Scholar] [CrossRef] [PubMed]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; McCleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E.; et al. AMPK Activation by Metformin Promotes Survival of Dormant ER+Breast Cancer Cells. Clin. Cancer Res. 2020, 26, 3707–3719. [Google Scholar] [CrossRef]
- Seo, Y.; Kim, J.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. Metformin Suppresses Cancer Stem Cells through AMPK Activation and Inhibition of Protein Prenylation of the Mevalonate Pathway in Colorectal Cancer. Cancers 2020, 12, 2554. [Google Scholar] [CrossRef] [PubMed]
- Vara-Ciruelos, D.; Dandapani, M.; Hardie, D.G. AMP-Activated Protein Kinase: Friend or Foe in Cancer? Annu. Rev. Cancer Biol. 2020, 4, 1–16. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 1–15. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Francescangeli, F.; Contavalli, P.; De Angelis, M.L.; Careccia, S.; Signore, M.; Haas, T.L.; Salaris, F.; Baiocchi, M.; Boe, A.; Giuliani, A.; et al. A pre-existing population of ZEB2+ quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, M.J.; Topol, L.; Anderson, S.M.; Yang, Y.; Bodine, D.M. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc. Natl. Acad. Sci. USA 2007, 104, 15436–15441. [Google Scholar] [CrossRef]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nat. Cell Biol. 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.K.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Meyskens, F.L.; Omenn, G.S.; Valanis, B.; Williams, J.H. The Beta-Carotene and Retinol Efficacy Trial: Incidence of Lung Cancer and Cardiovascular Disease Mortality During 6-Year Follow-up After Stopping -Carotene and Retinol Supplements. J. Natl. Cancer Inst. 2004, 96, 1743–1750. [Google Scholar] [CrossRef]
- Rehmana, A.; Collis, C.S.; Yangb, M.; Kellyb, M.; Diplock, A.T.; Halliwella, B.; Rice-Evans, C. The Effects of Iron and Vitamin C Co-supplementation on Oxidative Damage to DNA in Healthy Volunteers. Biochem. Biophys. Res. Commun. 1998, 246, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.J.; Dachs, G.U.; Morrin, H.R.; Davey, V.C.; Robinson, B.A.; Vissers, M.C.M. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer 2019, 19, 307. [Google Scholar] [CrossRef]
- Li, Y.; Sen, A.; Ren, J.; Askew, L.M.; Sidahmed, E.; Brenner, D.E.; Ruffin, M.T.; Turgeon, D.K.; Djuric, Z. Effects of vitamin E from supplements and diet on colonic α- and γ-tocopherol concentrations in persons at increased colon cancer risk. Nutr. Cancer 2014, 67, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Goossens, M.; Buntinx, F.; Joniau, S.; Ackaert, K.; Ameye, F.; Billiet, I.; Braeckman, J.; Breugelmans, A.; Darras, J.; Dilen, K.; et al. Designing the selenium and bladder cancer trial (SELEBLAT), a phase lll randomized chemoprevention study with selenium on recurrence of bladder cancer in Belgium. BMC Urol. 2012, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Castro, N.P.; Rangel, M.C.; Merchant, A.S.; MacKinnon, G.M.; Cuttitta, F.; Salomon, D.S.; Kim, Y.S. Sulforaphane Suppresses the Growth of Triple-negative Breast Cancer Stem-like Cells In vitro and In vivo. Cancer Prev. Res. 2019, 12, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Bijangi-Vishehsaraei, K.; Saadatzadeh, M.R.; Wang, H.; Nguyen, A.; Kamocka, M.M.; Cai, W.; Cohen-Gadol, A.; Halum, S.L.; Sarkaria, J.N.; Pollok, K.E.; et al. Sulforaphane suppresses the growth of glioblastoma cells, glioblastoma stem cell–like spheroids, and tumor xenografts through multiple cell signaling pathways. J. Neurosurg. 2017, 127, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Lamas, D.J.M.; Cortina, J.; Ventura, C.; Sterle, H.; Valli, E.; Balestrasse, K.B.; Blanco, H.; Cremaschi, G.; Rivera, E.S.; Medina, V. Enhancement of ionizing radiation response by histamine in vitro and in vivo in human breast cancer. Cancer Biol. Ther. 2014, 16, 137–148. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Redon, C.E.; Ferguson, N.F.; Kryston, T.B.; Parekh, P.; Dickey, J.S.; Nakamura, A.J.; Mitchell, J.B.; Bonner, W.M.; Martin, O.A. Systemic DNA damage accumulation under in vivo tumor growth can be inhibited by the antioxidant Tempol. Cancer Lett. 2014, 353, 248–257. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.S.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, E.H.; Mahmoud, H.T.; Al-Hudhud, M.Y.; Abdalla, M.Y.; Ahmad, I.M.; Yasin, S.R.; Elkarmi, A.Z.; Tahtamouni, L.H. 2-deoxy-d-Glucose Synergizes with Doxorubicin or l-Buthionine Sulfoximine to Reduce Adhesion and Migration of Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2015, 16, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Williams, R.R.; Birmingham, A.; Newman, D.J.; Kirkpatrick, D.L.; Powis, G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol. Cancer Ther. 2003, 2, 235–243. [Google Scholar]
- Evens, A.M.; Lecane, P.; Magda, D.; Prachand, S.; Singhal, S.; Nelson, J.; Miller, R.A.; Gartenhaus, R.B.; Gordon, L.I. Motexafin gadolinium generates reactive oxygen species and induces apoptosis in sensitive and highly resistant multiple myeloma cells. Blood 2005, 105, 1265–1273. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, Y.; Cheng, Y.-F.; Li, W.-J.; Zhang, Z.; Chen, S.-Y. Involvement of reactive oxygen species in 2-methoxyestradiol-induced apoptosis in human neuroblastoma cells. Cancer Lett. 2011, 313, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non–small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef]
- Allensworth, J.L.; Evans, M.; Bertucci, F.; Aldrich, A.J.; Festa, R.A.; Finetti, P.; Ueno, N.T.; Safi, R.; McDonnell, D.P.; Thiele, D.J.; et al. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol. Oncol. 2015, 9, 1155–1168. [Google Scholar] [CrossRef]
- Barr, P.M.; Miller, T.P.; Friedberg, J.W.; Peterson, D.R.; Baran, A.; Herr, M.; Spier, C.M.; Cui, H.; Roe, D.J.; Persky, D.O.; et al. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2014, 124, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.; Glorieux, C.; Stockis, J.; Sid, B.; Sandoval, J.M.; Felipe, K.B.; Kviecinski, M.R.; Verrax, J.; Calderon, P.B. Retinoic acid synergizes ATO-mediated cytotoxicity by precluding Nrf2 activity in AML cells. Br. J. Cancer 2014, 111, 874–882. [Google Scholar] [CrossRef]
- Mizutani, H.; Tada-Oikawa, S.; Hiraku, Y.; Kojima, M.; Kawanishi, S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci. 2005, 76, 1439–1453. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Chen, T.; Wang, W.; Liu, Q.; Li, H.; Yi, J.; Wang, J. Mechanisms of verapamil-enhanced chemosensitivity of gallbladder cancer cells to platinum drugs: Glutathione reduction and MRP1 downregulation. Oncol. Rep. 2012, 29, 676–684. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.K.; Shouman, S.; El-Demerdash, E.; Elgendy, M.; Abdel-Naim, A.B. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem. Interact. 2014, 217, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, R.; Tian, X.; Zhang, Z.; Zhang, P.; Li, C.; Feng, Z. Synergistic anti-tumor activity of Nimotuzumab in combination with Trastuzumab in HER2-positive breast cancer. Biochem. Biophys. Res. Commun. 2017, 489, 523–527. [Google Scholar] [CrossRef]
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 2014, 129, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Puppo, M.; Rapisarda, A.; Uranchimeg, B.; Cao, L.; Burger, A.M.; Ziche, M.; Melillo, G. Aminoflavone, a Ligand of the Aryl Hydrocarbon Receptor, Inhibits HIF-1α Expression in an AhR-Independent Fashion. Cancer Res. 2010, 70, 6837–6848. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Campia, I.; Polimeni, M.; Pescarmona, G.P.; Ghigo, D.; Bosia, A. Digoxin and ouabain induce P-glycoprotein by activating calmodulin kinase II and hypoxia-inducible factor-1α in human colon cancer cells. Toxicol. Appl. Pharmacol. 2009, 240, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Park, S.R.; Rapisarda, A.; Fer, N.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; et al. Weekly EZN-2208 (PEGylated SN-38) in combination with bevacizumab in patients with refractory solid tumors. Investig. New Drugs 2014, 32, 340–346. [Google Scholar] [CrossRef]
- Okuno, T.; Kawai, K.; Hata, K.; Murono, K.; Emoto, S.; Kaneko, M.; Sasaki, K.; Nishikawa, T.; Tanaka, T.; Nozawa, H. SN-38 Acts as a Radiosensitizer for Colorectal Cancer by Inhibiting the Radiation-induced Up-regulation of HIF-1α. Anticancer Res. 2018, 38, 3323–3331. [Google Scholar] [CrossRef]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.-H.; Fruehauf, J.P. HIF Inactivation of p53 in Ovarian Cancer Can Be Reversed by Topotecan, Restoring Cisplatin and Paclitaxel Sensitivity. Mol. Cancer Res. 2019, 17, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Alshaker, H.; Wang, Q.; Kawano, Y.; Arafat, T.; Böhler, T.; Winkler, M.; Cooper, C.; Pchejetski, D. Everolimus (RAD001) sensitizes prostate cancer cells to docetaxel by down-regulation of HIF-1α and sphingosine kinase 1. Oncotarget 2016, 7, 80943–80956. [Google Scholar] [CrossRef]
- Kaneko, M.; Nozawa, H.; Hiyoshi, M.; Tada, N.; Murono, K.; Nirei, T.; Emoto, S.; Kishikawa, J.; Iida, Y.; Sunami, E.; et al. Temsirolimus and chloroquine cooperatively exhibit a potent antitumor effect against colorectal cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Q.; Ma, S.; Yang, F.; Gong, Y.; Ke, C. Sirolimus Inhibits Human Pancreatic Carcinoma Cell Proliferation by a Mechanism Linked to the Targeting of mTOR/HIF-1 Alpha/VEGF Signaling. IUBMB Life 2007, 59, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.G.; Leonel, C.; Maschio-Signorini, L.B.; Borin, T.F.; Gelaleti, G.B.; Jardim-Perassi, B.V.; Ferreira, L.C.; Sonehara, N.M.; Carvalho, L.G.; Hellmén, E.; et al. Evaluation of Angiogenesis Process after Metformin and LY294002 Treatment in Mammary Tumor. Anti-Cancer Agents Med. Chem. 2019, 19, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, G.; Zhu, H.; Dong, X.; Zhao, D.; Jiang, X.; Li, J.; Qiao, H.; Ni, S.; Sun, X. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett. 2014, 355, 96–105. [Google Scholar] [CrossRef]
- Han, J.-Y.; Oh, S.H.; Morgillo, F.; Myers, J.N.; Kim, E.; Hong, W.K.; Lee, H.-Y. Hypoxia-inducible Factor 1α and Antiangiogenic Activity of Farnesyltransferase Inhibitor SCH66336 in Human Aerodigestive Tract Cancer. J. Natl. Cancer Inst. 2005, 97, 1272–1286. [Google Scholar] [CrossRef]
- Alqawi, O.; Moghaddas, M.; Singh, G. Effects of geldanamycin on HIF-1α mediated angiogenesis and invasion in prostate cancer cells. Prostate Cancer Prostatic Dis. 2006, 9, 126–135. [Google Scholar] [CrossRef]
- Fang, J.; Xia, C.; Cao, Z.; Zheng, J.Z.; Reed, E.; Jiang, B.-H. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005, 19, 342–353. [Google Scholar] [CrossRef]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone Deacetylase Inhibitors Induce VHL and Ubiquitin-Independent Proteasomal Degradation of Hypoxia-Inducible Factor 1α. Mol. Cell. Biol. 2006, 26, 2019–2028. [Google Scholar] [CrossRef]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef]
- Lee, K.; Qian, D.Z.; Rey, S.; Wei, H.; Liu, J.O.; Semenza, G.L. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2353–2358. [Google Scholar] [CrossRef]
- Befani, C.D.; Vlachostergios, P.J.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.N.; Liakos, P. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 2012, 90, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zang, Y.; Zhao, F.; Li, Z.; Zhang, J.; Fang, L.; Li, M.; Xing, L.; Xu, Z.; Yu, J. Inhibition of HIF-1α by PX-478 suppresses tumor growth of esophageal squamous cell cancer in vitro and in vivo. Am. J. Cancer Res. 2017, 7, 1198–1212. [Google Scholar] [PubMed]
- Park, M.K.; Ji, J.; Haam, K.; Han, T.-H.; Lim, S.; Kang, M.-J.; Lim, S.S.; Ban, H.S. Licochalcone A inhibits hypoxia-inducible factor-1α accumulation by suppressing mitochondrial respiration in hypoxic cancer cells. Biomed. Pharmacother. 2021, 133, 111082. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin Binds to High-Mobility Group Box 1 Protein and Inhibits Its Cytokine Activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Du, S.-Y.; Son, M.; Diamond, B. HIF-1α is a negative regulator of interferon regulatory factors: Implications for interferon production by hypoxic monocytes. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wang, G.; Hiramoto, K.; Ma, N.; Yoshikawa, N.; Ohnishi, S.; Murata, M.; Kawanishi, S. Glycyrrhizin Attenuates Carcinogenesis by Inhibiting the Inflammatory Response in a Murine Model of Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 2609. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef]
- Weller, M.; Nabors, L.; Gorlia, T.; Leske, H.; Rushing, E.; Bady, P.; Hicking, C.; Perry, J.; Hong, Y.-K.; Roth, P.; et al. Cilengitide in newly diagnosed glioblastoma: Biomarker expression and outcome. Oncotarget 2016, 7, 15018–15032. [Google Scholar] [CrossRef]
- Wong, K.M.; Horton, K.J.; Coveler, A.L.; Hingorani, S.; Harris, W.P. Targeting the Tumor Stroma: The Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr. Oncol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Moro, C.F.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Juárez, P.; Mohammad, K.S.; Yin, J.J.; Fournier, P.; McKenna, R.C.; Davis, H.W.; Peng, X.H.; Niewolna, M.; Javelaud, D.; Chirgwin, J.M.; et al. Halofuginone Inhibits the Establishment and Progression of Melanoma Bone Metastases. Cancer Res. 2012, 72, 6247–6256. [Google Scholar] [CrossRef] [PubMed]
- Bramhall, S.R.; Hallissey, M.T.; Whiting, J.; Scholefield, J.; Tierney, G.; Stuart, R.C.; Hawkins, R.; McCulloch, P.; Maughan, T.; Brown, P.D.; et al. Marimastat as maintenance therapy for patients with advanced gastric cancer: A randomised trial. Br. J. Cancer 2002, 86, 1864–1870. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Musteanu, M.; Garciagarcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Muñoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar] [PubMed]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014, 345, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, R.-D.; Al-Batran, S.-E.; Hartmann, F.; Hartung, G.; Jäger, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Oncol. Res. Treat. 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Mantoni, T.S.; Lunardi, S.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Pancreatic Stellate Cells Radioprotect Pancreatic Cancer Cells through β1-Integrin Signaling. Cancer Res. 2011, 71, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Sivendran, S.; Liu, Z.; Portas, L.J.; Yu, M.; Hahn, N.; Sonpavde, G.; Oh, W.K.; Galsky, M.D. Treatment-related mortality with vascular endothelial growth factor receptor tyrosine kinase inhibitor therapy in patients with advanced solid tumors: A meta-analysis. Cancer Treat. Rev. 2012, 38, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M.; et al. FGFR2 Gene Amplification in Gastric Cancer Predicts Sensitivity to the Selective FGFR Inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J.; Lim, S.H.; Sun, J.-M.; Choi, Y.-L.; Kim, H.R.; Ahn, S.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Kim, J.H.; et al. Efficacy and safety of dovitinib in pretreated advanced squamous non-small cell lung cancer with FGFR1 amplification: A single-arm, phase II study. J. Thorac. Oncol. 2016, 11, S16. [Google Scholar] [CrossRef][Green Version]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Chaudary, N.; Pintilie, M.; Jelveh, S.; Lindsay, P.; Hill, R.P.; Milosevic, M. Plerixafor Improves Primary Tumor Response and Reduces Metastases in Cervical Cancer Treated with Radio-Chemotherapy. Clin. Cancer Res. 2016, 23, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Podar, K.; Gupta, D.; Tai, Y.T.; Li, S.; Weller, E.; Hideshima, T.; Lentzsch, S.; Davies, F.; Li, C.; et al. The vascular en-dothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584 inhibits growth and migration of multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2002, 62, 5019–5026. [Google Scholar] [PubMed]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, C.-T.; Ma, T.-T.; Li, Z.-Y.; Zhou, L.-N.; Mu, B.; Leng, F.; Shi, H.-S.; Li, Y.-O.; Wei, Y.-Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib Mediates Reversal of Myeloid-Derived Suppressor Cell Accumulation in Renal Cell Carcinoma Patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Malecek, M.-K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef]
- Ma, S.; Song, W.; Xu, Y.; Si, X.; Zhang, D.; Lv, S.; Yang, C.; Ma, L.; Tang, Z.; Chen, X. Neutralizing tumor-promoting inflammation with polypeptide-dexamethasone conjugate for microenvironment modulation and colorectal cancer therapy. Biomaterials 2020, 232, 119676. [Google Scholar] [CrossRef] [PubMed]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef]
- Chiron, M.; Bagley, R.G.; Pollard, J.; Mankoo, P.K.; Henry, C.; Vincent, L.; Geslin, C.; Baltes, N.; Bergstrom, D.A. Differential Antitumor Activity of Aflibercept and Bevacizumab in Patient-Derived Xenograft Models of Colorectal Cancer. Mol. Cancer Ther. 2014, 13, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Nakatsuji, M.; Seno, H.; Ishizu, S.; Akitake-Kawano, R.; Kanda, K.; Ueo, T.; Komekado, H.; Kawada, M.; Minami, M.; et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis 2011, 32, 1333–1339. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Gao, N.; Zhang, Z.; Chen, G.; Budhraja, A.; Ke, Z.-J.; Son, Y.; Wang, X.; Luo, J.; Shi, X. Quercetin Induces Tumor-Selective Apoptosis through Downregulation of Mcl-1 and Activation of Bax. Clin. Cancer Res. 2010, 16, 5679–5691. [Google Scholar] [CrossRef]
- Ko, Y.-M.; Wu, T.-Y.; Wu, Y.-C.; Chang, F.-R.; Guh, J.-Y.; Chuang, L.-Y. Annonacin induces cell cycle-dependent growth arrest and apoptosis in estrogen receptor-α-related pathways in MCF-7 cells. J. Ethnopharmacol. 2011, 137, 1283–1290. [Google Scholar] [CrossRef]
- Subbiah, V.; Brown, R.E.; Buryanek, J.; Trent, J.; Ashkenazi, A.; Herbst, R.; Kurzrock, R. Targeting the Apoptotic Pathway in Chondrosarcoma Using Recombinant Human Apo2L/TRAIL (Dulanermin), a Dual Proapoptotic Receptor (DR4/DR5) Agonist. Mol. Cancer Ther. 2012, 11, 2541–2546. [Google Scholar] [CrossRef]
- Leong, S.; Cohen, R.B.; Gustafson, D.L.; Langer, C.J.; Camidge, D.R.; Padavic, K.; Gore, L.; Smith, M.; Chow, L.Q.; Von Mehren, M.; et al. Mapatumumab, an Antibody Targeting TRAIL-R1, in Combination with Paclitaxel and Carboplatin in Patients with Advanced Solid Malignancies: Results of a Phase I and Pharmacokinetic Study. J. Clin. Oncol. 2009, 27, 4413–4421. [Google Scholar] [CrossRef]
- Herbst, R.S.; Kurzrock, R.; Hong, D.S.; Valdivieso, M.; Hsu, C.-P.; Goyal, L.; Juan, G.; Hwang, Y.C.; Wong, S.; Hill, J.S.; et al. A First-in-Human Study of Conatumumab in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2010, 16, 5883–5891. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Infante, J.R.; Waterhouse, D.; Wong, L.; Vickers, S.; Arrowsmith, E.; He, A.R.; Hart, L.; Trent, D.; Wade, J.; et al. Phase 2, multicenter, open-label study of tigatuzumab (CS-1008), a humanized monoclonal antibody targeting death receptor 5, in combination with gemcitabine in chemotherapy-naive patients with unresectable or metastatic pancreatic cancer. Cancer Med. 2013, 2, 925–932. [Google Scholar] [CrossRef]
- Wakelee, H.A.; Patnaik, A.; Sikic, B.I.; Mita, M.; Fox, N.L.; Miceli, R.; Ullrich, S.J.; Fisher, G.A.; Tolcher, A.W. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Ann. Oncol. 2010, 21, 376–381. [Google Scholar] [CrossRef]
- Perimenis, P.; Galaris, A.; Voulgari, A.; Prassa, M.; Pintzas, A. IAP antagonists Birinapant and AT-406 efficiently synergise with either TRAIL, BRAF, or BCL-2 inhibitors to sensitise BRAFV600E colorectal tumour cells to apoptosis. BMC Cancer 2016, 16, 624. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Mérino, D.; Khaw, S.L.; Glaser, S.P.; Anderson, D.J.; Belmont, L.D.; Wong, C.; Yue, P.; Robati, M.; Phipson, B.; Fairlie, W.D.; et al. Bcl-2, Bcl-xL, and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood 2012, 119, 5807–5816. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nat. Cell Biol. 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Pal, K.; Roy, S.; Parida, P.K.; Dutta, A.; Bardhan, S.; Das, S.; Jana, K.; Karmakar, P. Folic acid conjugated curcumin loaded biopolymeric gum acacia microsphere for triple negative breast cancer therapy in invitro and invivo model. Mater. Sci. Eng. C 2019, 95, 204–216. [Google Scholar] [CrossRef]
- Wong, H.; Gould, S.E.; Budha, N.; Darbonne, W.C.; Kadel, E.E.; La, H.; Alicke, B.; Halladay, J.S.; Erickson, R.; Portera, C.; et al. Learning and Confirming with Preclinical Studies: Modeling and Simulation in the Discovery of GDC-0917, an Inhibitor of Apoptosis Proteins Antagonist. Drug Metab. Dispos. 2013, 41, 2104–2113. [Google Scholar] [CrossRef]
- Runckel, K.; Barth, M.; Mavis, C.; Gu, J.J.; Hernandez-Ilizaliturri, F.J. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018, 2, 3516–3525. [Google Scholar] [CrossRef]
- Makin; Holt, S.V.; Brookes, K.E.; Dive, C. Down-regulation of XIAP by AEG35156 in paediatric tumour cells induces apoptosis and sensitises cells to cytotoxic agents. Oncol. Rep. 2011, 25, 1177–1181. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Chloroquine Inhibits Autophagy to Potentiate Antiestrogen Responsiveness in ER+ Breast Cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef]
- Donohue, E.; Thomas, A.; Maurer, N.; Manisali, I.; Zeisser-Labouebe, M.; Zisman, N.; Anderson, H.J.; Ng, S.S.W.; Webb, M.; Bally, M.; et al. The Autophagy Inhibitor Verteporfin Moderately Enhances the Antitumor Activity of Gemcitabine in a Pancreatic Ductal Adenocarcinoma Model. J. Cancer 2013, 4, 585–596. [Google Scholar] [CrossRef]
- Wang, Y.; Peng, R.-Q.; Li, D.-D.; Ding, Y.; Wu, X.-Q.; Zeng, Y.-X.; Zhu, X.-F.; Zhang, X.-S. Chloroquine enhances the cytotoxicity of topotecan by inhibiting autophagy in lung cancer cells. Chin. J. Cancer 2011, 30, 690–700. [Google Scholar] [CrossRef]
- Qin, A.-C.; Li, Y.; Zhou, L.-N.; Xing, C.-G.; Lu, X.-S. Dual PI3K-BRD4 Inhibitor SF1126 Inhibits Colorectal Cancer Cell Growth in Vitro and in Vivo. Cell. Physiol. Biochem. 2019, 52, 758–768. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kögel, D. The Pan-Bcl-2 Inhibitor (−)-Gossypol Triggers Autophagic Cell Death in Malignant Glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Ghielmini, M.; Schmitz, S.F.; Bürki, K.; Pichert, G.; Betticher, D.C.; Stupp, R.; Wernli, M.; Lohri, A.; Schmitter, D.; Bertoni, F.; et al. The effect of Rituximab on patients with follicular and mantle-cell lymphoma. Swiss Group for Clinical Cancer Re-search (SAKK). Ann. Oncol. 2000, 11 (Suppl. S1), 123–126. [Google Scholar] [CrossRef]
- Cashen, A.; Lopez, S.; Gao, F.; Calandra, G.; MacFarland, R.; Badel, K.; DiPersio, J. A Phase II Study of Plerixafor (AMD3100) plus G-CSF for Autologous Hematopoietic Progenitor Cell Mobilization in Patients with Hodgkin Lymphoma. Biol. Blood Marrow Transplant. 2008, 14, 1253–1261. [Google Scholar] [CrossRef][Green Version]
- Sakamuri, D.; Glitza, I.C.; Cuellar, S.L.B.; Subbiah, V.; Fu, S.; Tsimberidou, A.M.; Wheler, J.J.; Hong, D.S.; Naing, A.; Falchook, G.S.; et al. Phase I Dose-Escalation Study of Anti–CTLA-4 Antibody Ipilimumab and Lenalidomide in Patients with Advanced Cancers. Mol. Cancer Ther. 2017, 17, 671–676. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Gu, J.-W.; Rizzo, P.; Pannuti, A.; Golde, T.; Osborne, B.; Miele, L. Notch signals in the endothelium and cancer “stem-like” cells: Opportunities for cancer therapy. Vasc. Cell 2012, 4, 7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.; Yang, J.; Wang, J.; Kopeček, J. Amplification of CD20 Cross-Linking in Rituximab-Resistant B-Lymphoma Cells Enhances Apoptosis Induction by Drug-Free Macromolecular Therapeutics. ACS Nano 2018, 12, 3658–3670. [Google Scholar] [CrossRef]
- Su, Y.-J.; Chang, Y.-W.; Lin, W.-H.; Liang, C.-L.; Lee, J.-L. An aberrant nuclear localization of E-cadherin is a potent inhibitor of Wnt/β-catenin-elicited promotion of the cancer stem cell phenotype. Oncogenesis 2015, 4, e157. [Google Scholar] [CrossRef]
- Zheng, F.; Dang, J.; Zhang, H.; Xu, F.; Ba, D.; Zhang, B.; Cheng, F.; Chang, A.E.; Wicha, M.S.; Li, Q. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J. Immunother. 2018, 41, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Deng, H.; Yao, H.; Liu, Q.; Su, F.; Song, E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 2010, 29, 4194–4204. [Google Scholar] [CrossRef] [PubMed]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Fouladi, M.; Stewart, C.F.; Olson, J.; Wagner, L.M.; Onar-Thomas, A.; Kocak, M.; Packer, R.J.; Goldman, S.; Gururangan, S.; Gajjar, A.; et al. Phase I Trial of MK-0752 in Children with Refractory CNS Malignancies: A Pediatric Brain Tumor Consortium Study. J. Clin. Oncol. 2011, 29, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Asghari, F.; Khademi, R.; Ranjbar, F.E.; Malekshahi, Z.V.; Majidi, R.F. Application of Nanotechnology in Targeting of Cancer Stem Cells: A Review. Int. J. Stem Cells 2019, 12, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Krishan, S.; Richardson, D.R.; Sahni, S. Adenosine Monophosphate–Activated Kinase and Its Key Role in Catabolism: Structure, Regulation, Biological Activity, and Pharmacological Activation. Mol. Pharmacol. 2014, 87, 363–377. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef]
- Das, B.; Neilsen, B.K.; Fisher, K.W.; Gehring, D.; Hu, Y.; Volle, D.J.; Kim, H.S.; McCall, J.L.; Kelly, D.L.; MacMillan, J.B.; et al. A Functional Signature Ontology (FUSION) screen detects an AMPK inhibitor with selective toxicity toward human colon tumor cells. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]



| ROS Modulating Strategies | Compounds Involved in Cancer Clinical Trials |
|---|---|
| Antioxidant approach | |
| intake of antioxidants | vitamins A [231], C [232] [233] and E [234], selenium [235] |
| NADPH oxidase inhibition | histamine [238] |
| GSH induction | sulforaphane [236,237] |
| nitroxide compound manipulation | tempol [239] |
| Pro-oxidant approach | |
| ROS generation | arsenic trioxide [249], imexon [248], doxorubicin, daunorubicin [250], cisplatin, oxaliplatin [251], sunitinib [252], gefitinib, erlotinib [253], trastuzumab [254], bevacizumab [255] |
| GSH depletion | β-phenylethyl isotiocyanate [241], buthionine sulfoximine [242] |
| thioredoxin inhibition | PX-12 [243], motexafin gadolinium [244] |
| superoxide dismutase inhibition | 2-methoxyestradiol [245], ATN-224 [246], disulfiram [247] |
| Mechanism of Action | Compounds Involved in Cancer Clinical Trials |
|---|---|
| inhibition of HIF-1α mRNA expression | aminoflavone [257] |
| inhibition of HIF-1α protein synthesis | topotecan [261], irinotecan [260], EZN-2208 [259], temsirolimus [263], everolimus [262], sirolimus [264], LY294002 [265], digoxin [258], 2-methoxyestradiol [266] |
| inhibition of HIF-1α stabilisation | geldanamycins [268], SCH66336 [267], apigenin [269], romidepsin [270] |
| inhibition of HIF-1α dimerisation | acriflavine [271] |
| inhibition of HIF/DNA binding | doxorubicin, daunorubicin, epirubicin [272] |
| inhibition of HIF-1 transcriptional activity | bortezomib [273] |
| inhibition of HIF-1α at multiple levels | PX-478 [274], glycyrrhizin [277,278,279], licochalcone A [275] |
| HIF-1α degradation | vorinostat [276] |
| Apoptosis and Autophagy Targeting Approaches | Compounds Involved in Cancer Clinical Trials |
|---|---|
| Stimulating the pro-apoptotic molecules | |
| BAX activator | quercetin [309], annonacin [310] |
| BAX upregulation | thioridazine [205] |
| DR4 agonist | mapatumumab [312] |
| DR5 agonist | conatumumab [313], lexatumumab [315], tigatuzumab [314] |
| DR4/5 agonist | dulanermin [311] |
| Inhibiting the anti-apoptotic molecules | |
| Bcl-2 antagonist | ABT-737 [319], navitoclax [318], venetoclax [317], AT101 [205,330], curcumin [320], annonacin [310] |
| Bcl-2 downregulation | thioridazine [205] |
| IAP antagonist | AT-406, birinapant [316], GDC-0917 [321], LCL161 [322] |
| XIAP antagonist | curcumin [320] |
| XIAP antisense oligonucleotide | AEG35156 [323] |
| Inhibiting autophagy | |
| autophagosome formation inhibition | SF1126 [328], verteporfin [326] |
| targeting lysosomes | chloroquine [327], hydroxychloroquine [325] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seebacher, N.A.; Krchniakova, M.; Stacy, A.E.; Skoda, J.; Jansson, P.J. Tumour Microenvironment Stress Promotes the Development of Drug Resistance. Antioxidants 2021, 10, 1801. https://doi.org/10.3390/antiox10111801
Seebacher NA, Krchniakova M, Stacy AE, Skoda J, Jansson PJ. Tumour Microenvironment Stress Promotes the Development of Drug Resistance. Antioxidants. 2021; 10(11):1801. https://doi.org/10.3390/antiox10111801
Chicago/Turabian StyleSeebacher, Nicole A., Maria Krchniakova, Alexandra E. Stacy, Jan Skoda, and Patric J. Jansson. 2021. "Tumour Microenvironment Stress Promotes the Development of Drug Resistance" Antioxidants 10, no. 11: 1801. https://doi.org/10.3390/antiox10111801
APA StyleSeebacher, N. A., Krchniakova, M., Stacy, A. E., Skoda, J., & Jansson, P. J. (2021). Tumour Microenvironment Stress Promotes the Development of Drug Resistance. Antioxidants, 10(11), 1801. https://doi.org/10.3390/antiox10111801