
Inhibition of Membrane-Associated Catalase, Extracellular ROS/RNS Signaling and Aquaporin/H2O2-Mediated Intracellular Glutathione Depletion Cooperate during Apoptosis Induction in the Human Gastric Carcinoma Cell Line MKN-45

Abstract
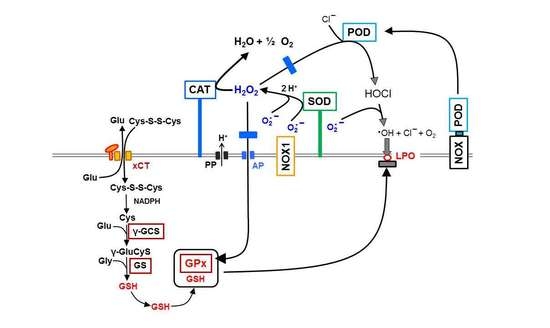
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells and Cell Culture
2.3. Quantification of Apoptosis Induction
2.4. Statistical Analysis
3. Results
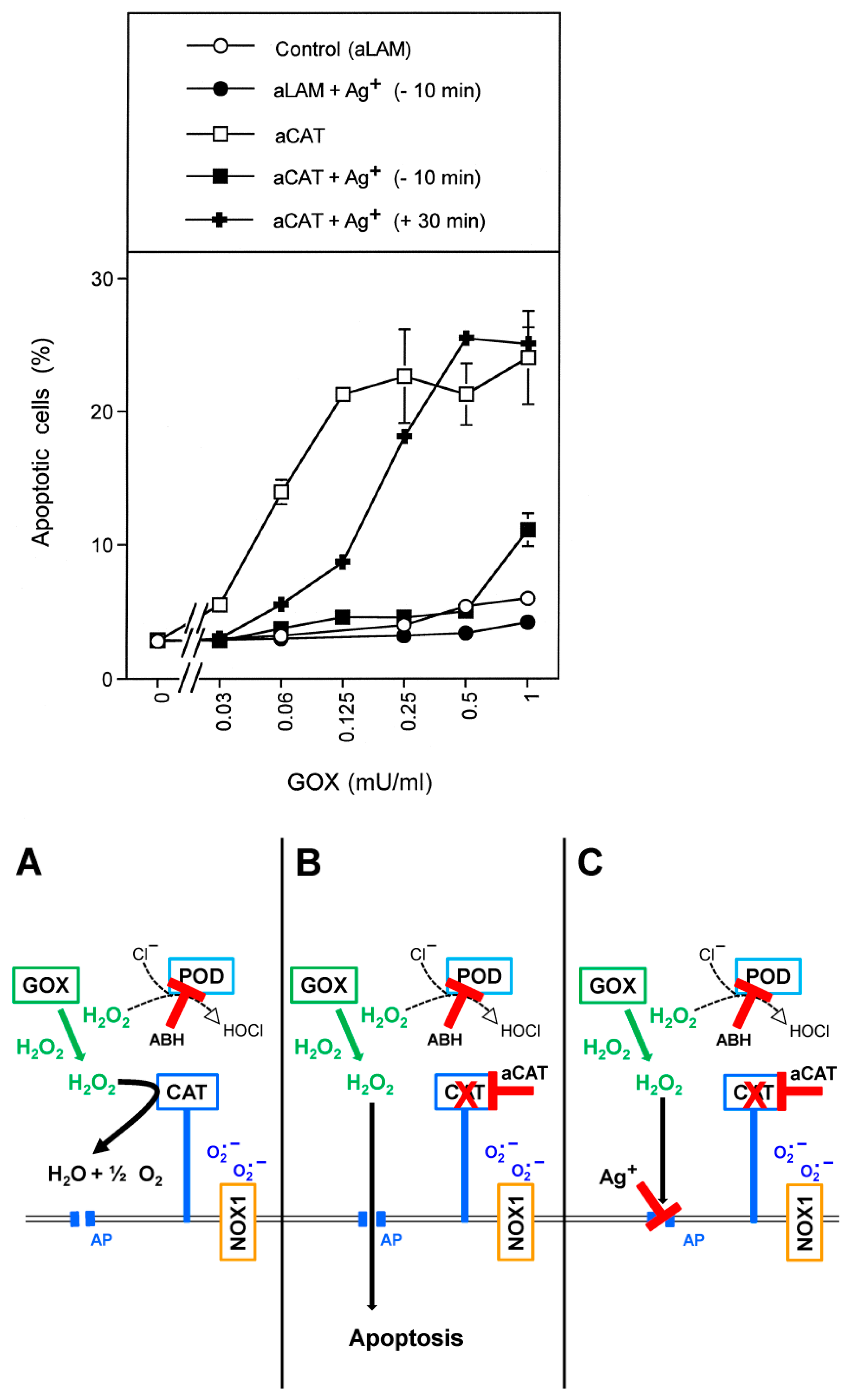
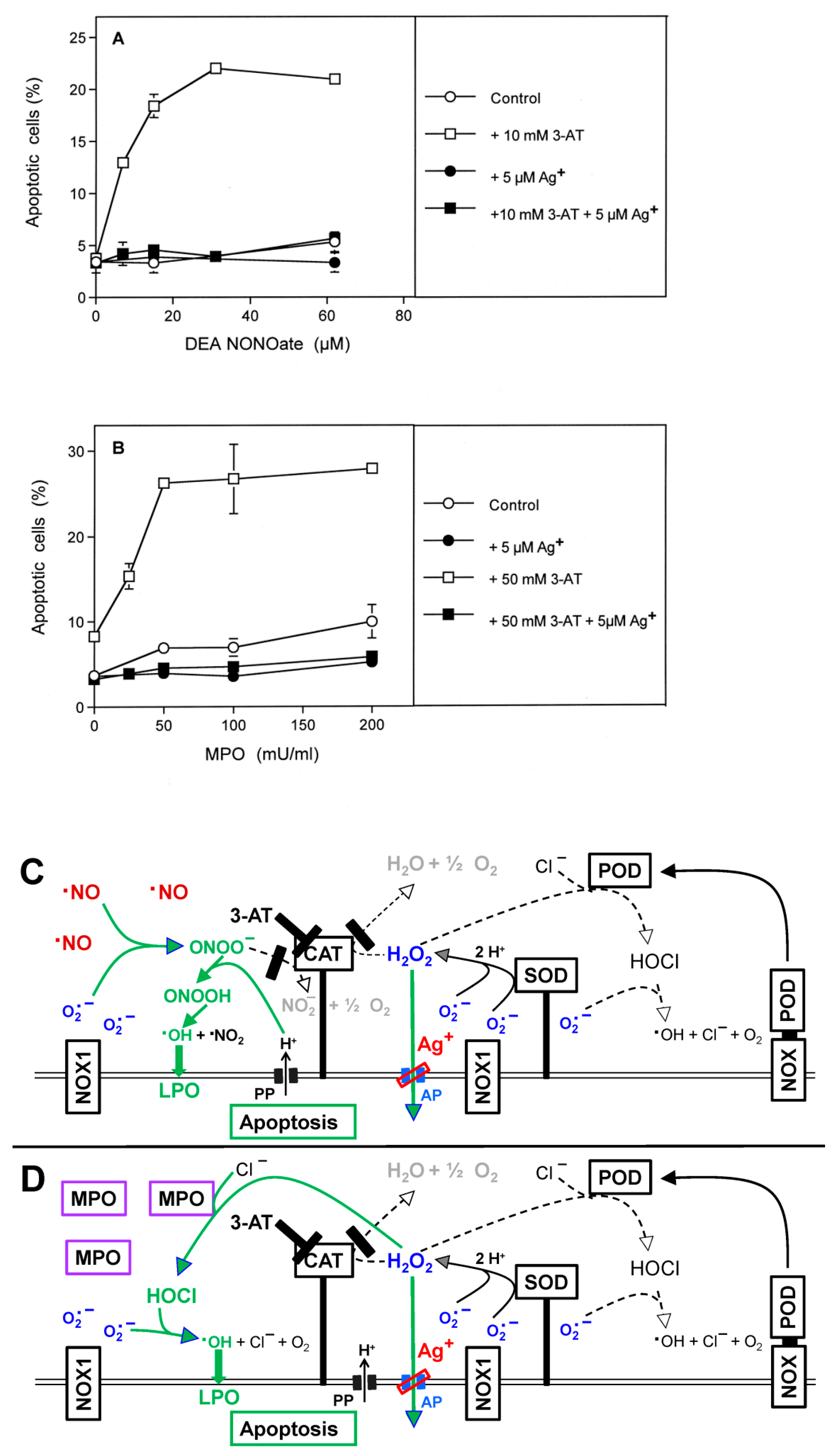
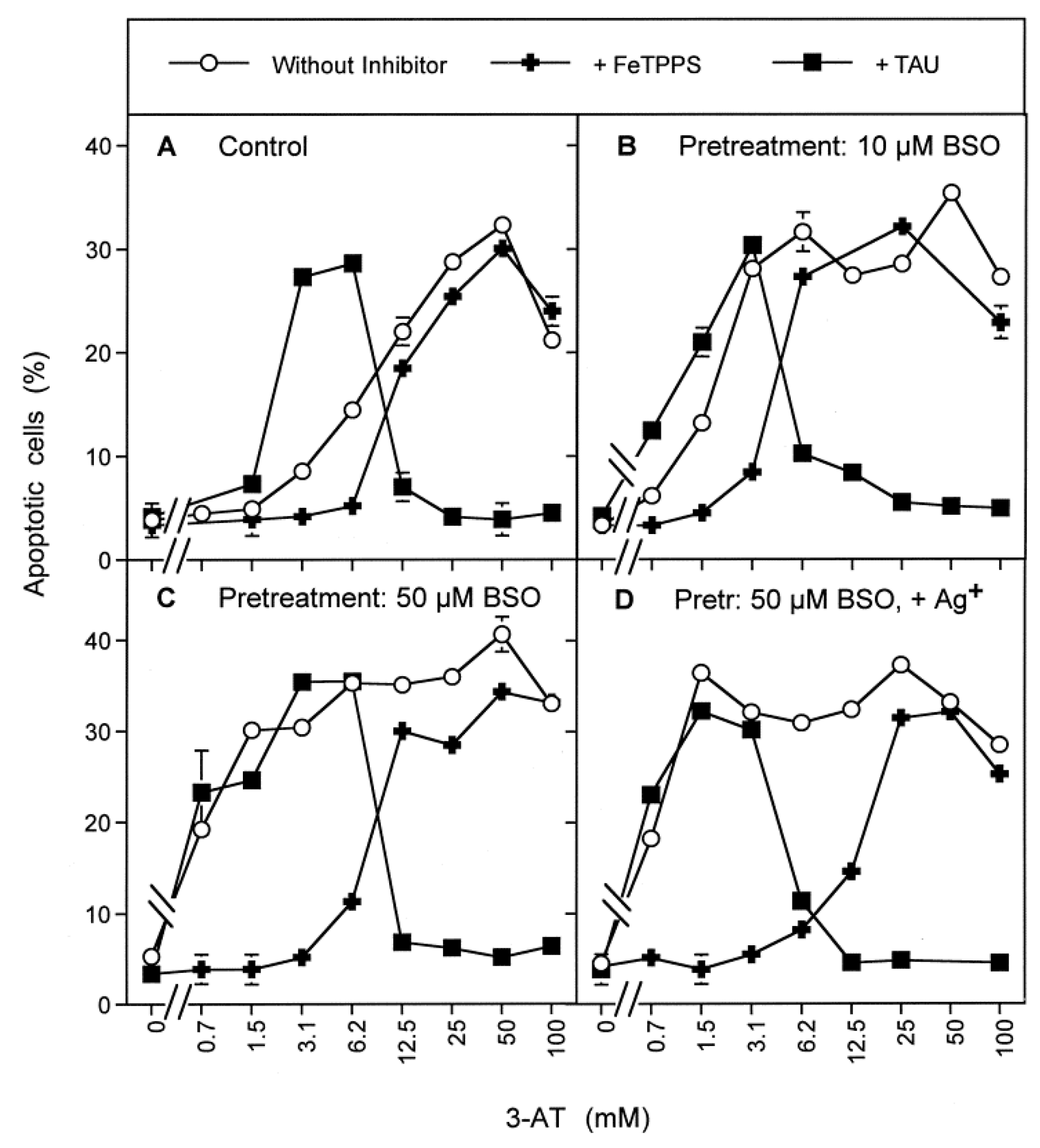
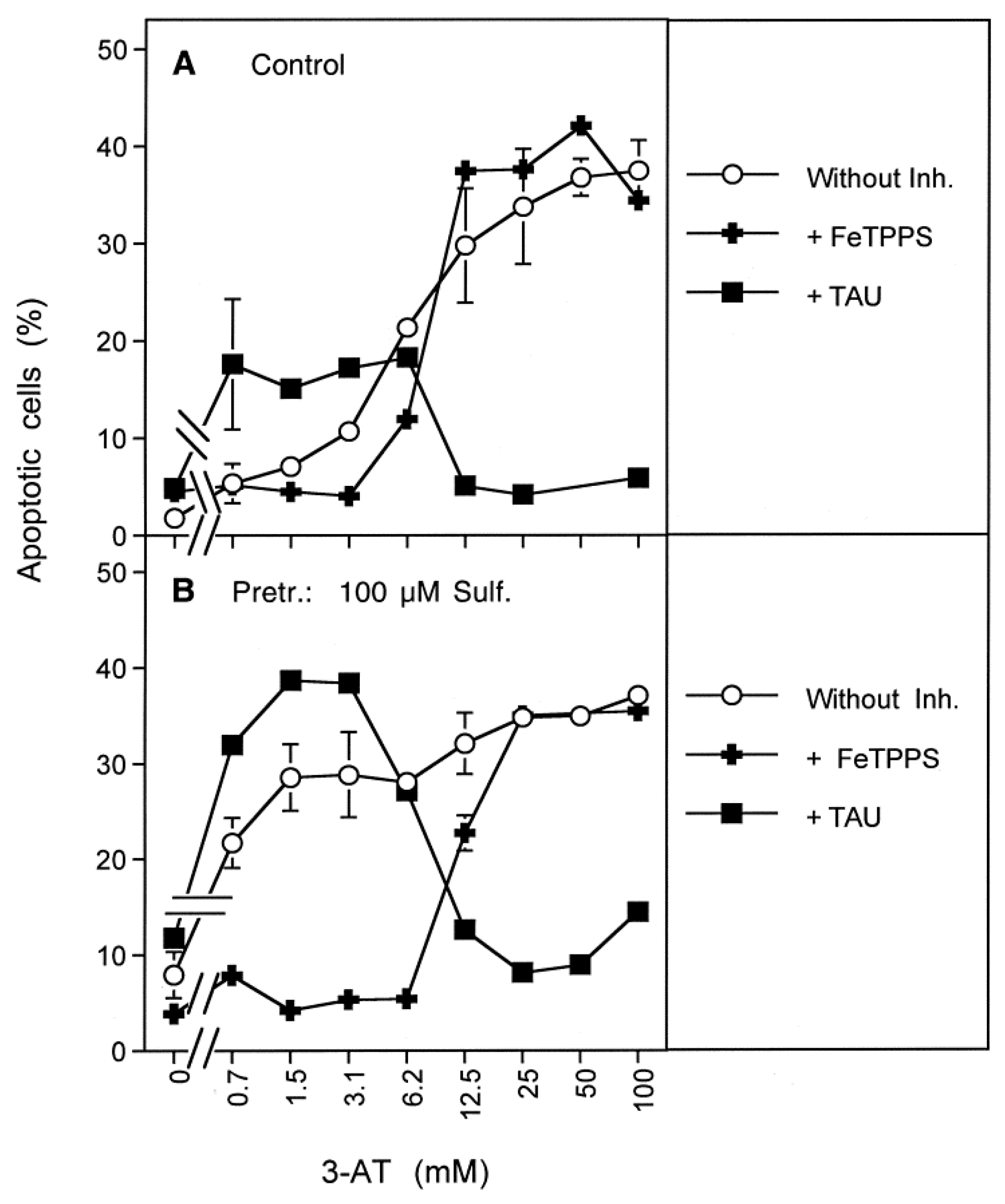
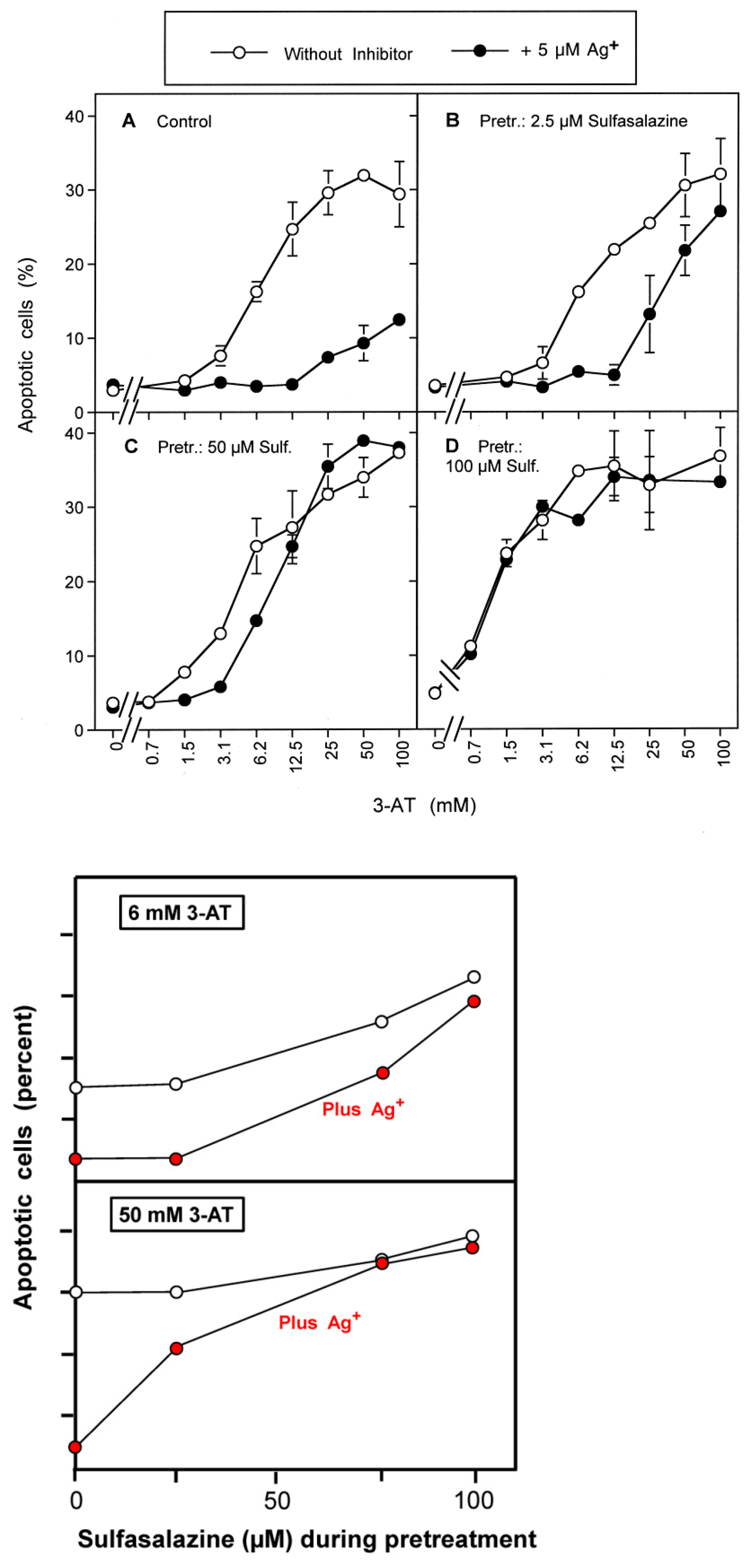
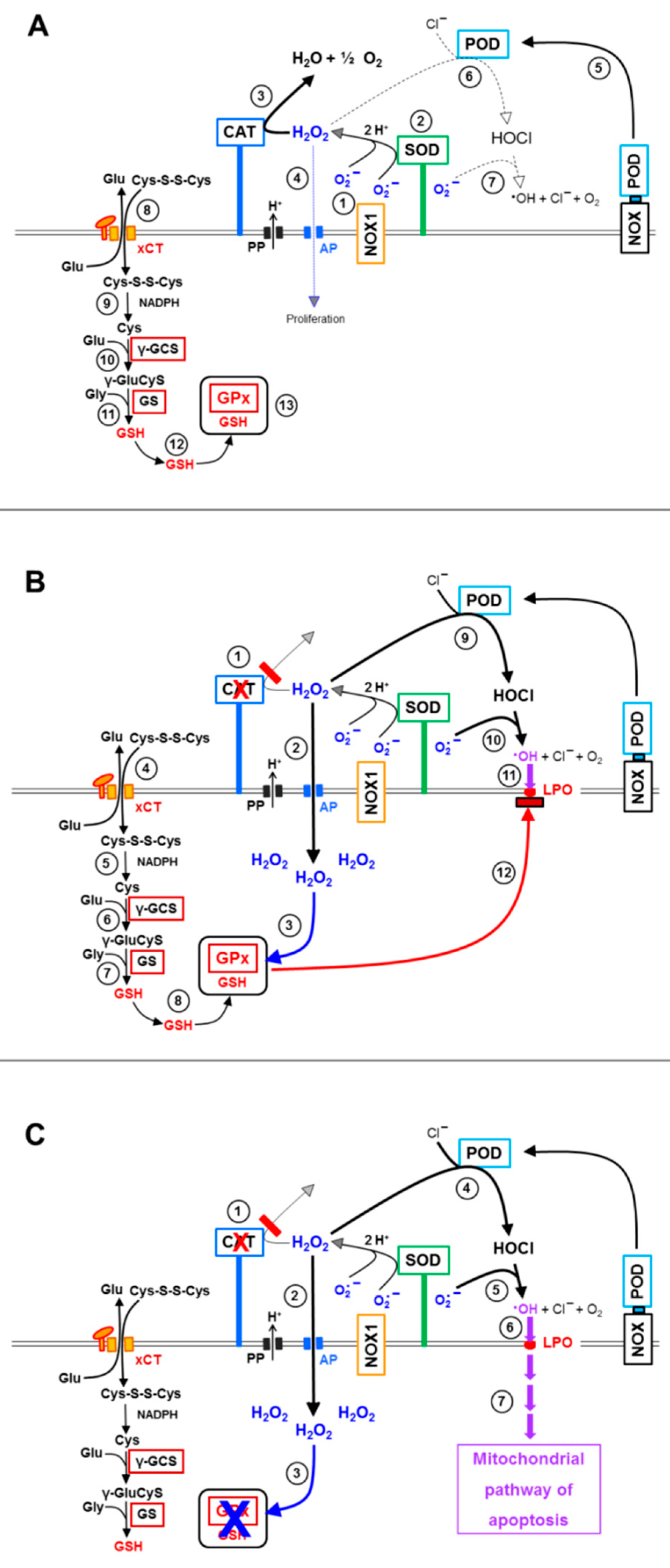
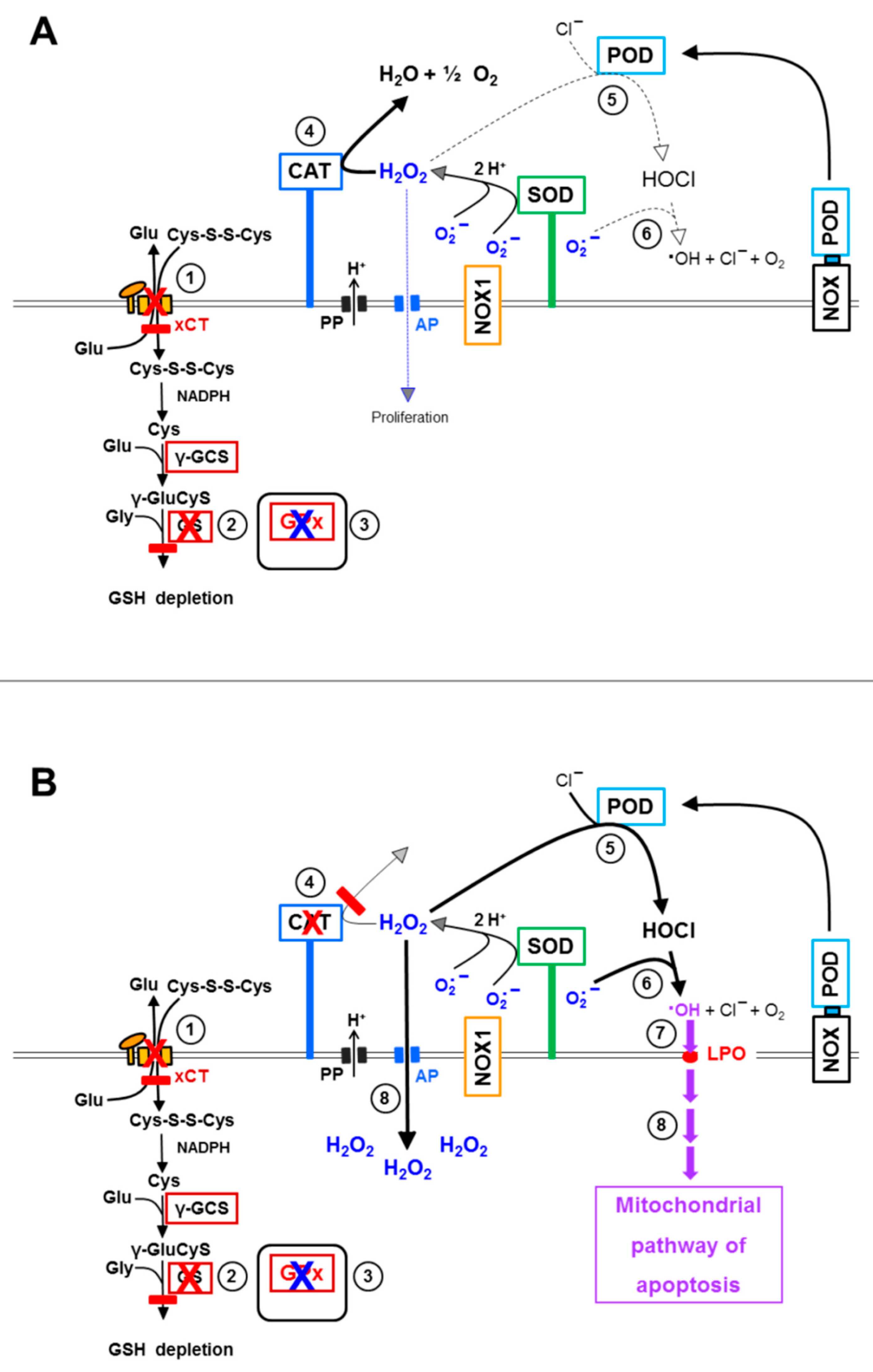
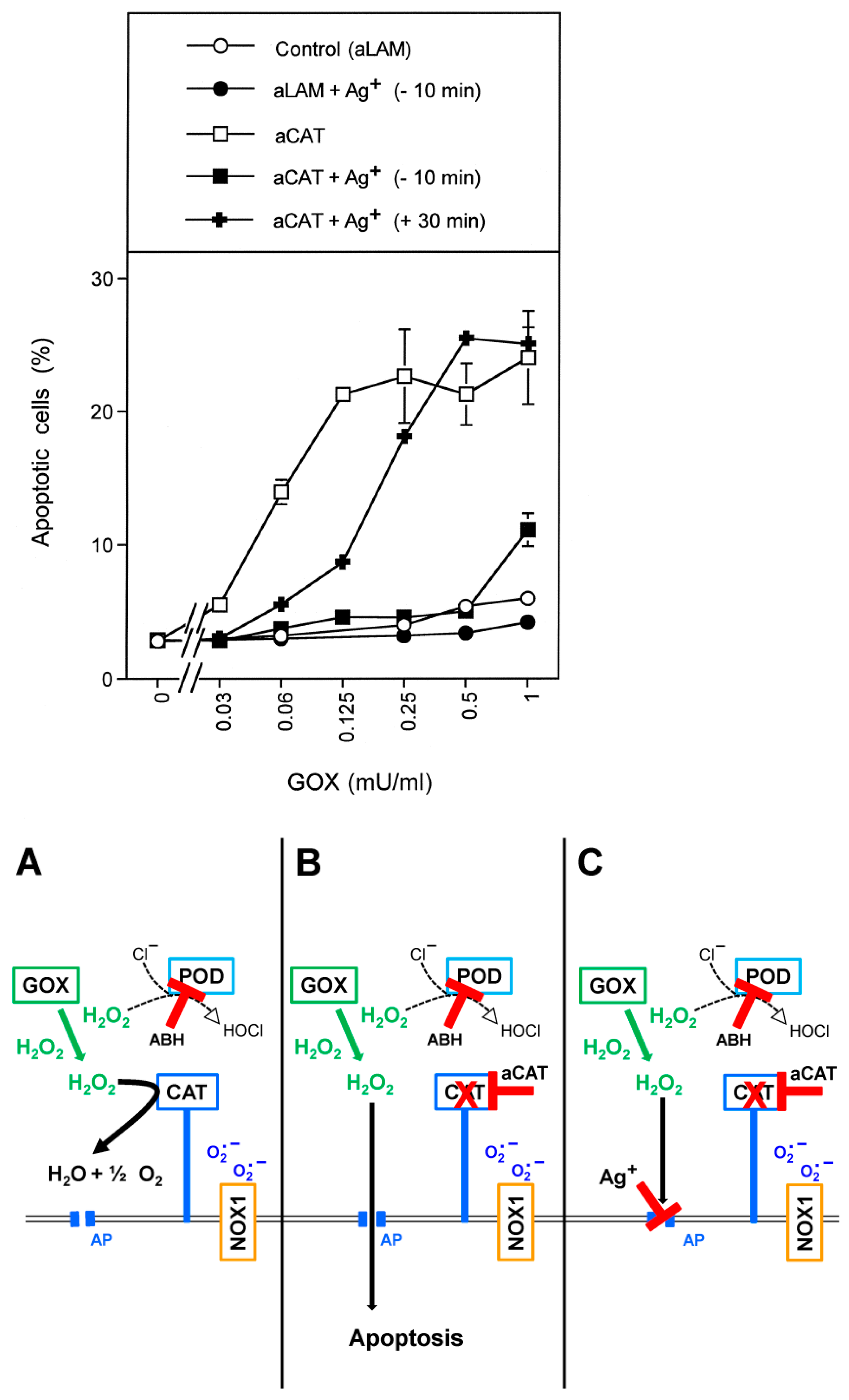
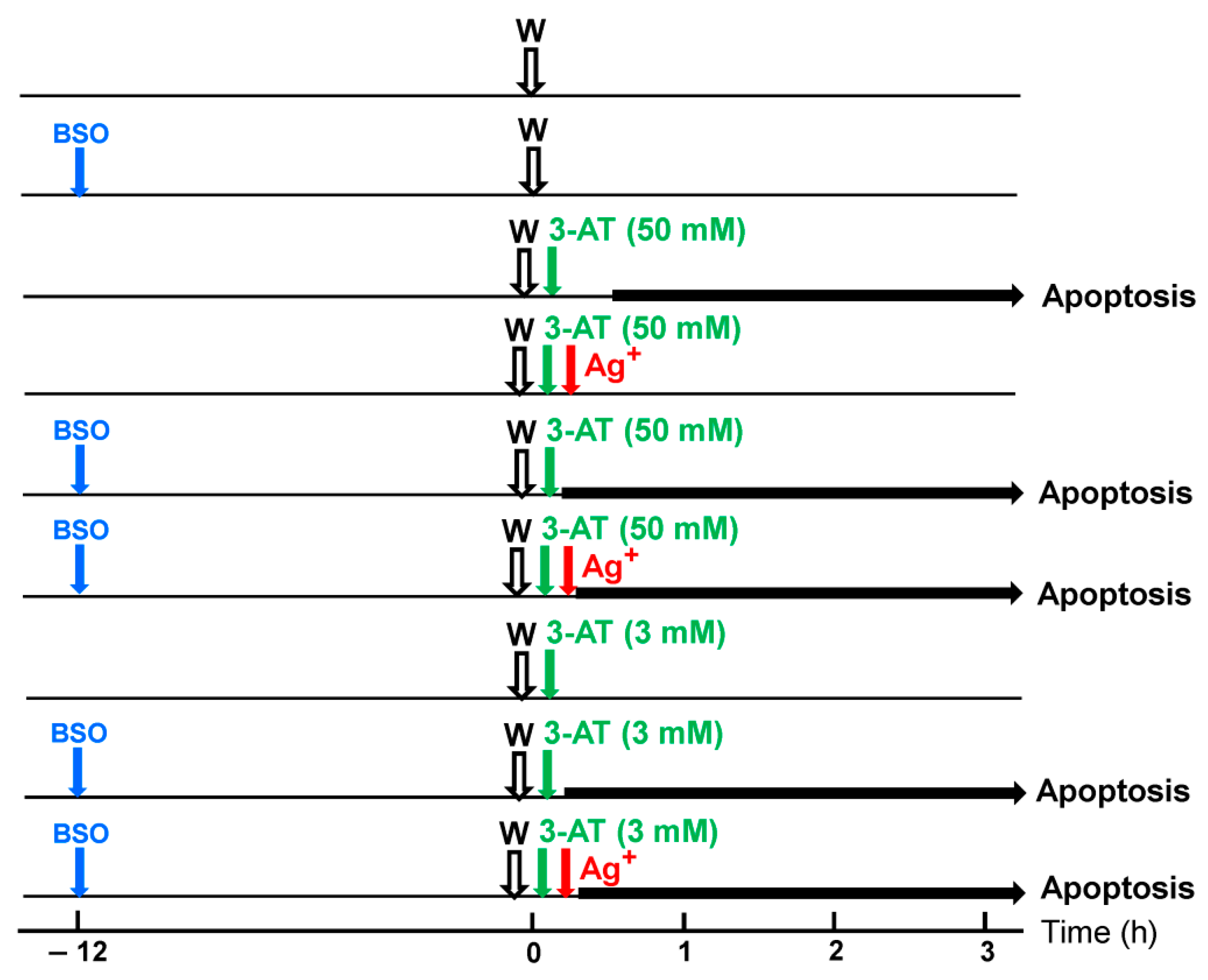
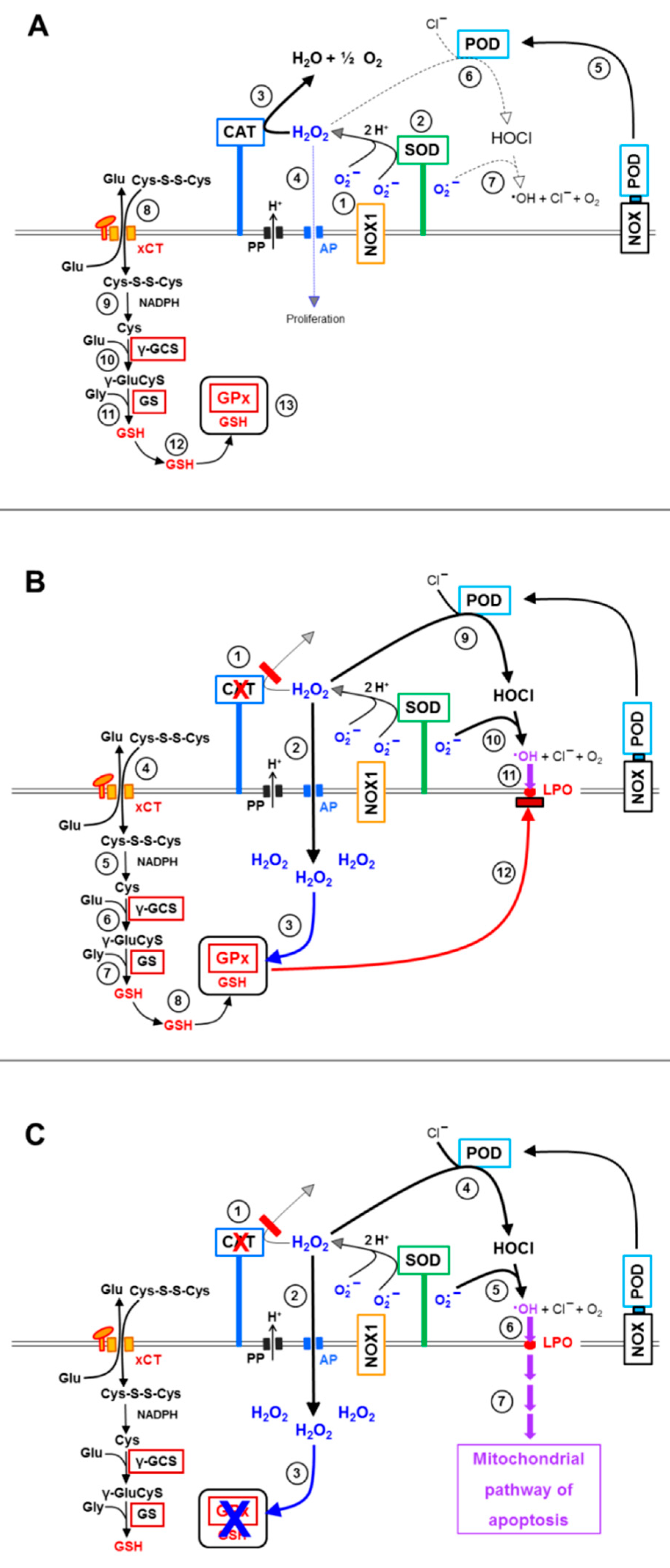
3.1. Membrane-Associated Catalase of Tumor Cells Prevents the Influx of H2O2 through Aquaporins and Subsequent H2O2-Mediated Apoptosis Induction
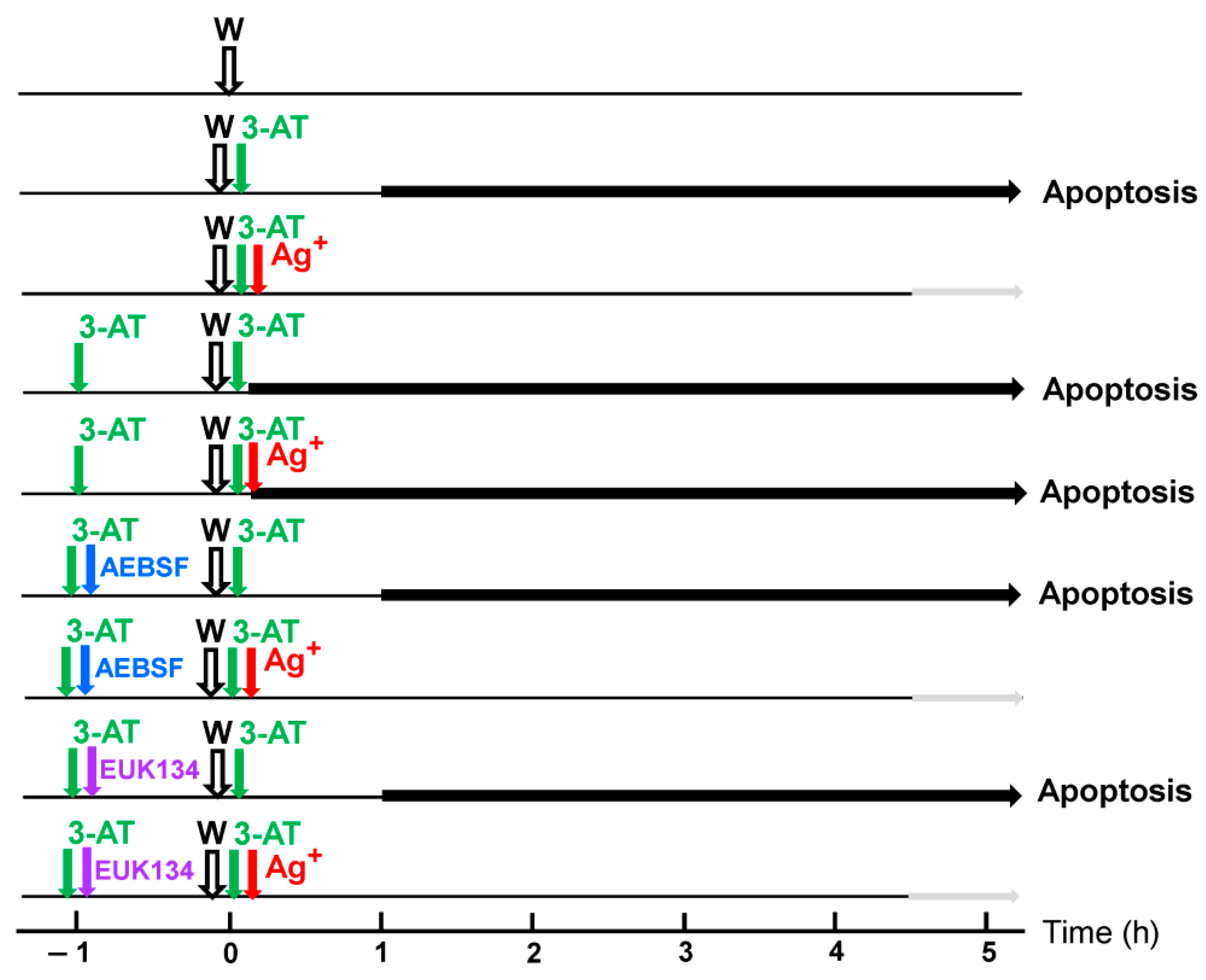
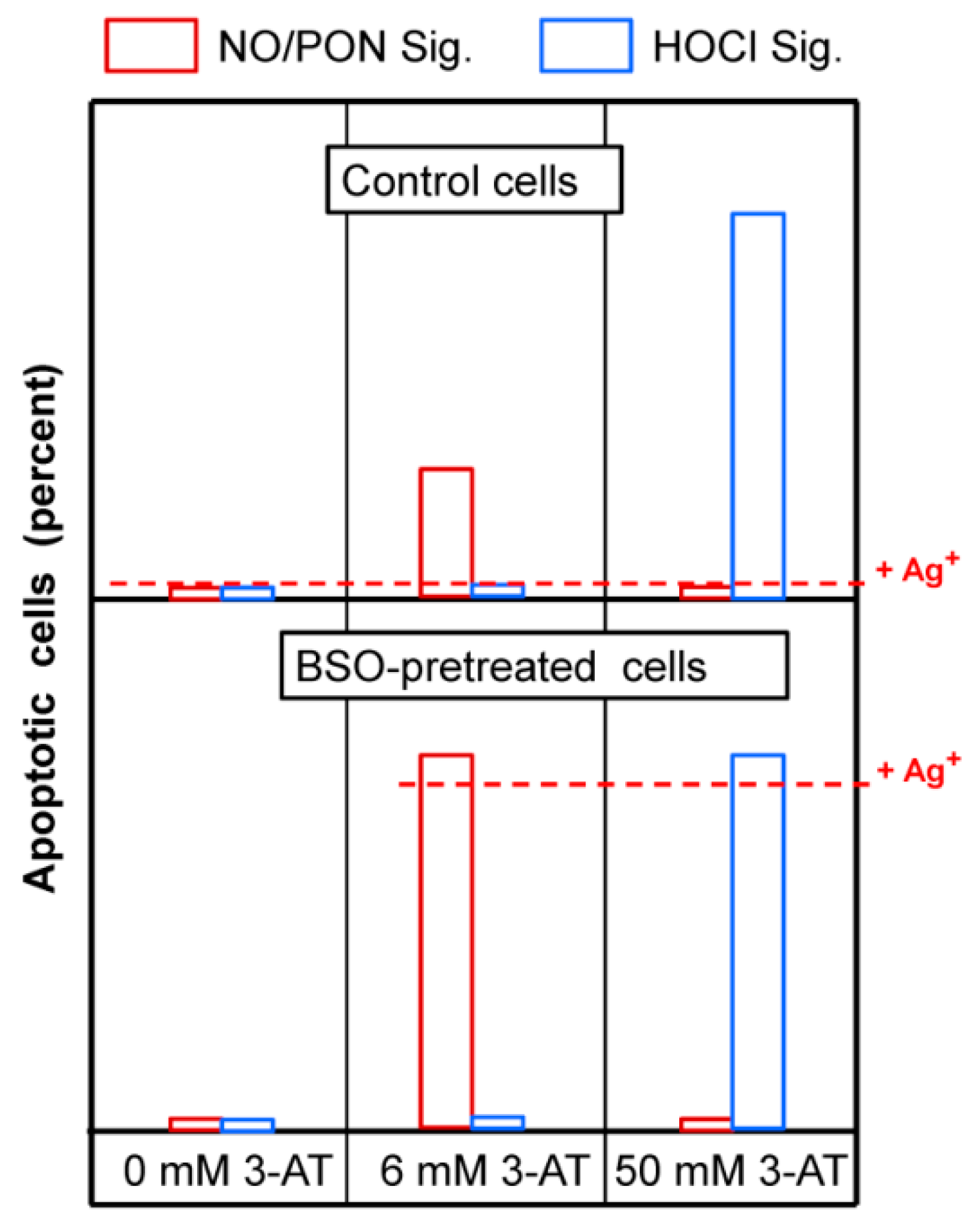
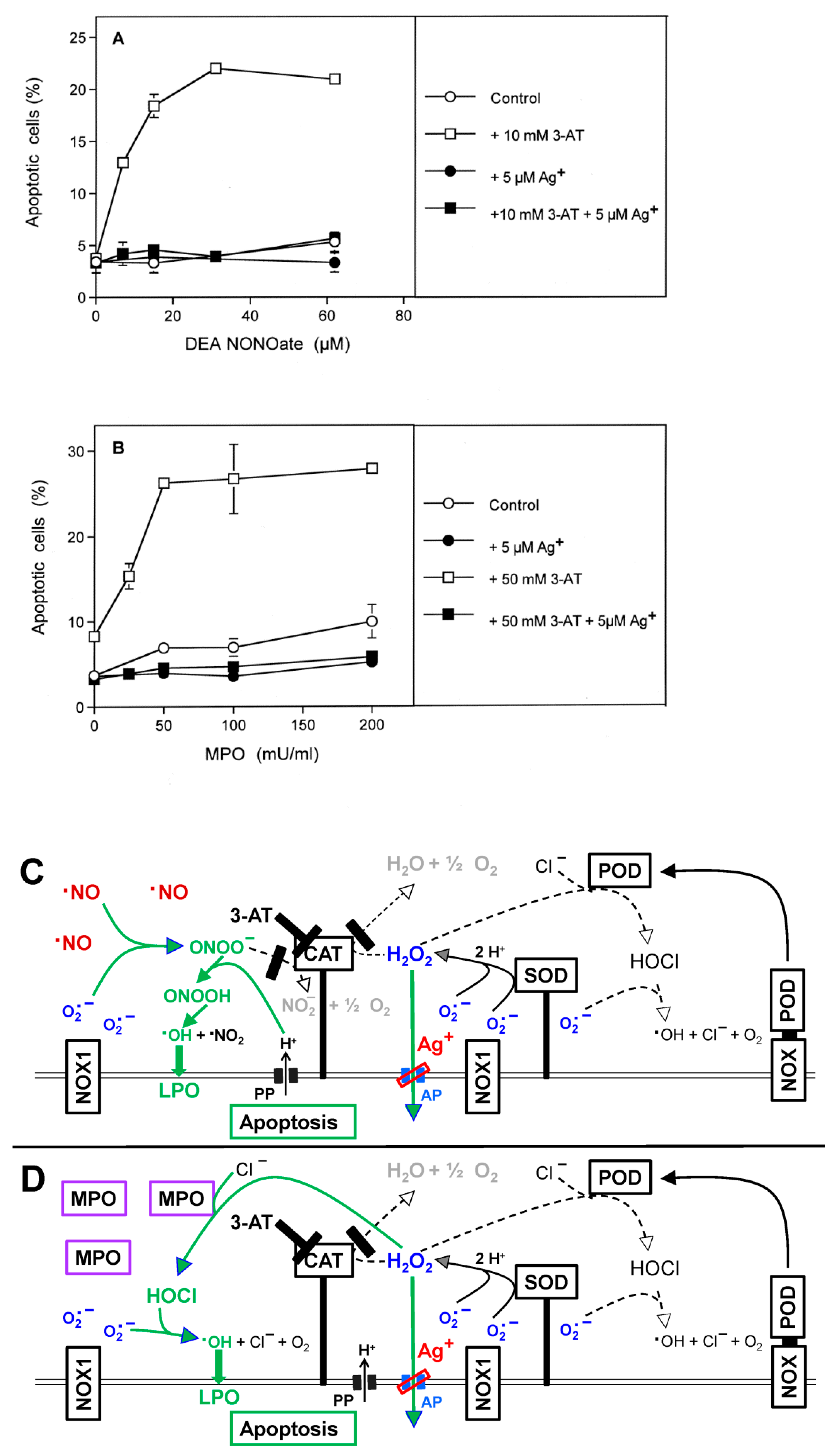
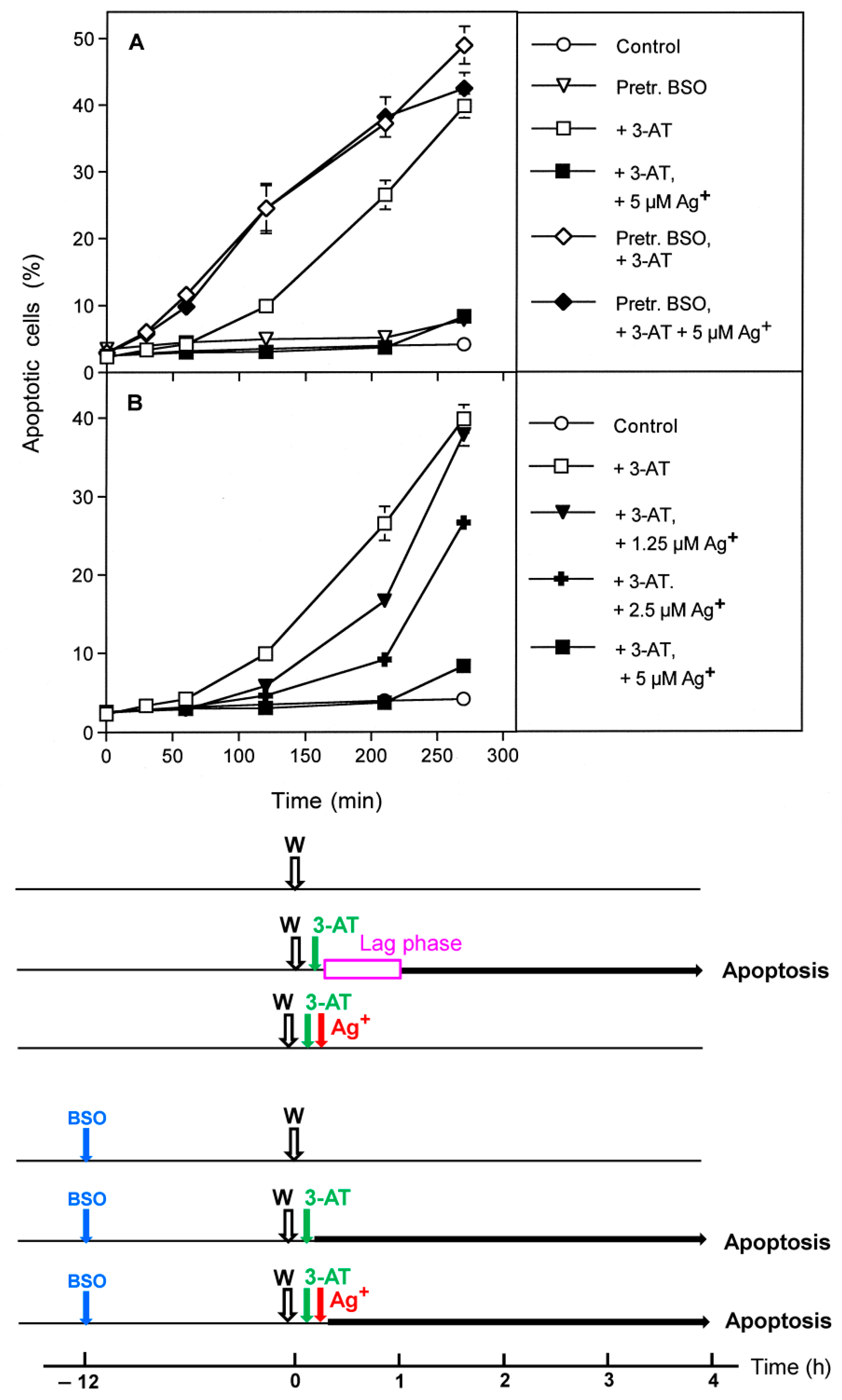
3.2. The Essential Cooperation between Extracellular ROS/RNS Signaling and Aquaporins
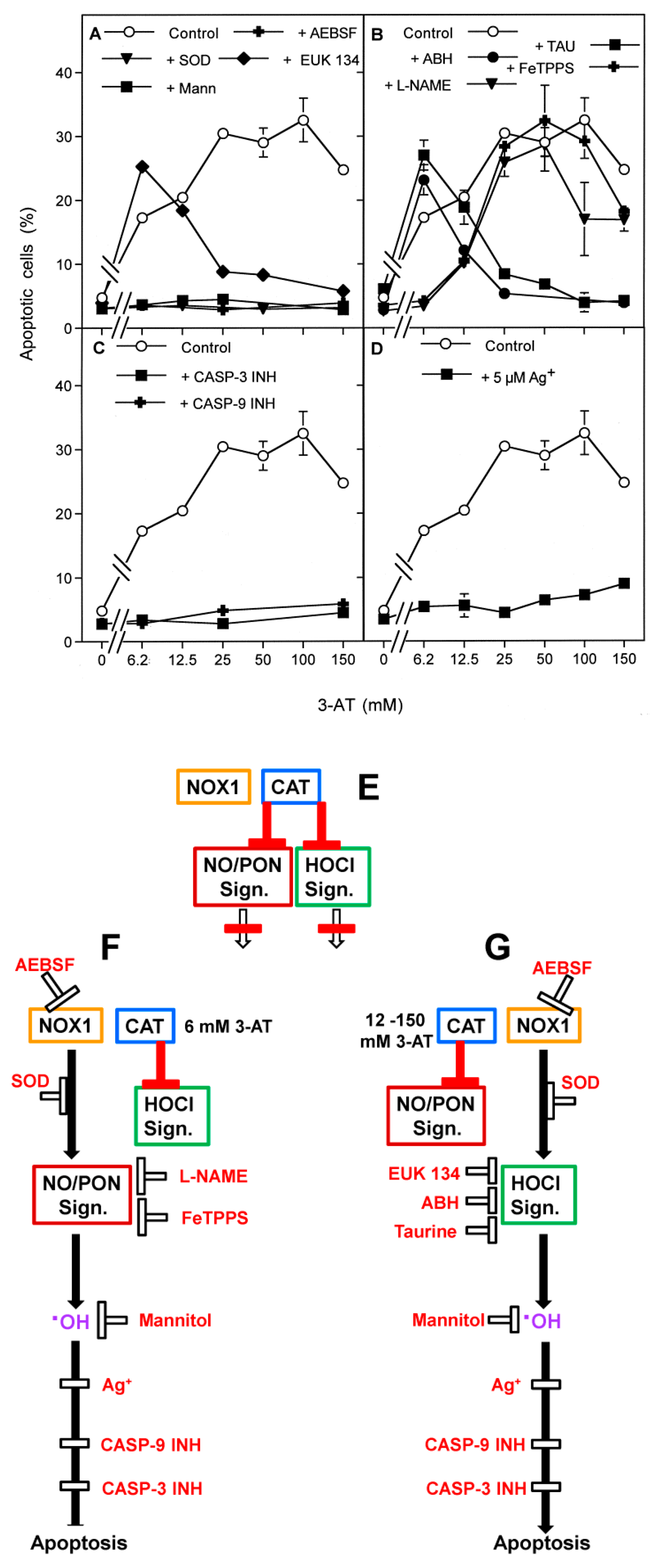
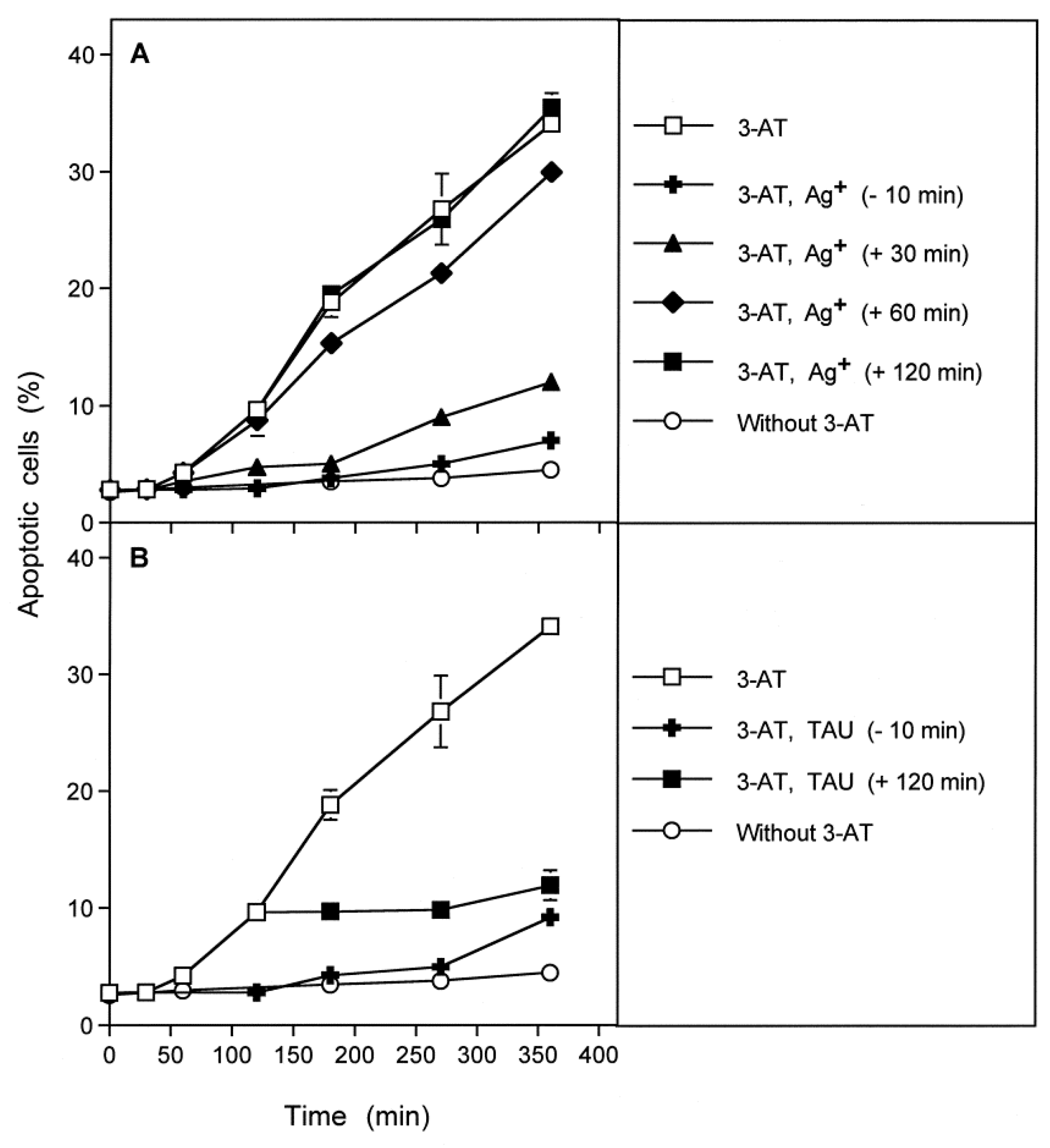
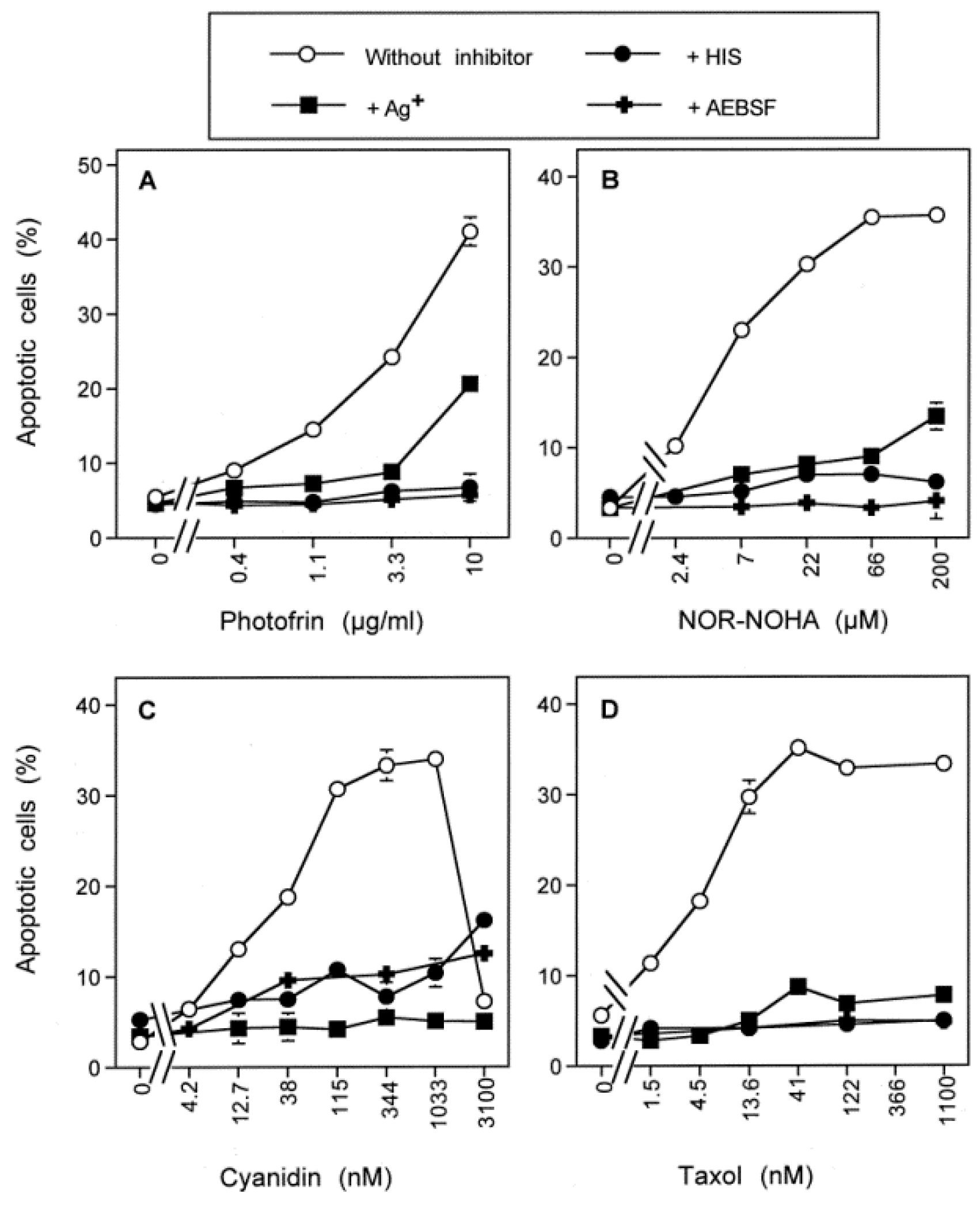
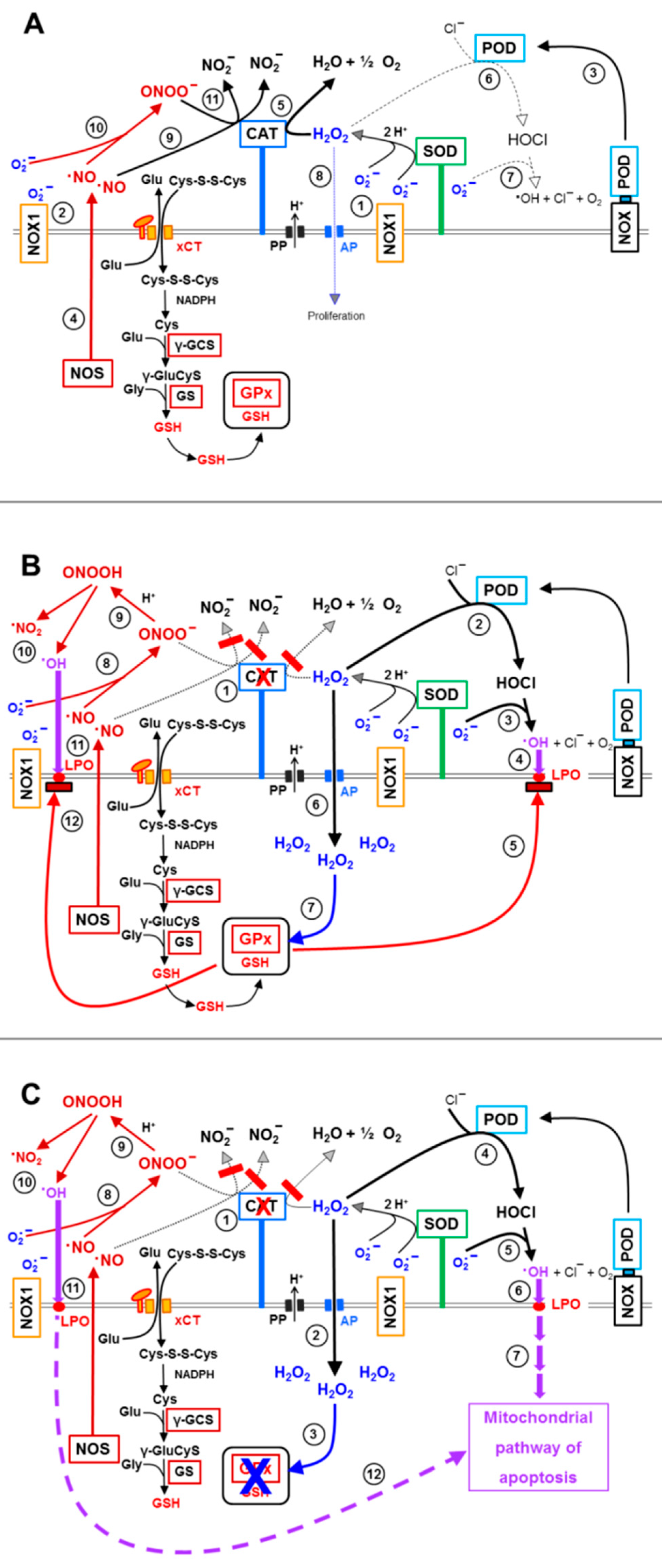
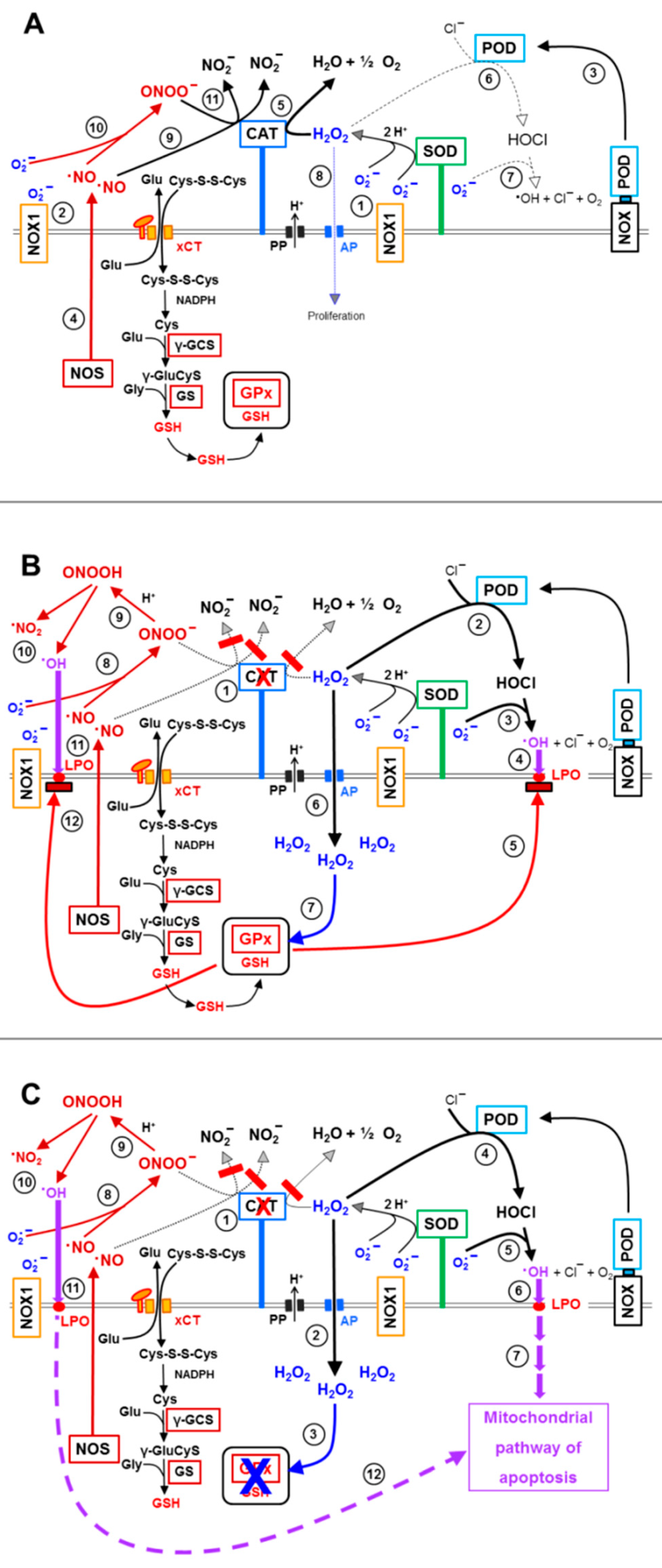
3.3. Aquaporin Action Is Required for Apoptosis-Inducing ROS- as Well as RNS Signaling of Tumor Cells
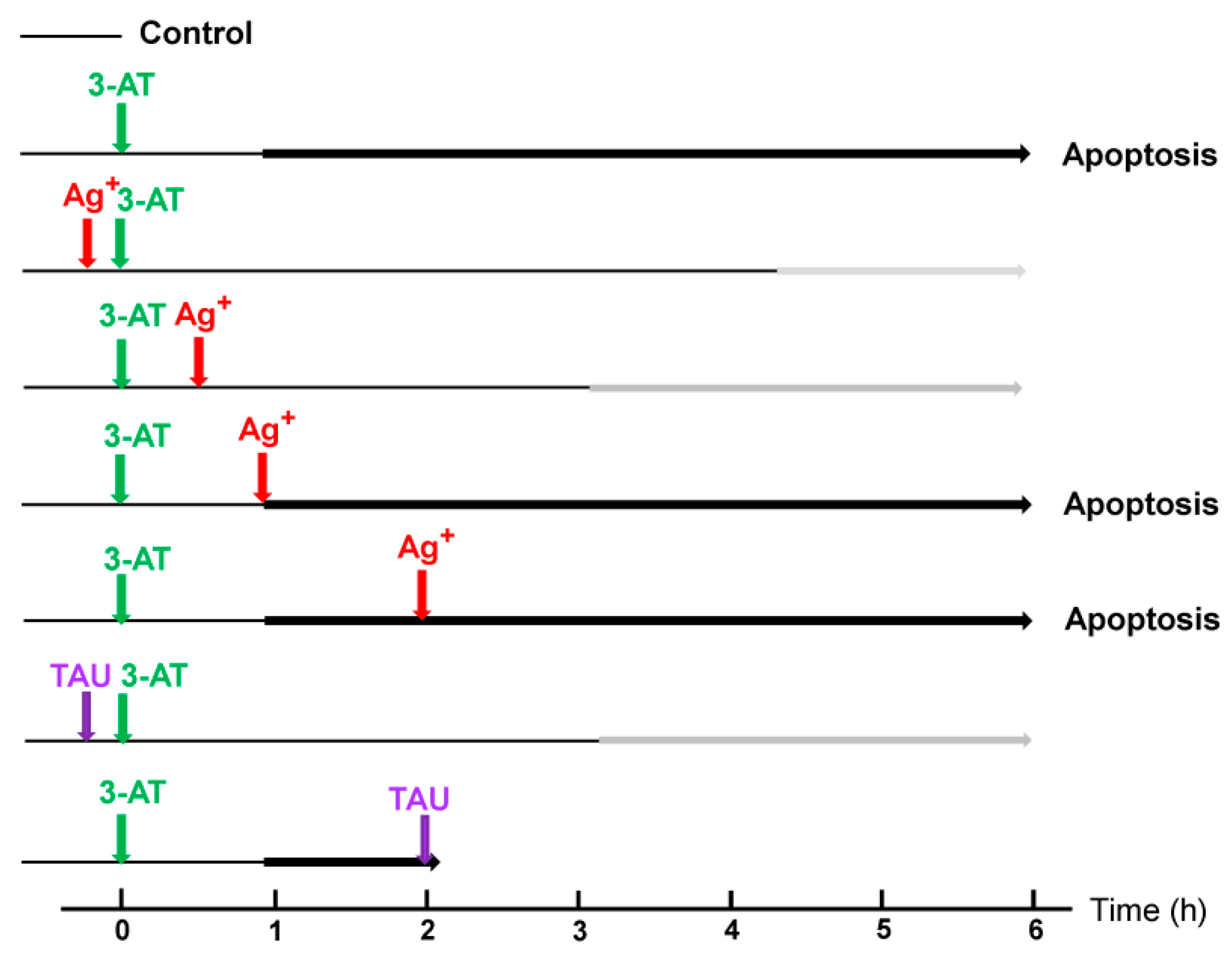
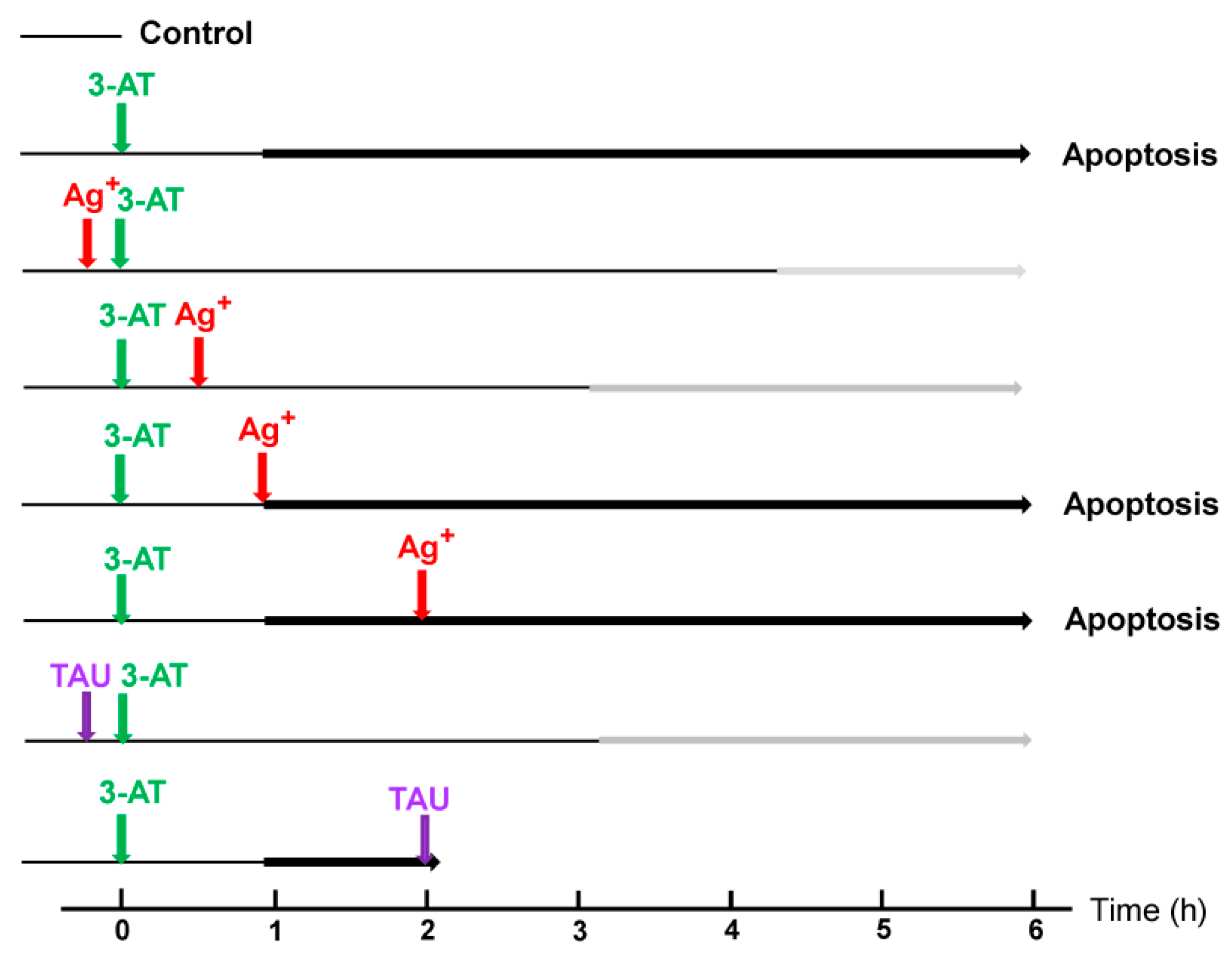
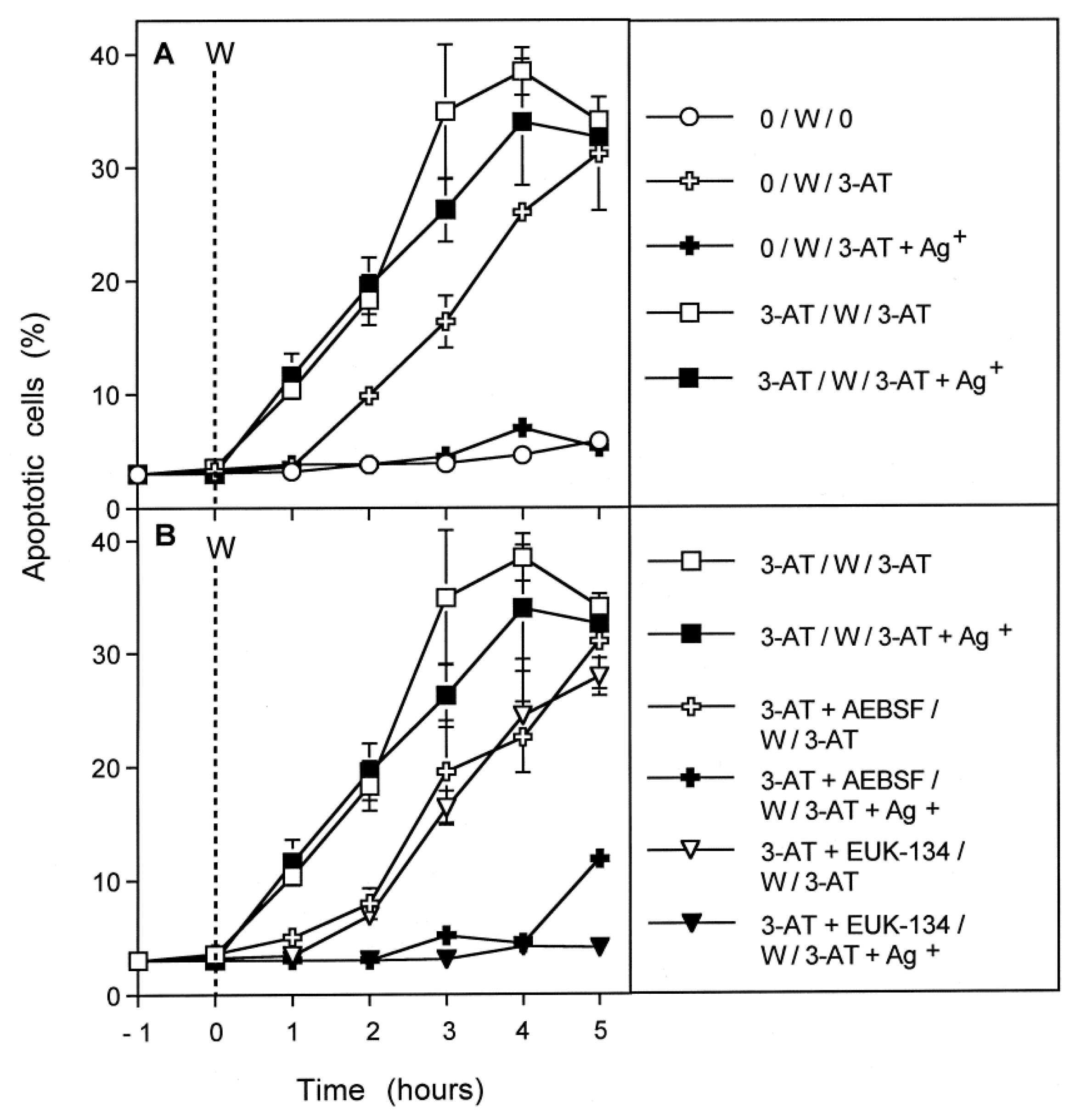
3.4. Aquaporin Action Is Only Initially Required
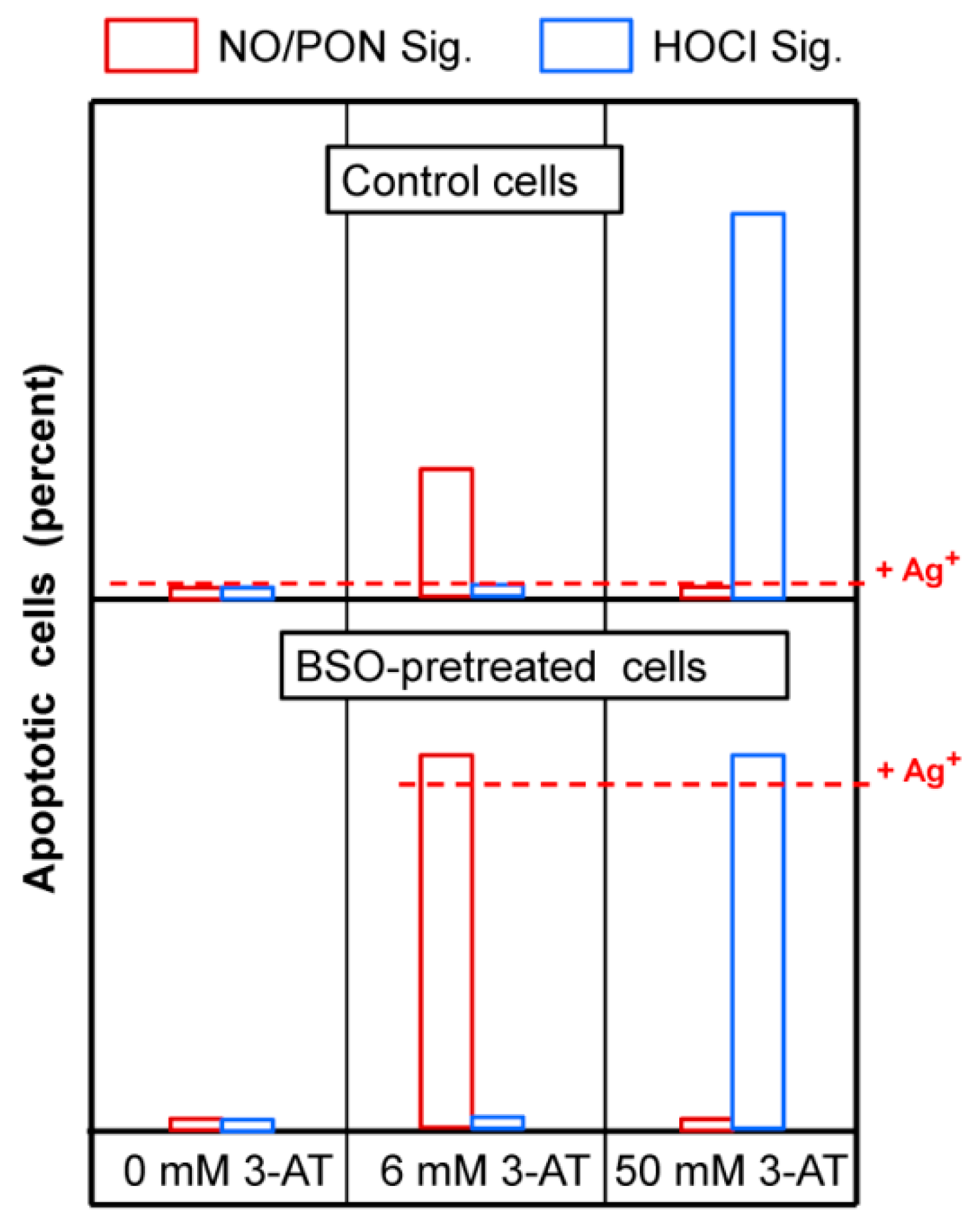
3.5. H2O2 Intruding through Aquaporins Is the Dismutation Product of NOX1-Derived Extracellular Superoxide Anions
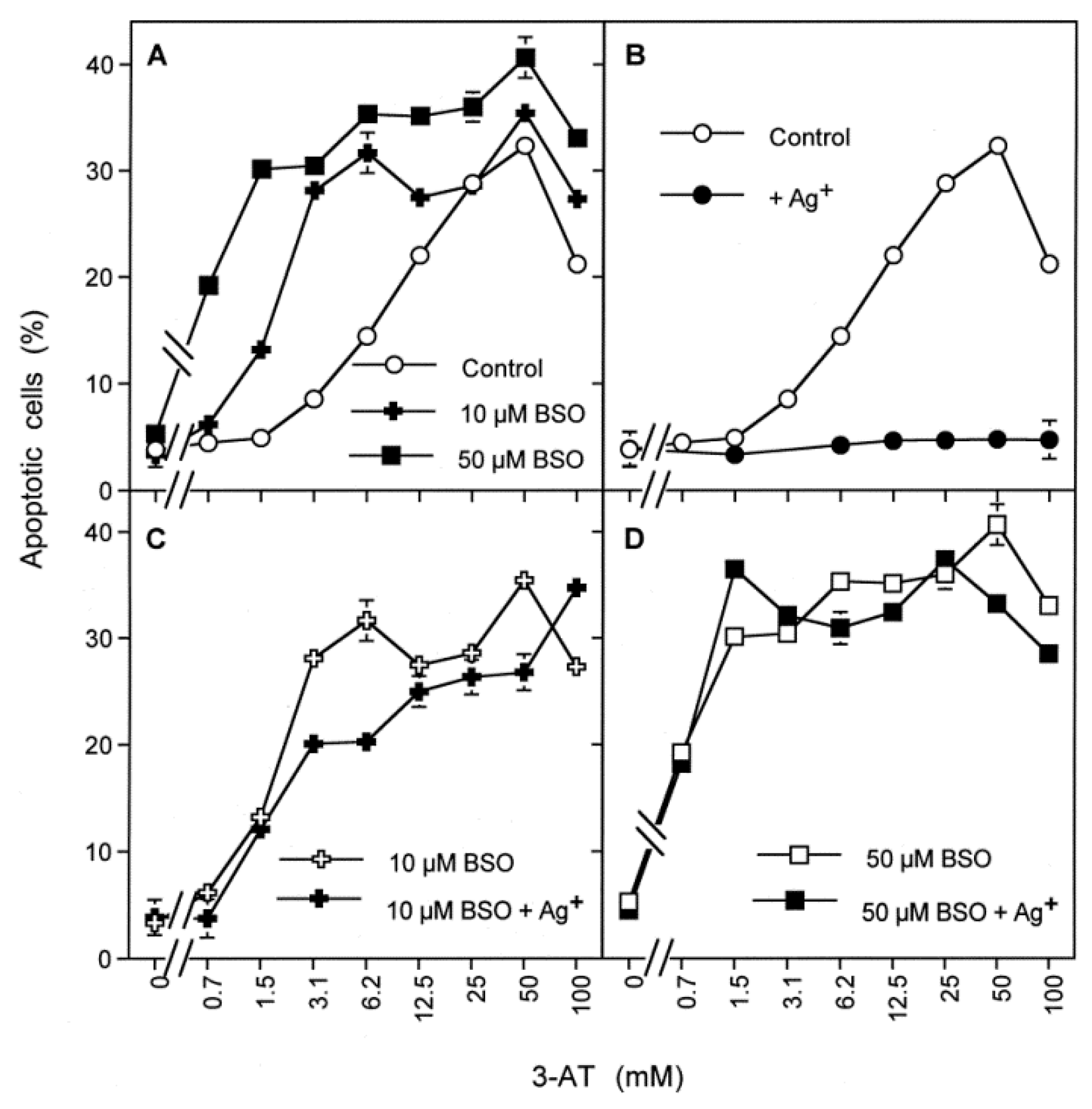
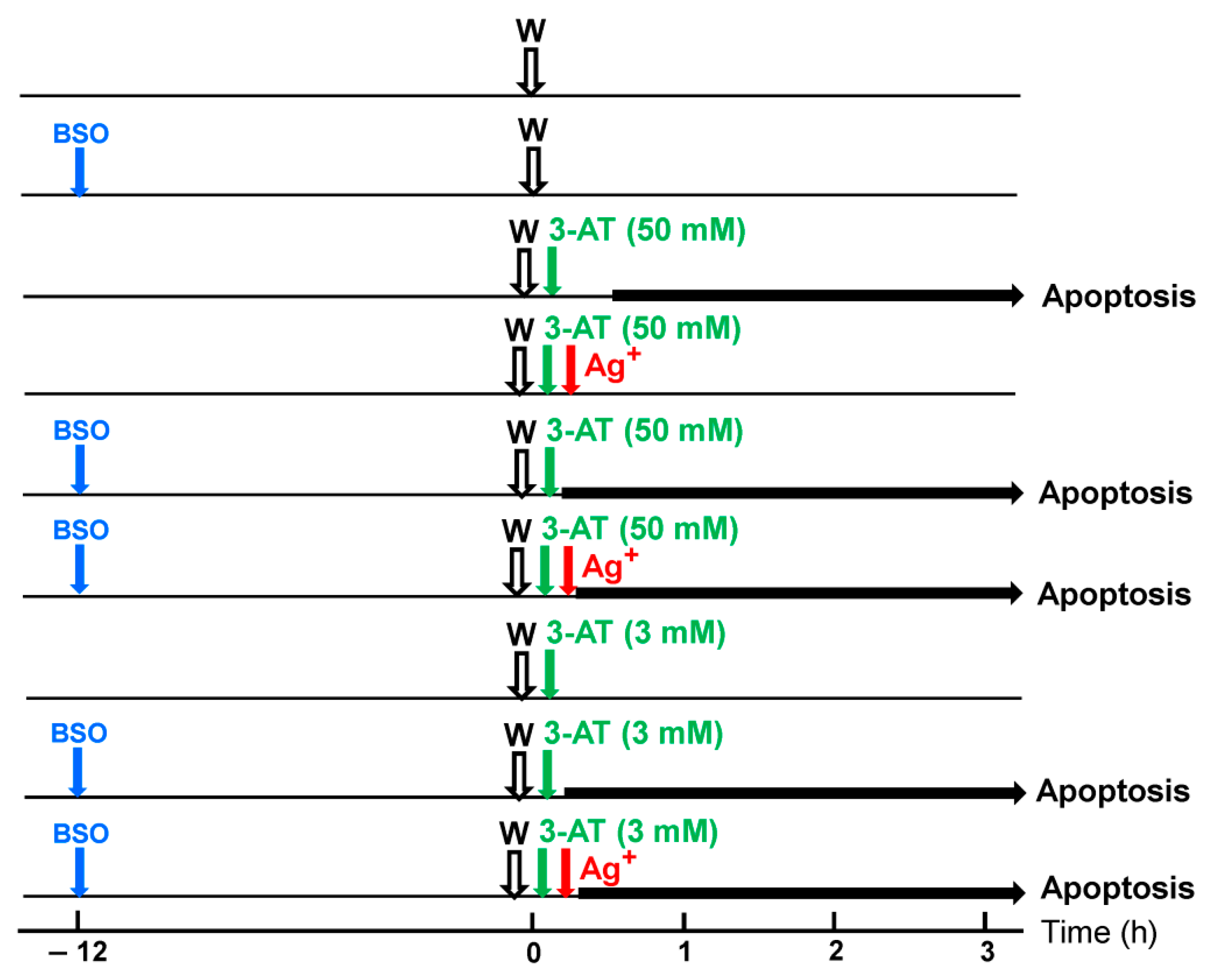
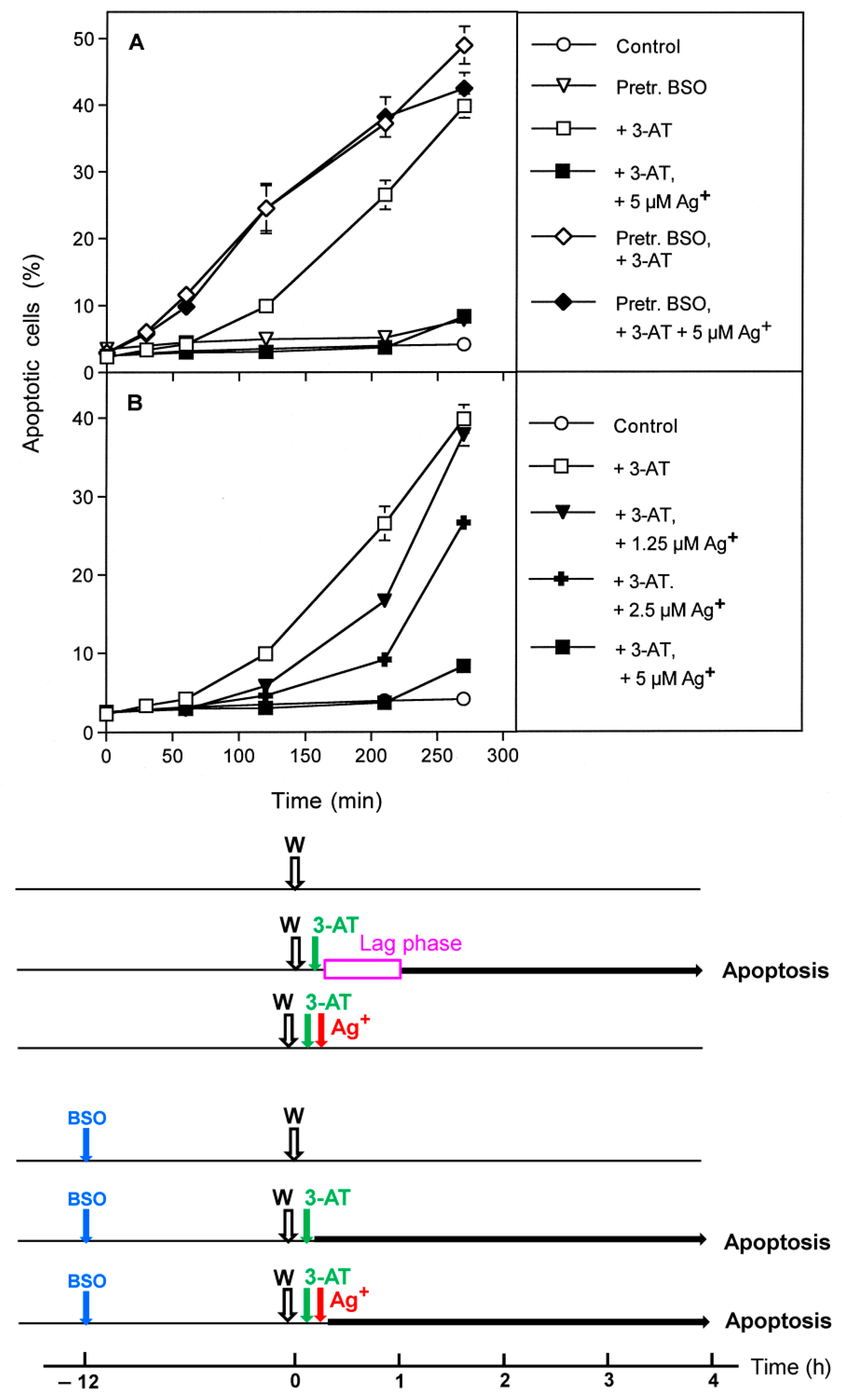
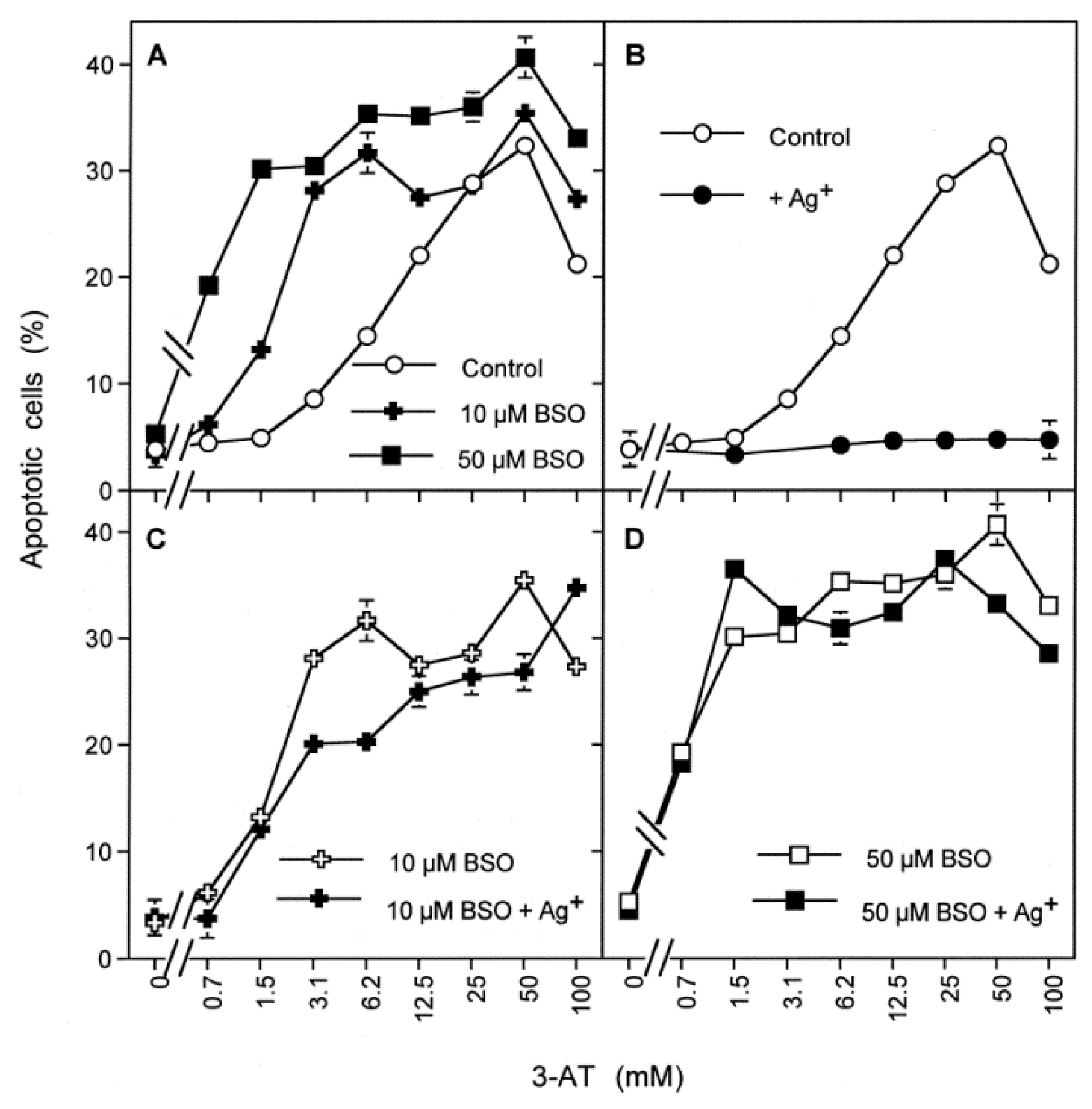
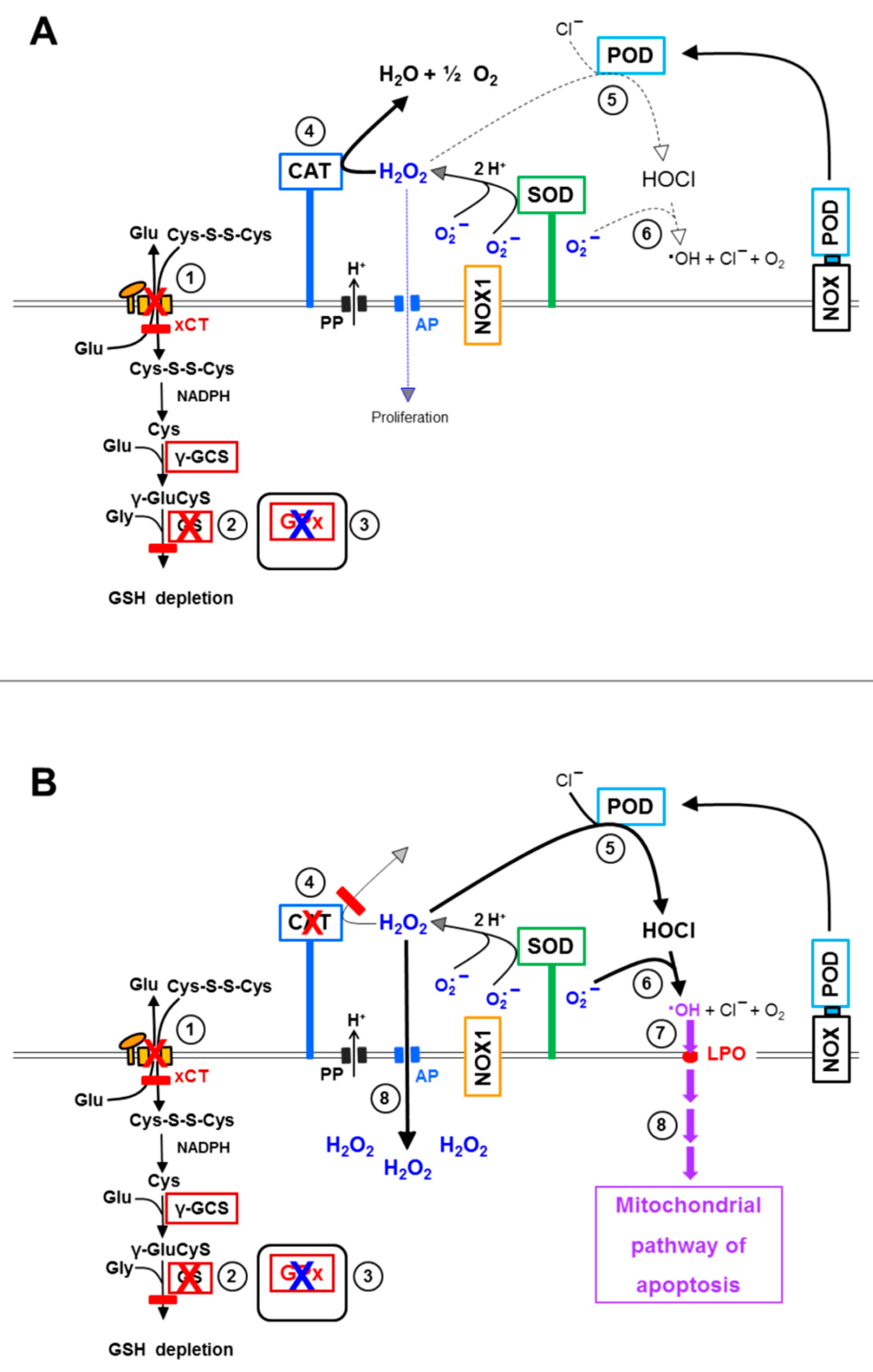
3.6. Targeting of Glutathione/Glutathione Peroxidase by Intruding H2O2
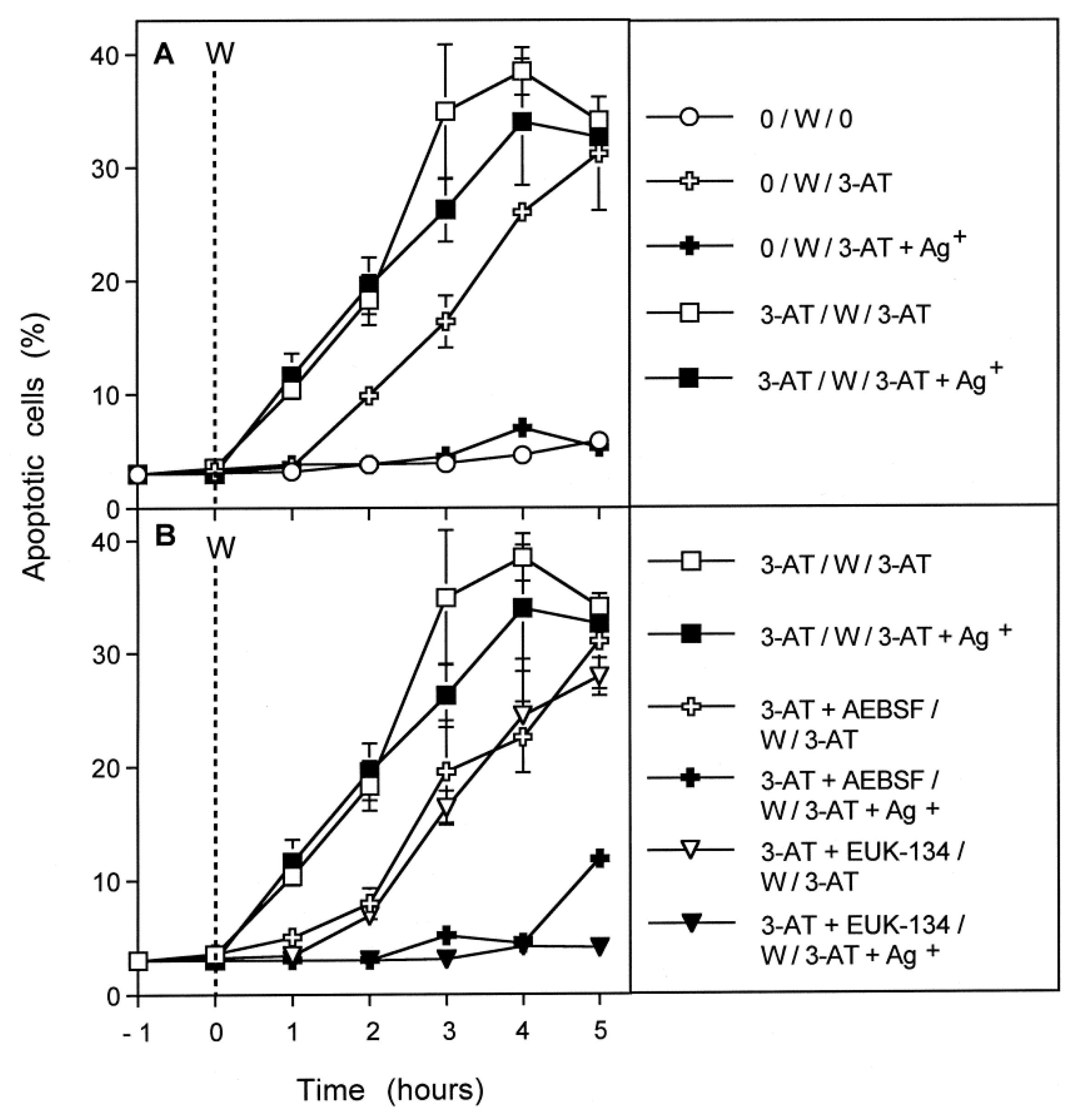
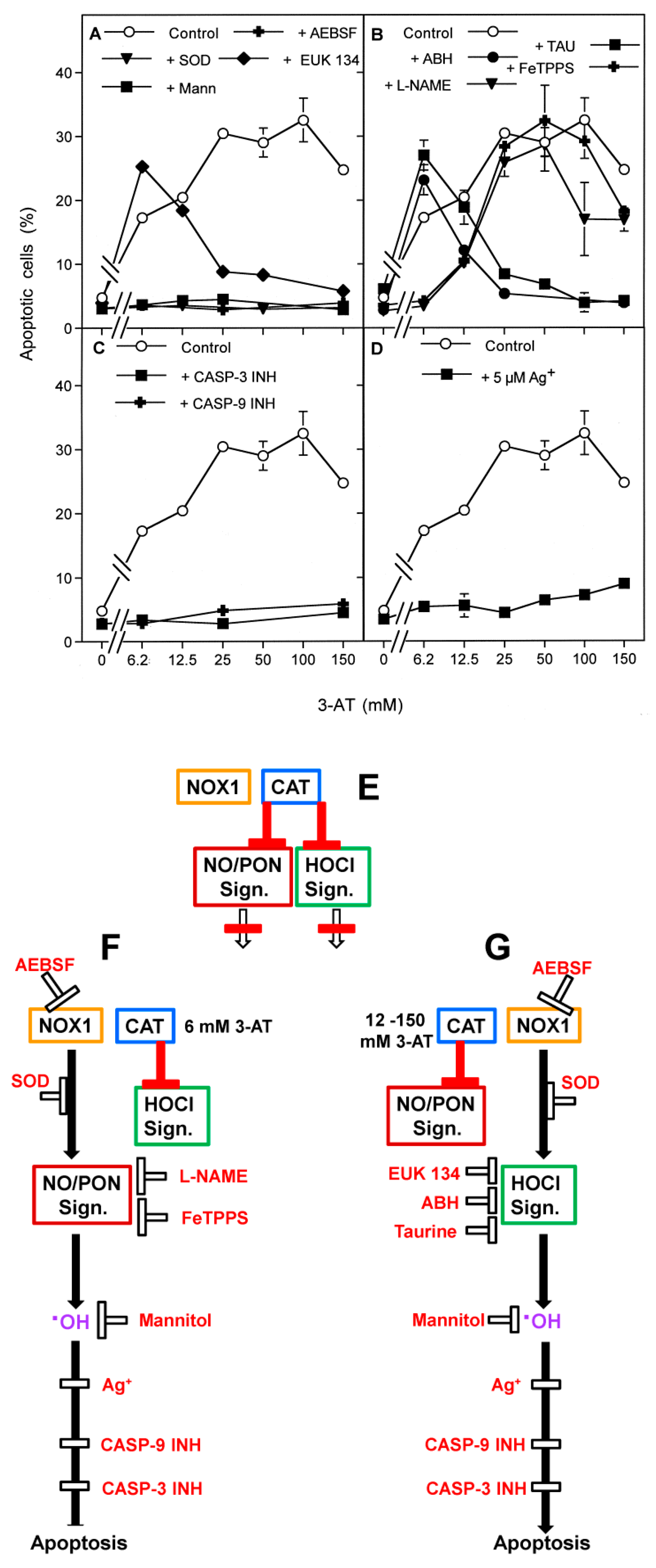
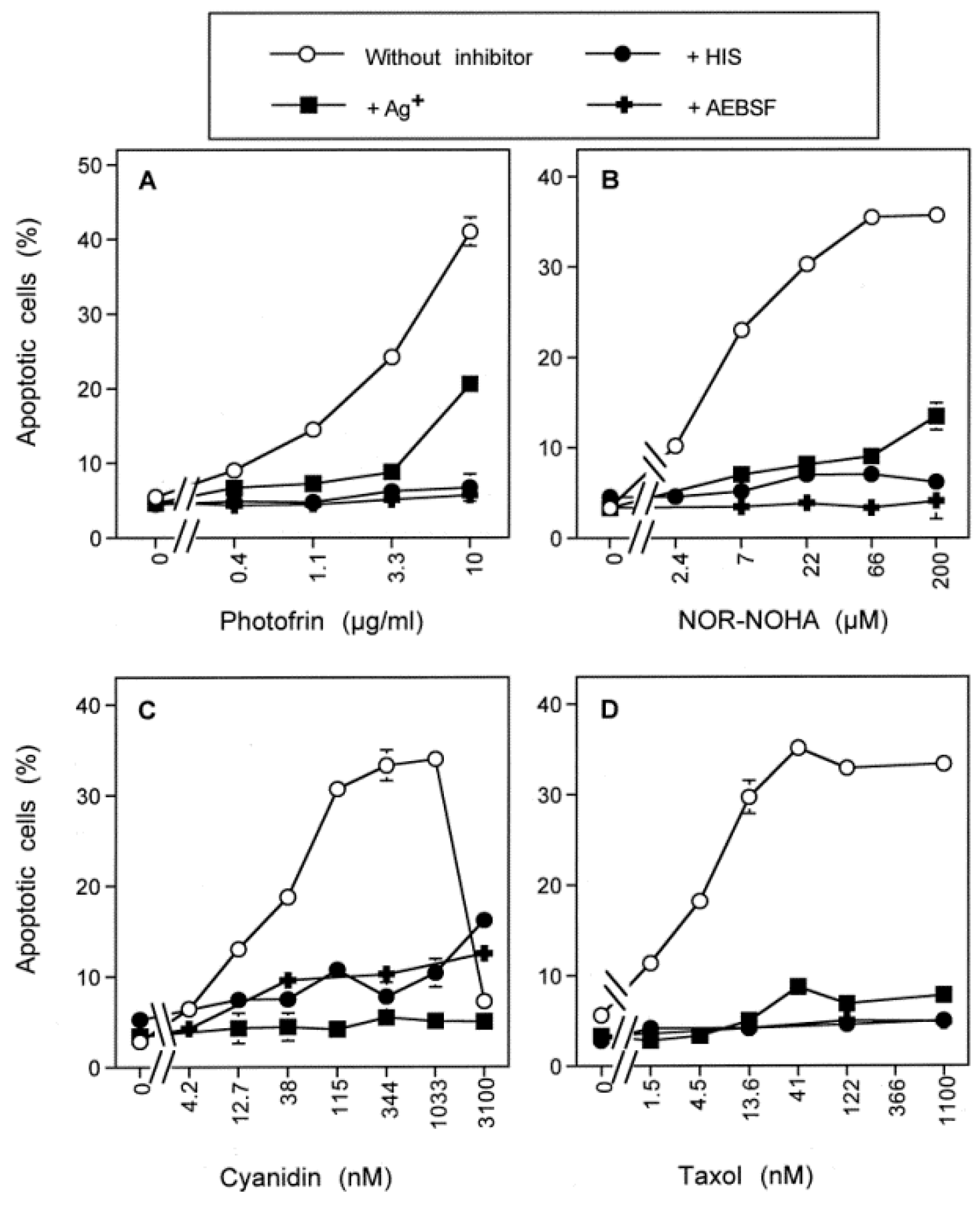
3.7. Dominant Role of Aquaporins after Singlet Oxygen-Mediated Inactivation of Membrane-Associated Catalase
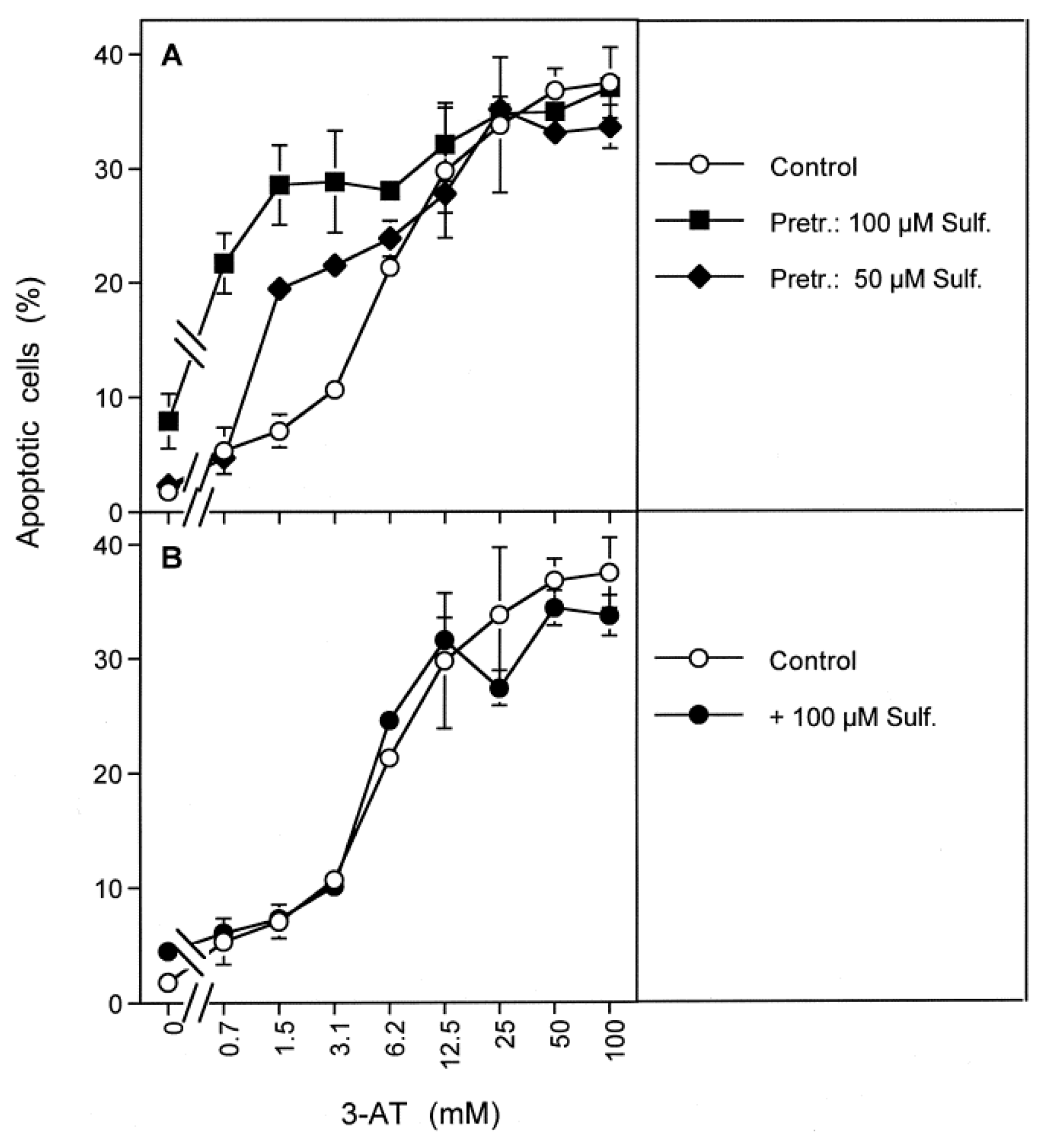
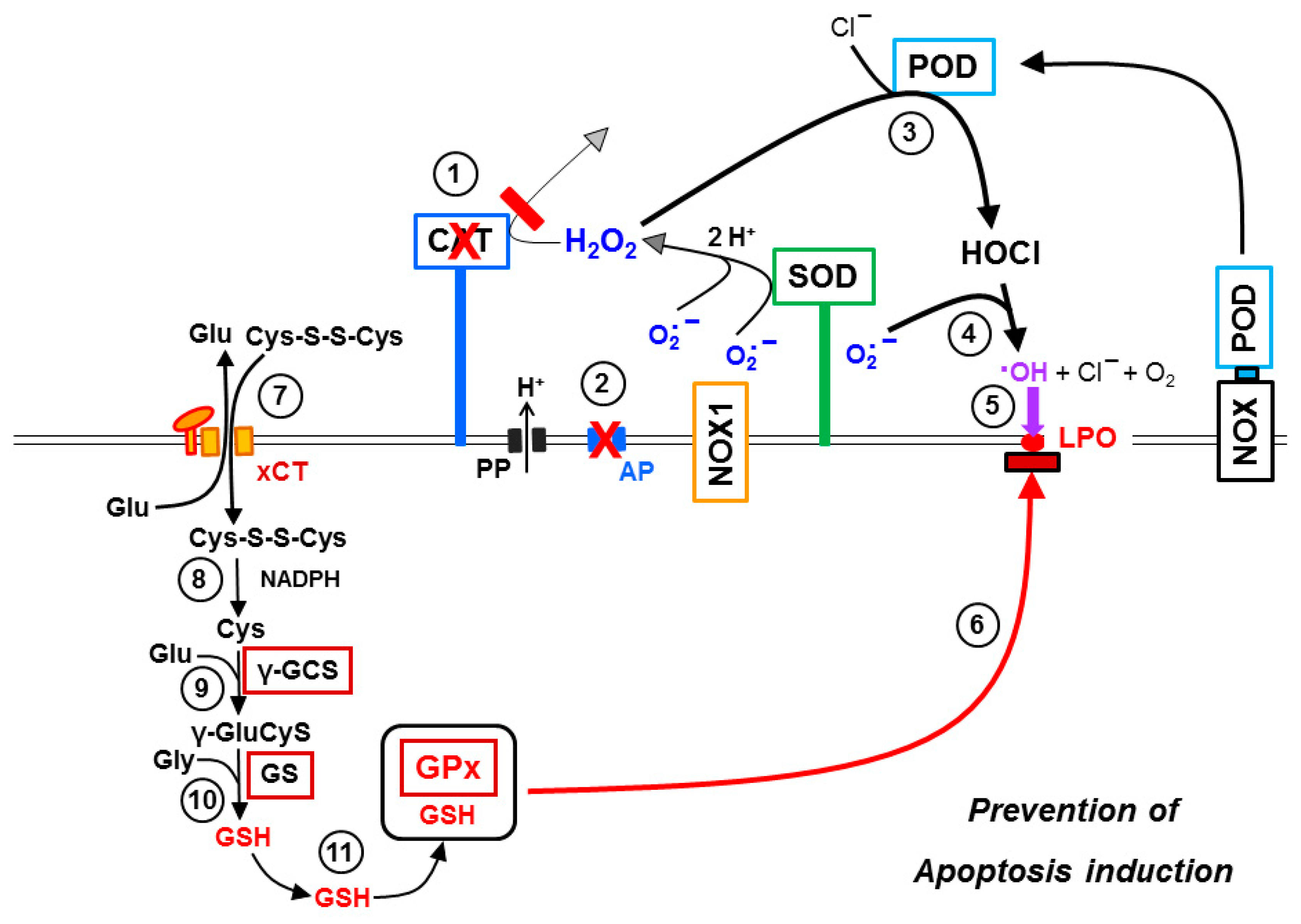
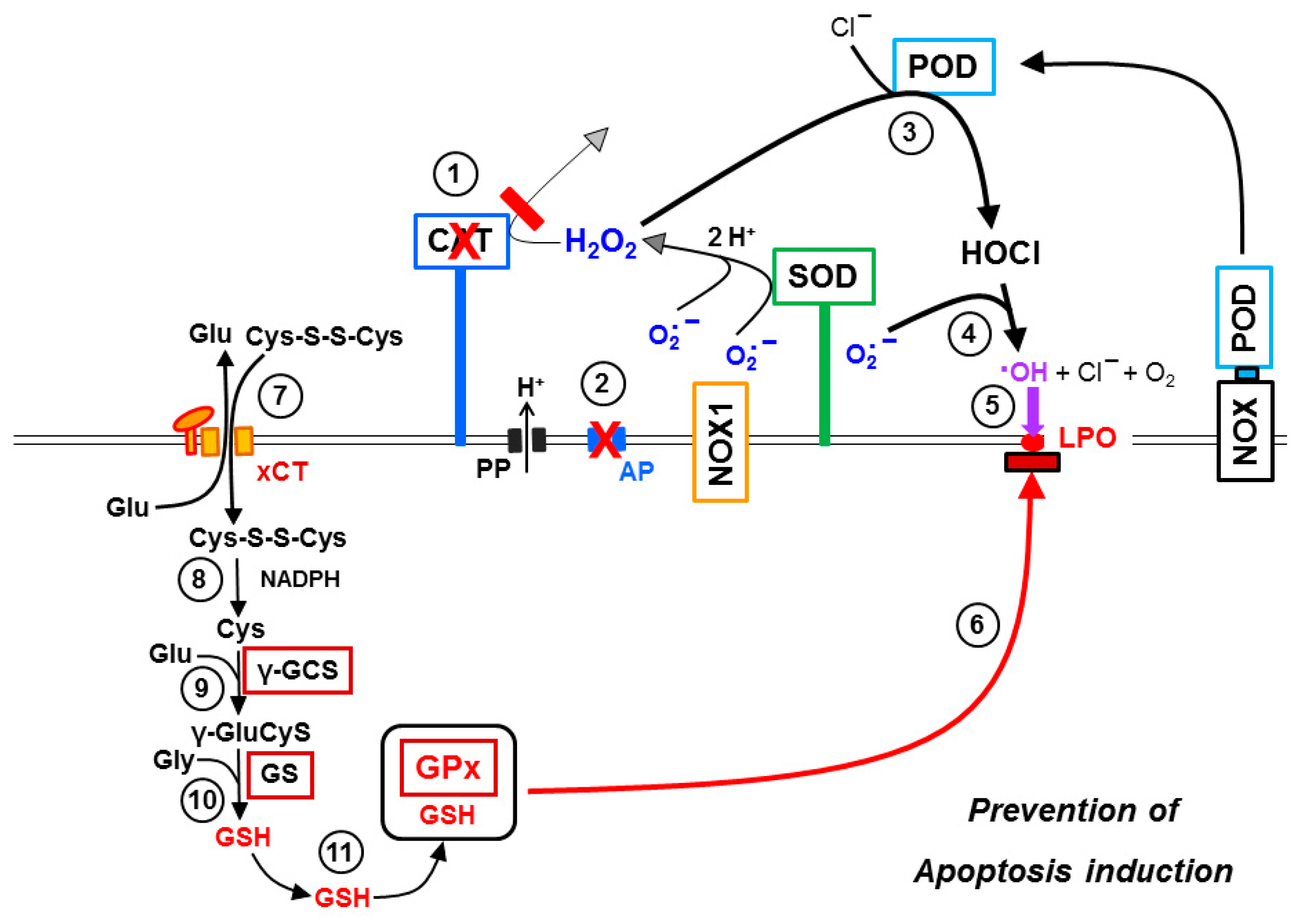
3.8. The Role of the xC Transporter for the Control of Apoptosis Induction
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Chance, B. The Steady State Level of Catalase Compound I in Isolated Hemoglobin-Free Perfused Rat Liver. FEBS Lett. 1970, 11, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide Metabolism in Mammalian Organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Findings in Redox Biology: From H2O2 to Oxidative Stress. J. Biol. Chem. 2020, 295, 13458–13473. [Google Scholar] [CrossRef] [PubMed]
- Parchment, R.E. Programmed Cell Death (Apoptosis) in Murine Blastocysts: Extracellular Free Radicals, Polyamines and Other Cytotoxic Agents. Vivo 1991, 5, 493–500. [Google Scholar]
- Pierce, G.B.; Parchment, R.E.; Lewellyn, A.L. Hydrogen Peroxide as A Mediator of Programmed Cell Death in the Blastocyst. Differentiation 1991, 46, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of Metabolic H2O2. Generation, Redox Signaling and Oxidative Stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of Iron and Hydrogen Peroxide: The Fenton Reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase: Friend and Foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Bauer, G. HOCl and the Control of Oncogenesis. J. Inorg. Biochem. 2018, 179, 10–23. [Google Scholar] [CrossRef]
- Saran, M.; Beck-Speier, I.; Fellerhoff, B.; Bauer, G. Phagocytic Killing of Microorganisms by Radical Processes:Consequences of the Reaction of Hydroxyl Radicals with Chloride Yielding Chlorine Atoms. Free. Radic. Biol. Med. 1999, 26, 482–490. [Google Scholar] [CrossRef]
- Folkes, L.K.; Candeias, L.P.; Wardman, P. Kinetics and Mechanisms of Hypochlorous Acid Reactions. Arch. Biochem. Biophys. 1995, 323, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Candeias, L.P.; Patel, K.B.; Stratford, M.R.; Wardman, P. Free Hydroxyl Radicals are Formed on Reaction Between the Neutrophil-Derived Species Superoxide Anion and Hypochlorous Acid. FEBS Lett. 1993, 333, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Herdener, M.; Heigold, S.; Saran, M.; Bauer, G. Target Cell-Derived Superoxide Anions Cause Efficiency and Selectivity of Intercellular Induction of Apoptosis. Free Rad. Biol. Med. 2000, 29, 1260–1271. [Google Scholar] [CrossRef]
- Engelmann, I.; Dormann, S.; Saran, M.; Bauer, G. Transformed Target Cell-Derived Superoxide Anions Drive Apoptosis Induction by Myeloperoxidase. Redox Rep. 2000, 5, 207–214. [Google Scholar] [CrossRef]
- Saran, M.; Bors, W. Oxygen Radicals as Chemical Messengers: A Hypothesis. Free. Rad Res Comm 1989, 7, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Saran, M.; Bors, W. Signalling by O2- and NO: How Far Can Either Radical, or Any Specific Reaction Product, Transmit a Message Under In Vivo Conditions? Chem. Biol. Interact. 1994, 90, 35–45. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic Signalling by Oxidants in Ras-Transformed Fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Irani, K.; Goldschmidt-Clermont, P.J. Ras, Superoxide and Signal Transduction. Biochem. Pharmacol. 1998, 55, 1339–1346. [Google Scholar]
- López-Lázaro, M. Excessive Superoxide Anion Generation Plays a Key Role in Carcinogenesis. Int. J. Cancer 2007, 120, 1378–1380. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. Dual Role of Hydrogen Peroxide in Cancer: Possible Relevance to Cancer Chemoprevention and Therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Tumor Cell-Protective Catalase as a Novel Target for Rational Therapeutic Approaches Based on Specific Intercellular ROS Signaling. Anticancer Res. 2012, 32, 2599–2624. [Google Scholar] [PubMed]
- Bauer, G. Targeting extracellular ROS signaling of tumor cells. Anticancer Res. 2014, 34, 1467–1482. [Google Scholar] [PubMed]
- Deichman, G.I.; Vendrov, E.L. Characteristics of In Vitro Transformed Cells Essential for their In Vivo Survival, Selection and Metastatic Activity. Int. J. Cancer 1986, 37, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Deichman, G.I.; Kluchareva, T.E.; Matveeva, V.A.; Kushlinsky, N.E.; Bassalyk, L.S.; Vendrov, E.L. Clustering of Discrete Cell Properties Essential for Tumorigenicity and Metastasis. I. Studies of Syrian Hamster Embryo Fibroblasts Spontaneously Transformedin Vitro. Int. J. Cancer 1989, 44, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Deichman, G.; Matveeva, V.A.; Kashkina, L.M.; Dyakova, N.A.; Uvarova, E.N.; Nikiforov, M.A.; Gudkov, A.V. Cell Trans-Forming Genes and Tumor Progression: In Vivo Unified Secondary Phenotypic Cell Changes. Int. J. Cancer 1998, 75, 277–283. [Google Scholar] [CrossRef]
- Deichman, I.G. Natural Selection and Early Changes of Phenotype of Tumor Cells In Vivo: Acquisition of New Defense Mechanisms. Biochem. 2000, 65, 78–94. [Google Scholar]
- Deichman, G. Early Phenotypic Changes of In Vitro Transformed Cells During In Vivo Progression: Possible Role of the Host Innate Immunity. Sem. Cancer. Biol. 2002, 12, 317–326. [Google Scholar] [CrossRef]
- Heinzelmann, S.; Bauer, G. Multiple Protective Functions of Catalase Against Intercellular Apoptosis-Inducing ROS Signaling of Human Tumor Cells. Biol. Chem. 2010, 391, 675–693. [Google Scholar] [CrossRef]
- Böhm, B.; Heinzelmann, S.; Motz, M.; Bauer, G. Extracellular Localization of Catalase is Associated with the Transformed State of Malignant Cells. Biol. Chem. 2015, 396, 1339–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, G. Signal Amplification by Tumor Cells: Clue to the Understanding of the Antitumor Effects of Cold Atmospheric Plasma and Plasma-Activated Medium. IEEE Trans. Radiat. Plasma Med Sci. 2018, 2, 87–98. [Google Scholar] [CrossRef]
- Bienert, G.P.; MØller, A.L.; Kristiansen, K.A.; Schulz, A.; MØller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific Aquaporins Facil-Itate the Diffusion of Hydrogen Peroxide Across Membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Beitz, E. Aquaporins with Selectivity for Unconventional Permeants. Cell. Mol. Life Sci. 2007, 64, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Hara-Chikuma, M.; Papadopoulos, M.C. Aquaporins—New Players in Cancer Biology. J. Mol. Med. 2008, 86, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Talbot, A.; Nourmohammadi, N.; Sherman, J.H.; Cheng, X.; Keidar, M. Toward Understanding the Selective Anticancer Capacity of Cold Atmospheric Plasma—A Model Based on Aquaporins (Review). Biointerphases 2015, 10, 040801. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Xiao, H.; Zhu, W.; Nourmohammadi, N.; Zhang, L.G.; Bian, K.; Keidar, M. The Role of Aquaporins in the An-Ti-Glioblastoma Capacity of the Cold Plasma-Stimulated Medium. J. Phys. D Appl. Phys. 2017, 50, 055401. [Google Scholar] [CrossRef]
- Bauer, G. The Synergistic Effect Between Hydrogen Peroxide and Nitrite, Two Long-Lived Molecular Species from Cold Atmos-Pheric Plasma, Triggers Tumor Cells to Induce their Own Cell Death. Redox Biol. 2019, 26, 101291. [Google Scholar] [CrossRef]
- Bauer, G. Intercellular Singlet Oxygen-Mediated Bystander Signaling Triggered by Long-Lived Species of Cold Atmospheric Plasma and Plasma-Activated Medium. Redox Biol. 2019, 26, 101301. [Google Scholar] [CrossRef]
- Bauer, G.; Sersenová, D.; Graves, D.B.; Machala, Z. Cold Atmospheric Plasma and Plasma-Activated Medium Trigger RONS-Based Tumor Cell Apoptosis. Sci. Rep. 2019, 9, 14210. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Sersenová, D.; Graves, D.B.; Machala, Z. Dynamics of Singlet Oxygen-Triggered, RONS-Based Apoptosis Induction after Treatment of Tumor Cells with Cold Atmospheric Plasma or Plasma-Activated Medium. Sci. Rep. 2019, 9, 13931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, A Potent Suppressor of Lymphoma Growth by Inhibition of the X(C)- Cystine Transporter: A New Action for an Old Drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine Transporter SLC7A11/Xct in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell 2020, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Eisenmann, S.; Sagwal, S.K.; Bodnar, Y.; Moritz, J.; Poschkamp, B.; Stoffels, I.; Emmert, S.; Madesh, M.; Weltmann, K.-D.; et al. xCT (SLC7A11) Expression Confers Intrinsic Resistance to Physical Plasma Treatment in Tumor Cells. Redox Biol. 2020, 30, 101423. [Google Scholar] [CrossRef]
- Heigold, S.; Sers, C.; Bechtel-Walz, W.; Ivanovas, B.; Schäfer, R.; Bauer, G. Nitric Oxide Mediates Apoptosis Induction Selectively in Transformed Fibroblasts Compared to Nontransformed Fibroblasts. Carcinogenesis 2002, 23, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Nitric Oxide’s Contribution to Selective Apoptosis Induction in Malignant Cells through Multiple Reaction Steps. Crit. Rev. Oncog. 2016, 21, 365–398. [Google Scholar] [CrossRef] [PubMed]
- Scheit, K.; Bauer, G. Direct and Indirect Inactivation of Tumor Cell Protective Catalase by Salicylic Acid and Anthocyanidins Reactivates Intercellular ROS Signaling and Allows for Synergistic Effects. Carcinogenesis 2015, 36, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, G.; Motz, M. The Antitumor Effect of Single-Domain Antibodies Directed towards Membrane-Associated Catalase and Superoxide Dismutase. Anticancer Res. 2016, 36, 5945–5956. [Google Scholar] [CrossRef] [Green Version]
- Bauer, G. Sirna-Based Analysis of The Abrogation of the Protective Function of Membrane-Associated Catalase of Tumor Cells. Anticancer Res. 2017, 37, 567–582. [Google Scholar] [CrossRef]
- Bauer, G. Increasing the Endogenous NO Level Causes Catalase Inactivation and Reactivation of Intercellular Apoptosis Sig-Naling Specifically in Tumor Cells. Redox Biol. 2015, 6, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Riethmüller, M.; Burger, N.; Bauer, G. Singlet Oxygen Treatment of Tumor Cells Triggers Extracellular Singlet Oxygen Generation, Catalase Inactivation and Reactivation of Intercellular Apoptosis-Inducing Signaling. Redox Biol. 2015, 6, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Bauer, G. Central Signaling Elements of Intercellular Reactive Oxygen/Nitrogen Species-Dependent Induction of Apoptosis in Malignant Cells. Anticancer Res. 2017, 37, 499–514. [Google Scholar] [CrossRef] [Green Version]
- Bechtel, W.; Bauer, G. Catalase Protects Tumor Cells Against Apoptosis Induction by Intercellular ROS Signaling. Anticancer Res 2009, 29, 4541–4557. [Google Scholar] [PubMed]
- Bechtel, W.; Bauer, G. Modulation of Intercellular ROS Signaling of Human Tumor Cells. Anticancer. Res. 2009, 29, 4559–4570. [Google Scholar] [PubMed]
- Bauer, G.; Zarkovic, N. Revealing Mechanisms of Selective, Concentration-Dependent Potentials of 4-Hydroxy-2-Nonenal to Induce Apoptosis in Cancer Cells through Inactivation of Membrane-Associated Catalase. Free. Radic. Biol. Med. 2015, 81, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Bereswill, S.; Aichele, P.; Glocker, E. Helicobacter Pylori Protects Oncogenically Transformed Cells from Reactive Oxygen Species-Mediated Intercellular Induction of Apoptosis. Carcinogenesis 2014, 35, 1582–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Jürgensmeier, J.; Schmitt, C.P.; Viesel, E.; Höfler, P.; Bauer, G. TGF-ß-Treated Normal Fibroblasts Eliminate Transformed Fibroblasts by Induction of Apoptosis. Cancer Res. 1994, 54, 393–398. [Google Scholar]
- Beck, E.; Schäfer, R.; Bauer, G. Sensitivity of Transformed Fibroblasts for Intercellular Induction of Apoptosis is Determined by their Transformed Phenotype. Exp. Cell Res. 1997, 234, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Krüger, H.; Bauer, G. Lactobacilli Enhance Reactive Oxygen Species-Dependent Apoptosis-Inducing Signaling. Redox Biol. 2017, 11, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Nakagawa, Y. Biological Significance of Phospholipid Hydroperoxide Glutathione Peroxidase (Phgpx, Gpx4) in Mammalian Cells. Free. Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef]
- Bauer, G. Autoamplificatory Singlet Oxygen Generation Sensitizes Tumor Cells for Intercellular Apoptosis-Inducing Signaling. Mech. Ageing Dev. 2018, 172, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Zucker, B.; Hanusch, J.; Bauer, G. Glutathione Depletion in Fibroblasts is the Basis for Induction of Apoptosis by Endogenous Reactive Oxygen Species. Cell Death Diff. 1997, 4, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Pigeolet, E.; Corbisier, P.; Houbion, A.; Lambert, D.; Michiels, C.; Raes, M.; Zachary, M.-D.; Remacle, J. Glutathione Peroxidase, Superoxide Dismutase, and Catalase Inactivation by Peroxides and Oxygen-Derived Free Radicals. Mech. Ageing Develop. 2020, 51, 283–297. [Google Scholar] [CrossRef]
- Gebicka, L.; Didik, J. Catalytic Scavenging of Peroxynitrite by Catalase. J. Inorg. Biochem. 2009, 103, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, L.; Yermilov, V.; Beckman, J.S. Modulation of Catalase Peroxidatic and Catalytic Activity by Nitric Oxide. Free Radic. Biol. Med. 2001, 30, 709–714. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical Biology of Nitric Oxide: Insights into Regulatory, Cytotoxic, and Cytoprotective Mechanisms of Nitric Oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, G. Inhibition of Membrane-Associated Catalase, Extracellular ROS/RNS Signaling and Aquaporin/H2O2-Mediated Intracellular Glutathione Depletion Cooperate during Apoptosis Induction in the Human Gastric Carcinoma Cell Line MKN-45. Antioxidants 2021, 10, 1585. https://doi.org/10.3390/antiox10101585
Bauer G. Inhibition of Membrane-Associated Catalase, Extracellular ROS/RNS Signaling and Aquaporin/H2O2-Mediated Intracellular Glutathione Depletion Cooperate during Apoptosis Induction in the Human Gastric Carcinoma Cell Line MKN-45. Antioxidants. 2021; 10(10):1585. https://doi.org/10.3390/antiox10101585
Chicago/Turabian StyleBauer, Georg. 2021. "Inhibition of Membrane-Associated Catalase, Extracellular ROS/RNS Signaling and Aquaporin/H2O2-Mediated Intracellular Glutathione Depletion Cooperate during Apoptosis Induction in the Human Gastric Carcinoma Cell Line MKN-45" Antioxidants 10, no. 10: 1585. https://doi.org/10.3390/antiox10101585
APA StyleBauer, G. (2021). Inhibition of Membrane-Associated Catalase, Extracellular ROS/RNS Signaling and Aquaporin/H2O2-Mediated Intracellular Glutathione Depletion Cooperate during Apoptosis Induction in the Human Gastric Carcinoma Cell Line MKN-45. Antioxidants, 10(10), 1585. https://doi.org/10.3390/antiox10101585