
Oxidative Stress in Optic Neuropathies

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Glaucoma
2.1. Oxidative Stress in Primary Glaucoma
2.1.1. Alterations in Antioxidant Defense Mechanisms in Primary Glaucoma
2.1.2. Overproduction of Reactive Oxygen and Nitrogen Species (RONS) in Primary Glaucoma
2.1.3. Oxidative Stress Markers in Primary Glaucoma
2.2. Oxidative Stress in Secondary Glaucoma
2.2.1. Oxidative Stress in Pseudoexfoliation Glaucoma
2.2.2. Oxidative Stress Associated with Ocular Surgery

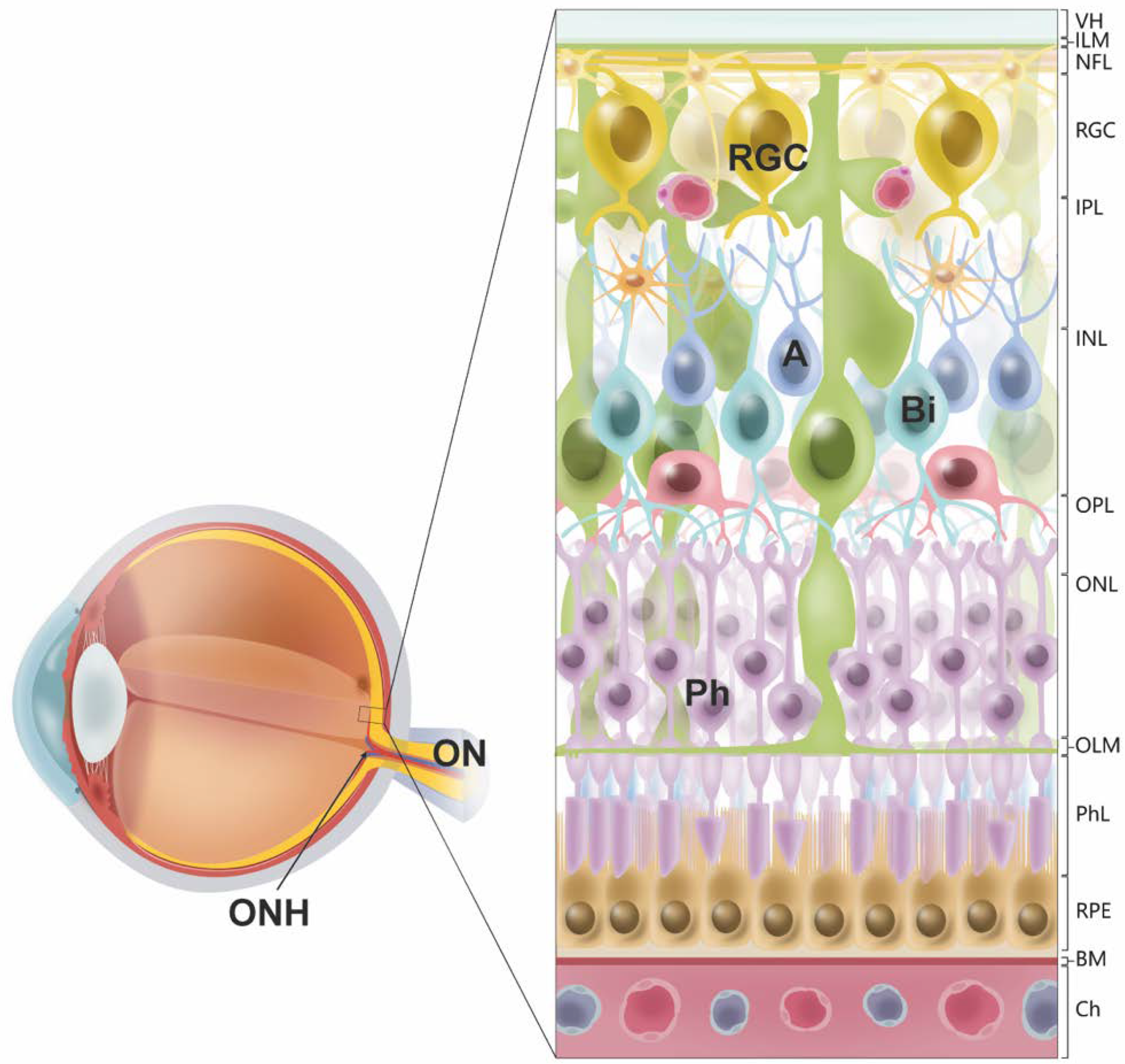
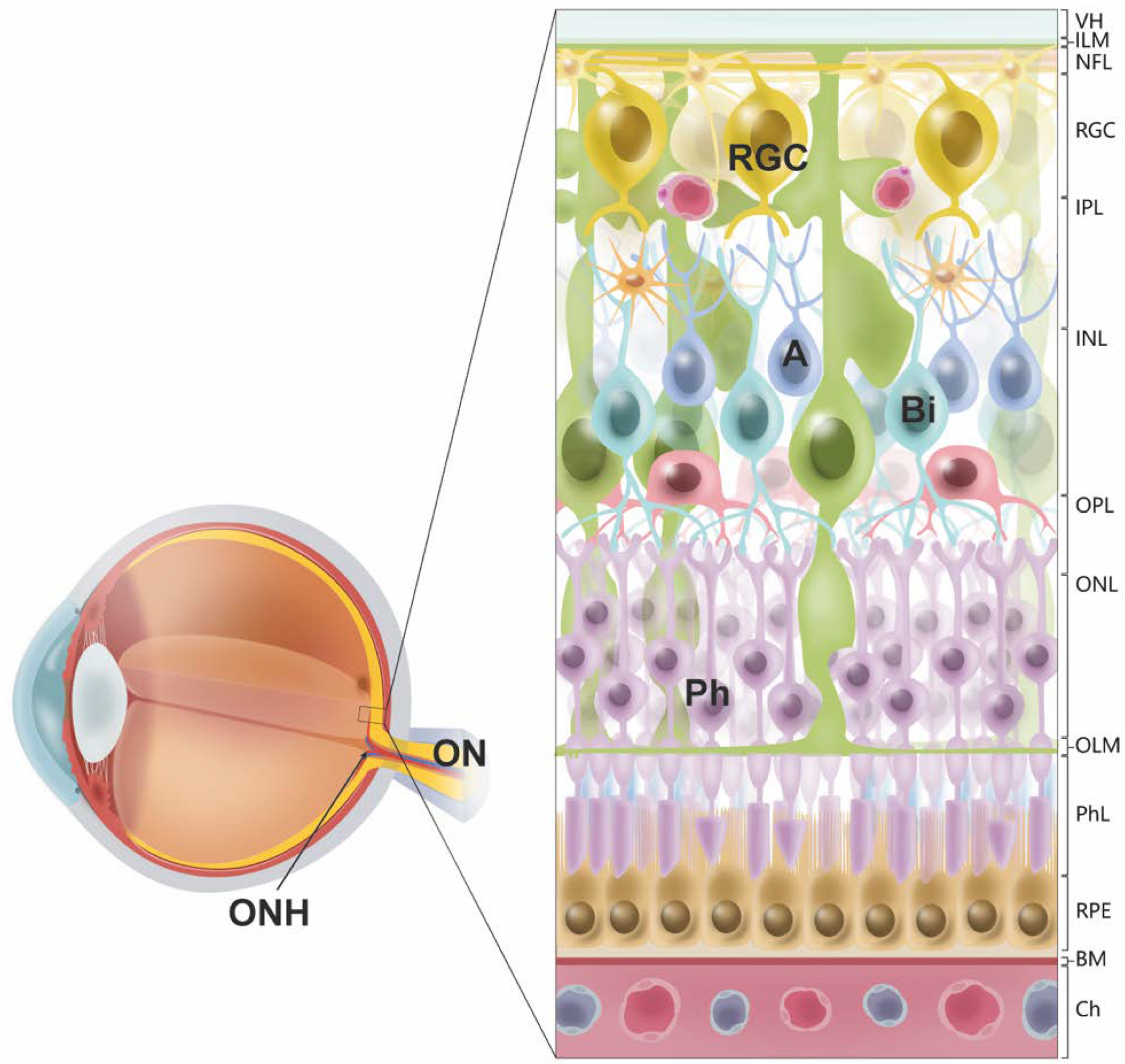{kind=link}
{kind=link}
| Type of Condition | Type of Sample | Outcome (Related to Control Group) * | Country | Authors |
|---|---|---|---|---|
| General antioxidant/oxidant status | ||||
| NAION | Plasma | No changes in TAS | Turkey | [134] |
| NAION | Plasma | No changes in TOS | Turkey | [134] |
| Optic neuritis | Blood | Positive correlation between the disulfide-to-native thiol ratio and P100 wave latency | Turkey | [135] |
| LHON (m.11778G>A, m.14484T>C, or m.3460G>A) | Plasma | Lower TAS | Serbia | [136] |
| LHON (m.11778G>A, m.14484T>C, or m.3460G>A) | Plasma | Higher TOS (only when comparing female LHON carriers against female controls) | Serbia | [136] |
| LHON (m.11778G>A, m.14484T>C, or m.3460G>A) | Plasma | Higher OSI | Serbia | [136] |
| LHON (m.11778G>A) | Peripheral blood cells | Higher cell death after incubation with 2-deoxy-d-ribose | Italy | [137] |
| ADOA | Lymphocytes | Increased susceptibility to oxidative stress and cell death after incubation with 2-deoxy-d-ribose | Italy | [138] |
| ADOA | Fibroblasts | Positive correlation between mitochondrial calcium uptake and cell death and symptom severity | Hungary | [139] |
| Antioxidant defense mechanisms | ||||
| Optic neuritis | Serum | Lower bilirubin levels | China | [140] |
| NMO | Serum | Lower bilirubin levels | USA | [141] |
| LHON (m.11778G>A) | Blood | Lower levels of α-tocopherol (vitamin E) | Hungary | [142] |
| LHON (m.11778G>A, m.14484T>C, or m.3460G>A) | Fibroblasts | Unaffected LHON carriers show higher expression of transcription factors and enzymes related to antioxidant pathways (compared to affected LHON carriers) * | Italy/Brazil | [143] |
| Toxic optic neuropathy (ethambutol) | Blood | Lower levels of SOD and catalase (especially in diabetic patients) | Saudi Arabia | [144] |
| Nutritional optic neuropathy | Blood | Decreased folate concentrations in subjects with optic neuropathy | Papua New Guinea | [145] |
| Nutritional optic neuropathy | Serum | Higher concentrations of thiamine (B12), riboflavin (B2), niacin (B3), and lycopene linked to a decreased risk of developing optic neuropathy | Cuba | [146] |
| Reactive oxygen and nitrogen species (RONS) and pro-oxidative enzymes | ||||
| NMO | Serum | Higher GGT levels | China | [147] |
| LHON (m.15927G>A) | Cybrid cell lines | Higher production of RONS | China | [148] |
| ADOA | Fibroblasts | Normal production of RONS | Italy | [149] |
| Oxidative stress markers | ||||
| NAION | Plasma | No changes in AOPP levels | Turkey | [134] |
| LHON (m.11778G>A, m.14484T>C, or m.3460G>A) | Plasma | Higher AOPP levels | Serbia | [136] |
| LHON (m.11778G>A, m.14484T>C, or m.3460G>A) | Fibroblast proteins | Increased S-glutathionylation | Singapore | [150] |
| LHON (m.11778G>A) | Leukocytes | Higher 8-OHdG levels | Taiwan | [151] |
| Genetic alterations related to oxidative stress | ||||
| NAION | Blood | Higher prevalence of loss-of-function deletion genotype in GSTM1 | Saudi Arabia | [152] |
| NAION | Blood | Higher prevalence of loss-of-function deletion genotype in GSTM1 | China | [153] |
| NAION | Blood | Higher levels of non-synonymous mutations in mtDNA and a higher content of relative mtDNA | USA | [154] |
| NAION | Leucocytes | Negative correlation between mtDNA relative content and visual acuity | Saudi Arabia | [155] |
| Optic neuritis | Blood | Higher prevalence of loss-of-function deletion genotype in GSTT1 | Saudi Arabia | [152] |
| ADOA | Fibroblasts | Increased depletion of mtDNA | UK | [156] |
3. Ischemic Optic Neuropathy
Oxidative Stress in Non-Arteritic Ischemic Optic Neuropathy
4. Optic Neuritis
Oxidative Stress in Optic Neuritis and NMO
5. Hereditary Optic Neuropathies
5.1. Leber’s Hereditary Optic Neuropathy (LHON)
Oxidative Stress in LHON
5.2. Autosomal Dominant Optic Atrophy (ADOA)
Oxidative Stress in ADOA
6. Optic Neuropathies Related to Environmental Health
6.1. Oxidative Stress in Toxic Optic Neuropathy
6.2. Oxidative Stress in Nutritional Optic Neuropathy
7. Optic Disc Drusen
Oxidative Stress in Optic Disc Drusen
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The Pathophysiology and Treatment of Glaucoma: A Review on the Topics Open-Angle Glaucoma and Angle-Closure Glaucoma. From the 4334 HHS Public Access. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Biousse, V.; Newman, N.J. Ischemic Optic Neuropathies. N. Engl. J. Med. 2015, 372, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.L. Optic Neuritis. Continuum 2019, 25, 1236–1264. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited Mitochondrial Optic Neuropathies. J. Med. Genet. 2009, 46, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.G.; Zimmerman, M.B. The Rate of Visual Field Loss in Optic Nerve Head Drusen. Am. J. Ophthalmol. 2005, 139, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Margolin, E.; Shemesh, A. Toxic and Nutritional Optic Neuropathy. Available online: https://pubmed.ncbi.nlm.nih.gov/29763154/ (accessed on 18 August 2021).
- Wichmann, W.; Müller-Forell, W. Anatomy of the Visual System. Eur. J. Radiol. 2004, 49, 8–30. [Google Scholar] [CrossRef]
- Schiefer, U.; Hart, W. Functional Anatomy of the Human Visual Pathway. In Clinical Neuro-Ophthalmology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 19–28. [Google Scholar]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O.L. Functional Architecture of the Retina: Development and Disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, G.D.; Fielder, A.R. Anatomy and Physiology of the Retina. In Pediatric Retina; Springer: Berlin/Heidelberg, Germany, 2011; pp. 39–65. [Google Scholar]
- Nguyen-Ba-Charvet, K.T.; Rebsam, A. Neurogenesis and Specification of Retinal Ganglion Cells. Int. J. Mol. Sci. 2020, 21, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Valença, V.M.; Bosco, A.; Vetter, M.L.; Silveira, M.S. On the Generation and Regeneration of Retinal Ganglion Cells. Front. Cell Dev. Biol. 2020, 8, 970. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative Stress and Mitochondrial Dysfunction in Glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Aslan, M.; Cort, A.; Yucel, I. Oxidative and Nitrative Stress Markers in Glaucoma. Free Radical Biol. Med. 2008, 45, 367–376. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Introductory Remarks. In Oxidative Stress; Academic Press: Cambidge, MA, USA, 1985; Volume 1. [Google Scholar]
- Sies, H. Oxidative Stress: Oxidants and Antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Halliwell, B. Free Radicals, Antioxidants, and Human Disease: Curiosity, Cause, or Consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Kaiser, C.A.; Amon, A.; Ploegh, H.; Bretscher, A.; Krieger, M.; Martin, K.C. Molecular Cell Biology, 8th ed.; Chegg: Santa Clara, CA, USA, 2016. [Google Scholar]
- Ji, L.L.; Yeo, D. Oxidative Stress: An Evolving Definition. Fac. Rev. 2021, 10, 13. [Google Scholar] [CrossRef]
- Burgoyne, C.; Buskirk, V. The Morphological Difference between Glaucoma and Other Optic Neuropathies the Optic Nerve Head (ONH) in Glaucoma. J. Neuroophthalmol. 2015, 35, S8–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Fingert, J.H.; Kuehn, M.H.; Alward, W.L.M. Primary Open-Angle Glaucoma. N. Engl. J. Med. 2009, 360, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, R.N.; Leung, C.K.S.; Crowston, J.G.; Medeiros, F.A.; Friedman, D.S.; Wiggs, J.L.; Martin, K.R. Primary Open-Angle Glaucoma. Nat. Rev. Dis. Prim. 2016, 2, 16067. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040 A Systematic Review and Meta-Analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Soh, Z.D.; Yu, M.; Betzler, B.K.; Majithia, S.; Thakur, S.; Tham, Y.C.; Wong, T.Y.; Aung, T.; Friedman, D.S.; Cheng, C.-Y. The Global Extent of Undetected Glaucoma in Adults: A Systematic Review and Meta-Analysis. Ophthalmology 2021, 128, 1393–1404. [Google Scholar] [CrossRef]
- Coleman, A.L.; Miglior, S. Risk Factors for Glaucoma Onset and Progression. Surv. Ophthalmol. 2008, 53, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Tielsch, J.M.; Katz, J.; Quigley, H.A.; Gottsch, J.D.; Javitt, J.; Singh, K. Relationship Between Intraocular Pressure and Primary Open Angle Glaucoma Among White and Black Americans: The Baltimore Eye Survey. Arch. Ophthalmol. 1991, 109, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.; Bengtsson, B.; Heijl, A. Lifetime Risk of Blindness in Open-Angle Glaucoma. Am. J. Ophthalmol. 2013, 156, 724–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Kook, M.S. Systemic and Ocular Hemodynamic Risk Factors in Glaucoma. BioMed Res. Int. 2015, 2015, 141905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, J.; Konieczka, K.; Bruno, R.M.; Virdis, A.; Flammer, A.J.; Taddei, S.; Ferrari, R. The Eye and the Heart. Eur. Heart J. 2009, 34, 1270–1278. [Google Scholar] [CrossRef]
- Resch, H.; Garhofer, G.; Fuchsjäger-Mayrl, G.; Hommer, A.; Schmetterer, L. Endothelial Dysfunction in Glaucoma. Acta Ophthalmol. 2009, 87, 4–12. [Google Scholar] [CrossRef]
- Pache, M.; Flammer, J. A Sick Eye in a Sick Body? Systemic Findings in Patients with Primary Open-Angle Glaucoma. Surv. Ophthalmol. 2006, 51, 179–212. [Google Scholar] [CrossRef]
- Evangelho, K.; Mogilevskaya, M.; Losada-Barragan, M.; Vargas-Sanchez, J.K. Pathophysiology of Primary Open-Angle Glaucoma from a Neuroinflammatory and Neurotoxicity Perspective: A Review of the Literature. Int. Ophthalmol. 2019, 39, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Vernazza, S.; Oddone, F.; Tirendi, S.; Bassi, A.M. Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention. Int. J. Mol. Sci. 2021, 22, 7994. [Google Scholar] [CrossRef] [PubMed]
- Opere, C.A.; Heruye, S.; Njie-Mbye, Y.-F.; Ohia, S.E.; Sharif, N.A. Regulation of Excitatory Amino Acid Transmission in the Retina: Studies on Neuroprotection. J. Ocul. Pharmacol. Ther. 2018, 34, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.Y.X.; van Bergen, N.J.; Trounce, I.A.; Crowston, J.G. Mitochondrial Dysfunction and Glaucoma. J. Glaucoma 2009, 18, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Toft-Kehler, A.K.; Skytt, D.M.; Svareb, A.; Lefeverec, E.; van Hovec, I.; Moonsc, L.; Waagepetersend, H.S.; Kolko, M. Mitochondrial Function in Müller Cells—Does It Matter? Mitochondrion 2017, 36, 43–51. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12, 699623. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A. Focus on Molecular Events in the Anterior Chamber Leading to Glaucoma. Cell. Mol. Life Sci. 2014, 71, 2197–2218. [Google Scholar] [CrossRef] [PubMed]
- Langbøl, M.; Saruhanian, S.; Baskaran, T.; Tiedemann, D.; Mouhammad, Z.A.; Toft-Kehler, A.K.; Jun, B.; Vohra, R.; Bazan, N.G.; Kolko, M. Increased Antioxidant Capacity and Pro-Homeostatic Lipid Mediators in Ocular Hypertension-A Human Experimental Model. J. Clin. Med. 2020, 9, 2979. [Google Scholar] [CrossRef]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell. Longev. 2017, 2017, 9208489. [Google Scholar] [CrossRef]
- Nucci, C.; di Pierro, D.; Varesi, C.; Ciuffoletti, E.; Russo, R.; Gentile, R.; Cedrone, C.; Duran, M.D.P.; Coletta, M.; Mancino, R. Increased Malondialdehyde Concentration and Reduced Total Antioxidant Capacity in Aqueous Humor and Blood Samples from Patients with Glaucoma. Mol. Vis. 2013, 19, 1841–1846. [Google Scholar]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A. Decreased Total Antioxidants in Patients with Primary Open Angle Glaucoma. Curr. Eye Res. 2013, 38, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Association between Systemic Oxidative Stress and Visual Field Damage in Open-Angle Glaucoma. Sci. Rep. 2016, 6, 25792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, Y.; Himori, N.; Kunikata, H.; Yamazaki, M.; Shiga, Y.; Omodaka, K.; Takahashi, H.; Nakazawa, T. Age- and Sex-Dependency of the Association between Systemic Antioxidant Potential and Glaucomatous Damage. Sci. Rep. 2017, 7, 8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A.O. Decreased Total Antioxidants Status in the Plasma of Patients with Pseudoexfoliation Glaucoma. Mol. Vis. 2011, 17, 2769–2775. [Google Scholar] [PubMed]
- Mousa, A.; Kondkar, A.A.; Al-Obeidan, S.A.; Azad, T.A.; Sultan, T.; Osman, E.; Abu-Amero, K.K. Association of Total Antioxidants Level with Glaucoma Type and Severity. Saudi Med. J. 2015, 36, 671–677. [Google Scholar] [CrossRef]
- Dursun, F.; Ozec, A.V.; Aydin, H.; Topalkara, A.; Dursun, A.; Toker, M.I.; Erdogan, H.; Arici, K.M. Total Oxidative Stress, Paraoxonase and Arylesterase Levels at Patients with Pseudoexfoliation Syndrome and Pseudoexfoliative Glaucoma. Int. J. Ophthalmol. 2015, 8, 985–990. [Google Scholar]
- Erdurmus, M.; Yagci, R.; Atıs, Ö.; Karadag, R.; Akbas, A.; Hepsen, İ.F. Antioxidant Status and Oxidative Stress in Primary Open Angle Glaucoma and Pseudoexfoliative Glaucoma. Curr. Eye Res. 2011, 36, 713–718. [Google Scholar] [CrossRef]
- Zanon-Moreno, V.; Garcia-Medina, J.J.; Gallego-Pinazo, R.; Vinuesa-Silva, I.; Moreno-Nadal, M.A.; Pinazo-Duran, M.D. Antioxidant Status Modifications by Topical Administration of Dorzolamide in Primary Open-Angle Glaucoma. Eur. J. Ophthalmol. 2009, 19, 565–571. [Google Scholar] [CrossRef]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative Stress Markers in Aqueous Humor of Glaucoma Patients. Am. J. Ophthalmol. 2004, 137, 62–69. [Google Scholar] [CrossRef]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Antioxidant Status in the Aqueous Humour of Patients with Glaucoma Associated with Exfoliation Syndrome. Eye 2009, 23, 1691–1697. [Google Scholar] [CrossRef] [Green Version]
- Ergan, E.; Ozturk, F.; Beyazyildiz, E.; Elgin, U.; Sen, E.; Cankaya, A.B.; Celik, T. Oxidant/Antioxidant Balance in the Aqueous Humor of Patients with Glaucoma. Int. J. Ophthalmol. 2016, 9, 249–252. [Google Scholar] [PubMed]
- Yabana, T.; Sato, K.; Shiga, Y.; Himori, N.; Omodaka, K.; Nakazawa, T. The Relationship between Glutathione Levels in Leukocytes and Ocular Clinical Parameters in Glaucoma. PLoS ONE 2019, 14, e0227078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherghel, D.; Griffiths, H.R.; Hilton, E.J.; Cunliffe, I.A.; Hosking, S.L. Systemic Reduction in Glutathione Levels Occurs in Patients with Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 877–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherghel, D.; Mroczkowska, S.; Qin, L. Reduction in Blood Glutathione Levels Occurs Similarly in Patients with Primary-Open Angle or Normal Tension Glaucoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3333–3339. [Google Scholar] [CrossRef]
- Yaz, Y.A.; Yıldırım, N.; Yaz, Y.; Tekin, N.; İnal, M.; Şahin, F.M. Role of Oxidative Stress in Pseudoexfoliation Syndrome and Pseudoexfoliation Glaucoma. Turk. J. Ophthalmol. 2019, 49, 61–67. [Google Scholar]
- Karakurt, Y.; Mertoglu, C.; Gok, G.; Ucak, T.; Tasli, N.; Erel, O.; Tasli, G. Thiol-Disulfide Homeostasis and Serum Ischemia Modified Albumin Levels in Patients with Primary Open-Angle Glaucoma. Curr. Eye Res. 2019, 44, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Canizales, L.; Rodriguez, L.; Rivera, C.; Martinez, A.; Mendez, F.; Castillo, A. Low-Level Expression of SOD1 in Peripheral Blood Samples of Patients Diagnosed with Primary Open-Angle Glaucoma. Biomark. Med. 2016, 10, 1218–1223. [Google Scholar] [CrossRef]
- Rokicki, W.; Zalejska-Fiolka, J.; Pojda-Wilczek, D.; Hampel, A.; Majewski, W.; Ogultekin, S.; Mrukwa-Kominek, E. Differences in Serum Oxidative Status between Glaucomatous and Nonglaucomatous Cataract Patients. BMC Ophthalmol. 2017, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Cetinkaya, E.; Duman, R.; Sabaner, M.C.; Erol, M.A.; Nural, C.; Erel, O. Evaluation of thiol-disulfide homeostasis in pseudoexfoliation glaucoma and primary open-angle glaucoma. Niger. J. Clin. Pract. 2020, 23, 1401–1406. [Google Scholar] [CrossRef]
- Koliakos, G.G.; Befani, C.D.; Mikropoulos, D.; Ziakas, N.G.; Konstas, A.G.P. Prooxidant-Antioxidant Balance, Peroxide and Catalase Activity in the Aqueous Humour and Serum of Patients with Exfoliation Syndrome or Exfoliative Glaucoma. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 1477–1483. [Google Scholar] [CrossRef]
- Ghanem, A.A.; Arafa, L.F.; El-Baz, A. Oxidative Stress Markers in Patients with Primary Open-Angle Glaucoma. Curr. Eye Res. 2010, 35, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Srivastava, A.; Sihota, R.; Kaur, J. Evaluation of Oxidative Stress Markers in Aqueous Humor of Primary Open Angle Glaucoma and Primary Angle Closure Glaucoma Patients. Curr. Eye Res. 2014, 39, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Gye, H.J.; Kim, J.M.; Yoo, C.; Shim, S.H.; Won, Y.S.; Sung, K.C.; Lee, M.Y.; Park, K.H. Relationship between High Serum Ferritin Level and Glaucoma in a South Korean Population: The Kangbuk Samsung Health Study. Ophthalmology 2016, 100, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Wang, S.Y.; Yoo, C.; Singh, K.; Lin, S.C. Association between Serum Ferritin and Glaucoma in the South Korean Population. JAMA Ophthalmol. 2014, 132, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Tsaia, D.-C.; Hsua, W.-M.; Chou, C.-K.; Chen, S.-J.; Peng, C.-H.; Chi, C.-W.; Ho, L.L.-T.; Liu, J.-H.; Chiou, S.-H. Significant Variation of the Elevated Nitric Oxide Levels in Aqueous Humor from Patients with Different Types of Glaucoma. Ophthalmologica 2002, 216, 346–350. [Google Scholar] [CrossRef]
- Fernandez-Durango, R.; Fernandez-Martinez, A.; Garcia-Feijoo, J.; Castillo, A.; de la Casa, J.M.; Garcia-Bueno, B.; Perez-Nievas, B.G.; Fernandez-Cruz, A.; Leza, J.C. Expression of Nitrotyrosine and Oxidative Consequences in the Trabecular Meshwork of Patients with Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2506–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondkar, A.A.; Azad, T.A.; Sultan, T.; Osman, E.A.; Almobarak, F.A.; Al-Obeidan, S.A. Elevated Plasma Level of 8-Hydroxy-2’-Deoxyguanosine Is Associated with Primary Open-Angle Glaucoma. J. Ophthalmol. 2020, 2020, 6571413. [Google Scholar] [CrossRef]
- Mohanty, K.; Dada, R.; Dada, T. Oxidative DNA Damage and Reduced Expression of DNA Repair Genes: Role in Primary Open Angle Glaucoma (POAG). Ophthalmic Genet. 2017, 38, 446–450. [Google Scholar] [CrossRef]
- Mumcu, U.Y.; Kocer, I.; Ates, O.; Alp, H.H. Decreased Paraoxonase1 Activity and Increased Malondialdehyde and Oxidative DNA Damage Levels in Primary Open Angle Glaucoma. Int. J. Ophthalmol. 2016, 9, 1518–1520. [Google Scholar]
- Kondkar, A.A.; Sultan, T.; Azad, T.A.; Tabussum, L.; Osman, E.A.; Al-Obeidan, S.A. Increased Plasma Levels of 8-Hydroxy-2’-Deoxyguanosine (8-OHdG) in Patients with Pseudoexfoliation Glaucoma. J. Ophthalmol. 2019, 2019, 8319563. [Google Scholar] [CrossRef]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Sacca, S.C. Mitochondrial Damage in the Trabecular Meshwork of Patients with Glaucoma. Arch. Ophthalmol. 2011, 128, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Saccà, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA Damage in the Human Trabecular Meshwork: Clinical Correlation in Patients with Primary Open-Angle Glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Umeno, A.; Tanito, M.; Kaidzu, S.; Takai, Y.; Horie, M.; Yoshida, Y. Comprehensive Measurements of Hydroxylinoleate and Hydroxyarachidonate Isomers in Blood Samples from Primary Open-Angle Glaucoma Patients and Controls. Sci. Rep. 2019, 9, 2171. [Google Scholar] [CrossRef]
- Zanon-Moreno, V.; Marco-Ventura, P.; Lleo-Perez, A.; Pons-Vazquez, S.; Garcia-Medina, J.J.; Vinuesa-Silva, I.; Moreno-Nadal, M.A.; Pinazo-Duran, M.D. Oxidative Stress in Primary Open-Angle Glaucoma. J. Glaucoma 2008, 17, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Feilchenfeld, Z.; Yücel, Y.H.; Gupta, N. Oxidative Injury to Blood Vessels and Glia of the Pre-Laminar Optic Nerve Head in Human Glaucoma. Exp. Eye Res. 2008, 87, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Lascaratos, G.; Chau, K.Y.; Zhu, H.; Gkotsi, D.; King, R.; Gout, I.; Kamal, D.; Luthert, P.J.; Schapira, A.H.V.; Garway-Heath, D.F. Resistance to the Most Common Optic Neuropathy Is Associated with Systemic Mitochondrial Efficiency. Neurobiol. Dis. 2015, 82, 78–85. [Google Scholar] [CrossRef]
- Jansen, E.H.J.M.; Ruskovska, T. Comparative Analysis of Serum (Anti)Oxidative Status Parameters in Healthy Persons. Int. J. Mol. Sci. 2013, 14, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Ruskovska, T.; Jansen, E.H.J.M.; Antarorov, R. Evaluation of Assays for Measurement of Serum (Anti)Oxidants in Hemodialysis Patients. BioMed Res. Int. 2014, 2014, 843157. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [Green Version]
- Michelet, F.; Gueguen, R.; Leroy, P.; Wellman, M.; Nicolas, A.; Siest, G. Blood and Plasma Glutathione Measured in Healthy Subjects by HPLC: Relation to Sex, Aging, Biological Variables, and Life Habits. Clin. Chem. 1995, 41, 1509–1517. [Google Scholar] [CrossRef]
- McCord, J.; Fridovich, I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Mills, G.C. Hemoglobin Catabolism: I. Glutathione Peroxidase, and Erythrocyte Enzyme Which Protects Hemoglobin from Oxidative Breakdown. J. Biol. Chem. 1957, 229, 189. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loew, O. A New Enzyme of General Occurrence in Organisms. Science 1900, 11, 701–702. [Google Scholar] [CrossRef] [Green Version]
- Lazarow, P.B.; de Duve, C. The Synthesis and Turnover of Rat Liver Peroxisomes: V. Intracellular Pathway of Catalase Synthesis. J. Cell Biol. 1973, 59, 507–524. [Google Scholar] [CrossRef]
- Middelkoop, E.; Wiemer, E.A.C.; Schoenmaker, D.E.T.; Strijland, A.; Tager, J.M. Topology of Catalase Assembly in Human Skin Fibroblasts. BBA Mol. Cell Res. 1993, 1220, 15–20. [Google Scholar] [CrossRef]
- Król, M.; Kepinska, M. Human Nitric Oxide Synthase-Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Reina-Torres, E.; de Ieso, M.L.; Pasquale, L.R.; Madekurozwaa, M.; van Batenburg-Sherwooda, J.; Overby, D.R.; Stamer, W.D. The Vital Role for Nitric Oxide in Intraocular Pressure Homeostasis. Prog. Retin. Eye Res. 2020, 83, 100922. [Google Scholar] [CrossRef]
- Schmetterer, L.; Polak, K. Role of Nitric Oxide in the Control of Ocular Blood Flow. Prog. Retin. Eye Res. 2001, 20, 823–847. [Google Scholar] [CrossRef]
- Orgül, S.; Gugleta, K.; Flammer, J. Physiology of Perfusion as It Relates to the Optic Nerve Head. Surv. Ophthalmol. 1999, 43 (Suppl. S1), S17–S26. [Google Scholar] [CrossRef]
- Salvatore, S.; Vingolo, E.M. Endothelin-1 Role in Human Eye: A Review. J. Ophthalmol. 2010, 2010, 354645. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Nitric Oxide and Cell Death. Biochim. Biophys. Acta Bioenerg. 1999, 1411, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Borutaite, V. Nitric Oxide, Mitochondria, and Cell Death. IUBMB Life 2001, 52, 189–195. [Google Scholar] [CrossRef]
- Ghanem, A.A.; Elewa, A.M.; Arafa, L.F. Endothelin-1 and Nitric Oxide Levels in Patients with Glaucoma. Ophthalmic Res. 2011, 46, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Polak, K.; Luksch, A.; Berisha, F.; Fuchsjaeger-Mayrl, G.; Dallinger, S.; Schmetterer, L. Altered Nitric Oxide System in Patients with Open-Angle Glaucoma. Arch. Ophthalmol. 2007, 125, 494–498. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, T.; Moriya, S.; Oku, H.; Azuma, I. Association of Endothelin-1 with Normal Tension Glaucoma: Clinical and Fundamental Studies. Surv. Ophthalmol. 1995, 39 (Suppl. S1), S49–S56. [Google Scholar] [CrossRef]
- Noske, W.; Hensen, J.; Wiederholt, M. Endothelin-like Immunoreactivity in Aqueous Humor of Patients with Primary Open-Angle Glaucoma and Cataract. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 551–552. [Google Scholar] [CrossRef]
- Cellini, M.; Strobbe, E.; Gizzi, C.; Balducci, N.; Toschi, P.G.; Campos, E.C. Endothelin-1 Plasma Levels and Vascular Endothelial Dysfunction in Primary Open Angle Glaucoma. Life Sci. 2012, 91, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Choritz, L.; Machert, M.; Thieme, H. Correlation of Endothelin-1 Concentration in Aqueous Humor with Intraocular Pressure in Primary Open Angle and Pseudoexfoliation Glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7336–7342. [Google Scholar] [CrossRef] [Green Version]
- Wareham, L.K.; Calkins, D.J. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R.; Miao, H.; Lukas, T. Astrocytes in Glaucomatous Optic Neuropathy. Prog. Brain Res. 2008, 173, 353–373. [Google Scholar] [PubMed]
- Garhöfer, G.; Zawinka, C.; Resch, H.; Huemer, K.H.; Schmetterer, L.; Dorner, G.T. Response of Retinal Vessel Diameters to Flicker Stimulation in Patients with Early Open Angle Glaucoma. J. Glaucoma 2004, 13, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Ford, G.; Harrison, P.; Rice, D.; Smith, J.; Treffry, A.; White, J.; Yariv, J. Ferritin: Design and Formation of an Iron-Storage Molecule. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1984, 304, 551–565. [Google Scholar] [PubMed]
- Lee, D.-H.; Zacharski, L.R.; Jacobs, D.R., Jr. Comparison of the Serum Ferritin and Percentage of Transferrin Saturation as Exposure Markers of Iron-Driven Oxidative Stress-Related Disease Outcomes. Am. Heart J. 2006, 151, 1247.e1–1247.e7. [Google Scholar] [CrossRef] [PubMed]
- Hori, A.; Mizoue, T.; Kasai, H.; Kawai, K.; Matsushita, Y.; Nanri, A.; Sato, M.; Ohta, M. Body Iron Store as a Predictor of Oxidative DNA Damage in Healthy Men and Women. Cancer Sci. 2010, 101, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Singh, K.; Lin, S.C. The Association between Glaucoma Prevalence and Supplementation with the Oxidants Calcium and Iron. Investig. Ophthalmol. Vis. Sci. 2012, 53, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Iyama, T.; Wilson, D.M. DNA Repair Mechanisms in Dividing and Non-Dividing Cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Cuchra, M.; Markiewicz, L.; Mucha, B.; Pytel, D.; Szymanek, K.; Szemraj, J.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. The Role of Base Excision Repair in the Development of Primary Open Angle Glaucoma in the Polish Population. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2015, 778, 26–40. [Google Scholar] [CrossRef]
- Gaweł, S.; Wardas, M.; Niedworok, E.; Wardas, P. Malondialdehyde (MDA) as a Lipid Peroxidation Marker. Wiad. Lek. 2004, 57, 453–455. [Google Scholar]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-Mediated Tyrosine Nitration Catalyzed by Superoxide Dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef]
- Ozaki, M. Mechanisms of Glaucoma in Exfoliation Syndrome. J. Glaucoma 2018, 27, S83–S86. [Google Scholar] [CrossRef] [PubMed]
- Ritch, R.; Schlötzer-Schrehardt, U.; Konstas, A.G.P. Why Is Glaucoma Associated with Exfoliation Syndrome? Prog. Retin. Eye Res. 2003, 22, 253–257. [Google Scholar] [CrossRef]
- Ritch, R. Ocular and Systemic Manifestations of Exfoliation Syndrome. J. Glaucoma 2014, 23, S1–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxidative Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumurcu, T.; Gunduz, A.; Ozyurt, H.; Nurcin, H.; Atis, O.; Egri, M. Increased oxidative stress in patients with pseudoexfoliation syndrome. Ophthalmic Res. 2010, 43, 169–172. [Google Scholar] [CrossRef]
- Song, Y.; Ding, W.; Bei, Y.; Xiao, Y.; da Tong, H.; Wang, L.B.; Ai, L.Y. Insulin Is a Potential Antioxidant for Diabetes-Associated Cognitive Decline via Regulating Nrf2 Dependent Antioxidant Enzymes. Biomed. Pharmacother. 2018, 104, 474–484. [Google Scholar] [CrossRef]
- Ahoor, M.H.; Ghorbanihaghjo, A.; Sorkhabi, R.; Kiavar, A. Klotho and Endothelin-1 in Pseudoexfoliation Syndrome and Glaucoma. J. Glaucoma 2016, 25, 919–922. [Google Scholar] [CrossRef]
- Koukoula, S.C.; Katsanos, A.; Tentes, I.K.; Labiris, G.; Kozobolis, V.P. Retrobulbar Hemodynamics and Aqueous Humor Levels of Endothelin-1 in Exfoliation Syndrome and Exfoliation Glaucoma. Clin. Ophthalmol. 2018, 12, 1199–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koliakos, G.G.; Konstas, A.G.; Schlötzer-Schrehardt, U.; Hollo, G.; Mitova, D.; Kovatchev, D.; Maloutas, S.; Georgiadis, N. Endothelin-1 Concentration Is Increased in the Aqueous Humour of Patients with Exfoliation Syndrome. Br. J. Ophthalmol. 2004, 88, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Chang, S. LXII Edward Jackson Lecture: Open Angle Glaucoma After Vitrectomy. Am. J. Ophthalmol. 2006, 141, 1033–1043.e1. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, N.; Kimura, H.; Matsumura, M.; Kuroda, S. Incidence of Late-Onset Ocular Hypertension Following Uncomplicated Pars Plana Vitrectomy in Pseudophakic Eyes. Am. J. Ophthalmol. 2015, 159, 727–732. [Google Scholar] [CrossRef]
- Siegfried, C.J.; Shui, Y.B.; Tian, B.; Nork, T.M.; Heatley, G.A.; Kaufman, P.L. Effects of Vitrectomy and Lensectomy on Older Rhesus Macaques: Oxygen Distribution, Antioxidant Status, and Aqueous Humor Dynamics. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4003–4014. [Google Scholar] [CrossRef] [Green Version]
- Siegfried, C.J.; Shui, Y.-B. Intraocular Oxygen and Antioxidant Status: New Insights on the Effect of Vitrectomy and Glaucoma Pathogenesis. Am. J. Ophthalmol. 2019, 203, 12–25. [Google Scholar] [CrossRef] [Green Version]
- Richer, S.P.; Rose, R.C. Water Soluble Antioxidants in Mammalian Aqueous Humor: Interaction with UV B and Hydrogen Peroxide. Vis. Res. 1998, 38, 2881–2888. [Google Scholar] [CrossRef] [Green Version]
- Birer, S.; Arda, H.; Kilic, D.; Baskol, G. Systemic Oxidative Stress in Non-Arteritic Anterior Ischemic Optic Neuropathy. Eye 2019, 33, 1140–1144. [Google Scholar] [CrossRef]
- Vural, G.; Gümüşyayla, Ş.; Deniz, O.; Neşelioğlu, S.; Erel, Ö. Relationship between Thiol-Disulphide Homeostasis and Visual Evoked Potentials in Patients with Multiple Sclerosis. Neurol. Sci. 2019, 40, 385–391. [Google Scholar] [CrossRef]
- Rovcanin, B.; Jancic, J.; Pajic, J.; Rovcanin, M.; Samardzic, J.; Djuric, V.; Nikolic, B.; Ivancevic, N.; Novakovic, I.; Kostic, V. Oxidative Stress Profile in Genetically Confirmed Cases of Leber’s Hereditary Optic Neuropathy. J. Mol. Neurosci. 2021, 71, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Battisti, C.; Formichi, P.; Cardaioli, E.; Bianchi, S.; Mangiavacchi, P.; Tripodi, S.A.; Tosi, P.; Federico, A. Cell Response to Oxidative Stress Induced Apoptosis in Patients with Leber’s Hereditary Optic Neuropathy. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1731–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formichi, P.; Radi, E.; Giorgi, E.; Gallus, G.N.; Brunetti, J.; Battisti, C.; Rufa, A.; Dotti, M.T.; Franceschini, R.; Bracci, L.; et al. Analysis of Opa1 Isoforms Expression and Apoptosis Regulation in Autosomal Dominant Optic Atrophy (ADOA) Patients with Mutations in the Opa1 Gene. J. Neurol. Sci. 2015, 351, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, L.; Rajki, A.; Maka, E.; Molnár, M.J.; Spät, A. Mitochondrial Ca2+ Uptake Correlates with the Severity of the Symptoms in Autosomal Dominant Optic Atrophy. Cell Calcium 2015, 57, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Liang, X.-M.; Zhang, X.-L.; Ling, S.; Yang, T.; Li, M.; Peng, F. Relationship between Serum Bilirubin Levels and Optic Neuritis. Chin. Med. J. 2013, 126, 3307–3310. [Google Scholar] [PubMed]
- Peng, F.; Yang, Y.; Liu, J.; Jiang, Y.; Zhu, C.; Deng, X.; Hu, X.; Chen, X.; Zhong, X. Low Antioxidant Status of Serum Uric Acid, Bilirubin and Albumin in Patients with Neuromyelitis Optica. Eur. J. Neurol. 2012, 19, 277–283. [Google Scholar] [CrossRef]
- Klivenyi, P.; Karg, E.; Rozsa, C.; Horvath, R.; Komoly, S.; Nemeth, I.; Turi, S.; Vecsei, L. α-Tocopherol/Lipid Ratio in Blood Is Decreased in Patients with Leber’s Hereditary Optic Neuropathy and Asymptomatic Carriers of the 11778 MtDNA Mutation. J. Neurol. Neurosurg. Psychiatry 2001, 70, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Giordano, L.; Deceglie, S.; D’adamo, P.; Valentino, M.L.; Morgia, L.; Fracasso, F.; Roberti, M.; Cappellari, M.; Petrosillo, G.; Ciaravolo, S.; et al. Cigarette Toxicity Triggers Leber’s Hereditary Optic Neuropathy by Affecting MtDNA Copy Number, Oxidative Phosphorylation and ROS Detoxification Pathways. Cell Death Dis. 2015, 6, e2021. [Google Scholar] [CrossRef] [Green Version]
- Rasool, M.; Malik, A.; Manan, A.; Aziz, K.; Mahmood, A.; Zaheer, S.; Shuja, N.; Qazi, M.H.; Kamal, M.A.; Karim, S. Determination of Potential Role of Antioxidative Status and Circulating Biochemical Markers in the Pathogenesis of Ethambutol Induced Toxic Optic Neuropathy among Diabetic and Non-Diabetic Patients. Saudi J. Biol. Sci. 2015, 22, 739–743. [Google Scholar] [CrossRef] [Green Version]
- Tousignant, B.; Brian, G.; Venn, B.J.; Gould, C.; McKay, R.; Williams, S. Optic Neuropathy among a Prison Population in Papua New Guinea. Ophthalmic Epidemiol. 2013, 20, 4–12. [Google Scholar] [CrossRef]
- The Cuba Neuropathy Field Investigation Team. Epidemic Optic Neuropathy in Cuba—Clinical Characterization and Risk Factors. N. Engl. J. Med. 1995, 333, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Shua, Y.; Lia, R.; Qiua, W.; Changa, Y.; Suna, X.; Fanga, L.; Chena, C.; Yanga, Y.; Lua, Z.; Hua, X.; et al. Association of Serum Gamma-Glutamyltransferase and C-Reactive Proteins with Neuromyelitis Optica and Multiple Sclerosis. Mult. Scler. Relat. Disord. 2017, 18, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jic, Y.; Liua, X.; Chenb, J.; Wangb, B.; Zhange, M.; Guan, M.-X. Leber’s Hereditary Optic Neuropathy Caused by a Mutation in Mitochondrial TRNA Thr in Eight Chinese Pedigrees. Mitochondrion 2018, 42, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Cazzola, S.; Bortolozzi, M.; Baracca, A.; Loro, E.; Casarin, A.; Solaini, G.; Sgarbi, G.; Casalena, G.; Cenacchi, G.; et al. A Novel Deletion in the GTPase Domain of OPA1 Causes Defects in Mitochondrial Morphology and Distribution, but Not in Function. Hum. Mol. Genet. 2008, 17, 3291–3302. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Chun, J.; Chan, Y.; Chupin, S.; Gueguen, N.; Desquiret-Dumas, V.; Koh, S.K.; Li, J.; Gao, Y.; Deng, L.; et al. Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON). Int. J. Mol. Sci. 2020, 21, 3027. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.Y.; Kao, S.H.; Wang, A.G.; Wei, Y.H. Increased 8-Hydroxy-2′-Deoxyguanosine in Leukocyte DNA in Leber’s Hereditary Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1688–1691. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Milcarek, B.; Bosley, T.M. GSTM1 and GSTT1 Deletion Genotypes in Various Spontaneous Optic Neuropathies in Arabs. Br. J. Ophthalmol. 2009, 93, 1101–1104. [Google Scholar] [CrossRef]
- Wan, W.; Peng, T.; Jin, X.; Li, Q.; Zhang, F.; Zheng, G.; Lv, Y.; Wan, G.; Zhu, Y. Glutathione-S-Transferase Deletions and Non-Arteritic Anterior Ischemic Optic Neuropathy. Mol. Neurobiol. 2016, 53, 2361–2367. [Google Scholar] [CrossRef]
- Bosley, T.M.; Abu-Amero, K.K.; Ozand, P.T. Mitochondrial DNA Nucleotide Changes in Non-Arteritic Ischemic Optic Neuropathy. Neurology 2004, 63, 1305–1308. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Bosley, T.M. Increased Relative Mitochondrial DNA Content in Leucocytes of Patients with NAION. Br. J. Ophthalmol. 2006, 90, 823–825. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated Mitophagy and Mitochondrial Organization in Optic Atrophy Due to OPA1 Mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, A.C. Pathogenesis of Nonarteritic Anterior Ischemic Optic Neuropathy. J. Neuro-Ophthalmol. 2003, 23, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Hayreh, S.S. Ischemic Optic Neuropathies—Where Are We Now? Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 1873–1884. [Google Scholar] [CrossRef]
- Hattenhauer, M.G.; Leavitt, J.A.; Hodge, D.O.; Grill, R.; Gray, D.T. Incidence of Nonarteritic Anteripr Ischemic Optic Neuropathy. Am. J. Ophthalmol. 1997, 123, 103–107. [Google Scholar] [CrossRef]
- Preechawat, P.; Bruce, B.B.; Newman, N.J.; Biousse, V. Anterior Ischemic Optic Neuropathy in Patients Younger than 50 Years. Am. J. Ophthalmol. 2007, 144, 953–960. [Google Scholar] [CrossRef]
- Newman, N.J.; Dickersin, K.; Kaufman, D.; Kelman, S.; Scherer, R.; Kennerdell, J.; Tyutyunikov, A.; Edwards, R.; Goodglick, T.; Lang, D.; et al. Characteristics of Patients with Nonarteritic Anterior Ischemic Optic Neuropathy Eligible for the Ischemic Optic Neuropathy Decompression Trial. Arch. Ophthalmol. 1996, 114, 1366–1374. [Google Scholar]
- Doro, S.; Lessell, S. Cup-Disc Ratio and Ischemic Optic Neuropathy. Arch. Ophthalmol. 1985, 103, 1143–1144. [Google Scholar] [CrossRef]
- Beck, R.W.; Servais, G.E.; Hayreh, S.S. Anterior Ischemic Optic Neuropathy: IX. Cup-to-Disc Ratio and Its Role in Pathogenesis. Ophthalmology 1987, 94, 1503–1508. [Google Scholar] [CrossRef]
- Burde, R.M. Optic Disk Risk Factors for Nonarteritic Anterior Ischemic Optic Neuropathy. Am. J. Ophthalmol. 1993, 116, 759–764. [Google Scholar] [CrossRef]
- Hamann, S.; Malmqvist, L.; Wegener, M.; Fard, M.A.; Biousse, V.; Bursztyn, L.; Citirak, G.; Costello, F.; Crum, A.V.; Digre, K.; et al. Young Adults with Anterior Ischemic Optic Neuropathy: A Multicenter Optic Disc Drusen Study. Am. J. Ophthalmol. 2020, 217, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Rueløkke, L.L.; Malmqvist, L.; Hamann, S. Prevalence of Optic Disc Drusen in Young Patients with Nonarteritic Anterior Ischemic Optic Neuropathy: A 10-Year Retrospective Study. J. Neuroophthalmol. 2021, 41, 200–205. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, T.D. Special Article the Glutathione S-transferases: An Update. Hepatology 1989, 9, 486–496. [Google Scholar] [CrossRef]
- Lee, H.C.; Lu, C.Y.; Fahn, H.J.; Wei, Y.H. Aging- and Smoking-Associated Alteration in the Relative Content of Mitochondrial DNA in Human Lung. FEBS Lett. 1998, 441, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Wei, Y.H. Mitochondrial Biogenesis and Mitochondrial DNA Maintenance of Mammalian Cells under Oxidative Stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Rader, J.; Feuer, W.J.; Anderson, D.R. Peripapillary Vasoconstriction in the Glaucomas and the Anterior Ischemic Optic Neuropathies. Am. J. Ophthalmol. 1994, 117, 72–80. [Google Scholar] [CrossRef]
- Hoorbakht, H.; Bagherkashi, F. Optic Neuritis, Its Differential Diagnosis and Management. Open Ophthalmol. J. 2012, 6, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Han, J.; Yang, M.; Oh, S.Y. Population-Based Incidence of Pediatric and Adult Optic Neuritis and the Risk of Multiple Sclerosis. Ophthalmology 2020, 127, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Braithwaite, T.; Subramanian, A.; Petzold, A.; Galloway, J.; Adderley, N.J.; Mollan, S.P.; Plant, G.T.; Nirantharakumar, K.; Denniston, A.K. Trends in Optic Neuritis Incidence and Prevalence in the UK and Association with Systemic and Neurologic Disease. JAMA Neurol. 2020, 77, 1514. [Google Scholar] [CrossRef]
- Zeid, N.A.; Bhatti, M.T. Acute Inflammatory Demyelinating Optic Neuritis: Evidence-Based Visual and Neurological Considerations. Neurologist 2008, 14, 207–223. [Google Scholar] [CrossRef]
- Pau, D.; al Zubidi, N.; Yalamanchili, S.; Plant, G.T.; Lee, A.G. Optic Neuritis. Eye 2011, 25, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative Stress in Multiple Sclerosis: Central and Peripheral Mode of Action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Ibitoye, R.; Kemp, K.; Rice, C.; Hares, K.; Scolding, N.; Wilkins, A. Oxidative Stress-Related Biomarkers in Multiple Sclerosis: A Review. Biomark. Med. 2016, 10, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobore, T.O. Oxidative/Nitroxidative Stress and Multiple Sclerosis. J. Mol. Neurosci. 2020, 71, 506–514. [Google Scholar] [CrossRef]
- Glass, G.A.; Stark, A.-A. Promotion of Glutathione-g-Glutamyl Transpeptidase-Dependent Lipid Peroxidation by Copper and Ceruloplasmin: The Requirement for Iron and the Effects of Antioxidants and Antioxidant Enzymes. Environ. Mol. Mutagenesis 1997, 29, 73–80. [Google Scholar] [CrossRef]
- Stark, A.A.; Russell, J.J.; Langenbach, R.; Pagano, D.A.; Zeiger, E.; Huberman, E. Localization of Oxidative Damage by a Glutathione-γ-Glutamyl Transpeptidase System in Preneoplastic Lesions in Sections of Livers from Carcinogen-Treated Rats. Carcinogenesis 1994, 15, 343–348. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin Is an Antioxidant of Possible Physiological Importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wong, Y.H.; Wu, K.; ZhangBao, J.; Zhou, L.; Zong, Y.; Zhou, X.; Quan, C.; Wang, M. Alterations in the Retinal Vascular Network and Structure in Myelin Oligodendrocyte Glycoprotein Antibody-Associated Optic Neuritis: A Longitudinal OCTA Study. Ocul. Immunol. Inflamm. 2021, 1–5. [Google Scholar] [CrossRef]
- Chen, T.-C.; Yeh, C.-Y.; Lin, C.-W.; Yang, C.-M.; Yang, C.-H.; Lin, I.-H.; Chen, P.-Y.; Cheng, J.-Y.; Hu, F.-R. Vascular Hypoperfusion in Acute Optic Neuritis Is a Potentially New Neurovascular Model for Demyelinating Diseases. PLoS ONE 2017, 12, e0184927. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.-I.; Park, K.-A.; Oh, S.Y.; Min, J.-H.; Kim, B.J. Differential Patterns of Parafoveal and Peripapillary Vessel Density in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder. Mult. Scler. Relat. Disord. 2021, 49, 102780. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of Aquaporin 4 in Lesions of Neuromyelitis Optica: Distinction from Multiple Sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Parratt, J.D.E.; Prineas, J.W. Neuromyelitis Optica: A Demyelinating Disease Characterized by Acute Destruction and Regeneration of Perivascular Astrocytes. Mult. Sceloris J. 2010, 16, 1156–1172. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puomila, A.; Hämäläinen, P.; Kivioja, S.; Savontaus, M.-L.L.; Koivumäki, S.; Huoponen, K.; Nikoskelainen, E. Epidemiology and Penetrance of Leber Hereditary Optic Neuropathy in Finland. Eur. J. Human Genet. 2007, 15, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J. Treatment of Hereditary Optic Neuropathies. Nat. Rev. Neurol. 2012, 8, 545–556. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F. Treatment Strategies for Inherited Optic Neuropathies: Past, Present and Future. Eye 2014, 28, 521–537. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, L.; Kolbach, D.N.; de Coo, R.F.; Plomp, A.S.; Bauer, N.J.; Smeets, H.J.; de Die-Smulders, C.E.M. Influence of Mutation Type on Clinical Expression of Leber Hereditary Optic Neuropathy. Am. J. Ophthalmol. 2006, 141, 676–676.e8. [Google Scholar] [CrossRef]
- Howell, N.; Bindoff, L.A.; McCullough, D.A.; Kubacka, I.; Poulton, J.; Mackey, D.; Taylor, L.; Turnbull, D.M. Leber Hereditary Optic Neuropathy: Identification of the Same Mitochondrial NDI Mutation in Six Pedigrees. Am. J. Hum. Genet. 1991, 49, 939–950. [Google Scholar]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.S.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA Mutation Associated with Leber’s Hereditary Optic Neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Kirkman, M.A.; Yu-Wai-Man, P.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; de Coo, I.F.; Klopstock, T.; Chinnery, P.F. Gene-Environment Interactions in Leber Hereditary Optic Neuropathy. Brain 2009, 132, 2317–2326. [Google Scholar] [CrossRef]
- Pisano, A.; Preziuso, C.; Iommarini, L.; Perli, E.; Grazioli, P.; Campese, A.F.; Maresca, A.; Montopoli, M.; Masuelli, L.; Sadun, A.A.; et al. Targeting Estrogen Receptor β as Preventive Therapeutic Strategy for Leber’s Hereditary Optic Neuropathy. Hum. Mol. Genet. 2015, 24, 6921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balducci, N.; Cascavilla, M.L.; Ciardella, A.; la Morgia, C.; Triolo, G.; Parisi, V.; Bandello, F.; Sadun, A.A.; Carelli, V.; Barboni, P. Peripapillary Vessel Density Changes in Leber’s Hereditary Optic Neuropathy: A New Biomarker. Clin. Experiment. Ophthalmol. 2018, 46, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Hoyt, W.F.; Susac, J.O. Ocular Fundus in Acute Leber Optic Neuropathy. Arch. Ophthalmol. 1973, 90, 349–354. [Google Scholar] [CrossRef]
- Kızıltunç, P.B.; Bilen, F.T.; Atilla, H. Optical Coherence Tomography Angiography Findings in Long-Term Follow-up of Leber’s Hereditary Optic Neuropathy: Report of Two Cases. Turk. J. Ophthalmol. 2020, 50, 313–316. [Google Scholar] [CrossRef]
- Asanad, S.; Meer, E.; Tian, J.J.; Fantini, M.; Nassisi, M.; Sadun, A.A. Leber’s Hereditary Optic Neuropathy: Severe Vascular Pathology in a Severe Primary Mutation. Intractable Rare Dis. Res. 2019, 8, 52–55. [Google Scholar] [CrossRef] [Green Version]
- Nikoskelainen, E.; Hoyt, W.F.; Nummelin, K.; Schatz, H. Fundus Findings in Leber’s Hereditary Optic Neuroretinopathy: III. Fluorescein Angiographic Studies. Arch. Ophthalmol. 1984, 102, 981–989. [Google Scholar] [CrossRef]
- Yang, T.-C.; Yarmishyn, A.A.; Yang, Y.-P.; Lu, P.-C.; Chou, S.-J.; Wang, M.-L.; Lin, T.-C.; Hwang, D.-K.; Chou, Y.-B.; Chen, S.-J.; et al. Mitochondrial Transport Mediates Survival of Retinal Ganglion Cells in Affected LHON Patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef]
- Ghezzi, P. Protein Glutathionylation in Health and Disease. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- Qian, Y.; Zhou, X.; Liang, M.; Qu, J.; Guan, M.-X. The Altered Activity of Complex III May Contribute to the High Penetrance of Leber’s Hereditary Optic Neuropathy in a Chinese Family Carrying the ND4 G11778A Mutation. Mitochondrion 2011, 11, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Puomila, A.; Huoponen, K.; Mäntyjärvi, M.; Hämäläinen, P.; Paananen, R.; Sankila, E.-M.; Savontaus, M.-L.; Somer, M.; Nikoskelainen, E. Dominant Optic Atrophy: Correlation between Clinical and Molecular Genetic Studies. Acta Ophthalmol. Scand. 2005, 83, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Eiberg, H.; Kjer, B.; Kjer, P.; Rosenberg, T. Dominant Optic Atrophy (OPA1) Mapped to Chromosome 3q Region. I. Linkage Analysis. Hum. Mol. Genet. 1994, 3, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Burke, A.; Sellar, P.W.; Clarke, M.P.; Gnanaraj, L.; Ah-Kine, D.; Hudson, G.; Czermin, B.; Taylor, R.W.; et al. The Prevalence and Natural History of Dominant Optic Atrophy Due to OPA1 Mutations. Ophthalmology 2010, 117, 1538–1546.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjer, B.; Eiberg, H.; Kjer, P.; Rosenberg, T. Dominant Optic Atrophy Mapped to Chromosome 3q Region. Acta Ophthalmol. Scand. 1996, 74, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.E.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.M.; Patel, N.; et al. Spectrum, Frequency and Penetrance of OPA1 Mutations in Dominant Optic Atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Liang, X.; Lu, Y.; Zhu, L.; Fu, R.; Ji, Y.; Fan, W.; Chen, J.; Lin, B.; et al. A Novel ADOA-Associated OPA1 Mutation Alters the Mitochondrial Function, Membrane Potential, ROS Production and Apoptosis OPEN. Sci. Rep. 2017, 18, 5704. [Google Scholar] [CrossRef]
- Kao, S.H.; Yen, M.Y.; Wang, A.G.; Yeh, Y.L.; Lin, A.L. Changes in Mitochondrial Morphology and Bioenergetics in Human Lymphoblastoid Cells with Four Novel Opa1 Mutations. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 Links Human Mitochondrial Genome Maintenance to MtDNA Replication and Distribution. Genome Res. 2011, 21, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Ferré, M.; Caignard, A.; Milea, D.; Leruez, S.; Cassereau, J.; Chevrollier, A.; Amati-Bonneau, P.; Verny, C.; Bonneau, D.; Procaccio, V.; et al. Improved Locus-Specific Database for OPA1 Mutations Allows Inclusion of Advanced Clinical Data. Hum. Mutat. 2015, 36, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Eiberg, H.; Hansen, L.; Kjer, B.; Hansen, T.; Pedersen, O.; Bille, M.; Rosenberg, T.; Tranebjærg, L. Autosomal Dominant Optic Atrophy Associated with Hearing Impairment and Impaired Glucose Regulation Caused by a Missense Mutation in the WFS1 Gene. J. Med. Genet. 2006, 43, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 Causes Dominant Optic Atrophy with External Ophthalmoplegia, Ataxia, Deafness and Multiple Mitochondrial DNA Deletions: A Novel Disorder of MtDNA Maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-System Neurological Disease Is Common in Patients with OPA1 Mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Agier, V.; Oliviero, P.; Lainé, J.; L’Hermitte-Stead, C.; Girard, S.; Fillaut, S.; Jardel, C.; Bouillaud, F.; Bulteau, A.L.; Lombès, A. Defective Mitochondrial Fusion, Altered Respiratory Function, and Distorted Cristae Structure in Skin Fibroblasts with Heterozygous OPA1 Mutations. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1570–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Country, M.W. Retinal Metabolism: A Comparative Look at Energetics in the Retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Prokosch, V. Energy Metabolism in the Inner Retina in Health and Glaucoma. Int. J. Mol. Sci. 2021, 22, 3689. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.A.; di Polo, A. Mitochondrial Dynamics, Transport, and Quality Control: A Bottleneck for Retinal Ganglion Cell Viability in Optic Neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Balducci, N.; Ciardella, A.; Gattegna, R.; Zhou, Q.; Cascavilla, M.L.; la Morgia, C.; Savini, G.; Parisi, V.; Bandello, F.; Carelli, V.; et al. Optical Coherence Tomography Angiography of the Peripapillary Retina and Optic Nerve Head in Dominant Optic Atrophy. Mitochondrion 2017, 36, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Cesareo, M.; Giannini, C.; di Marino, M.; Aloe, G.; Martucci, A.; Aiello, F.; Cusumano, A.; Mancino, R.; Ricci, F.; Sorge, R.P.; et al. Optical Coherence Tomography Angiography in the Multimodal Assessment of the Retinal Posterior Pole in Autosomal Dominant Optic Atrophy. Acta Ophthalmol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Rodrigues, T.M.; Soares, M.; Dolan, M.-J.; Murta, J.N.; Silva, R.; Marques, J.P. Peripapillary and Macular Morpho-Vascular Changes in Patients with Genetic or Clinical Diagnosis of Autosomal Dominant Optic Atrophy: A Case-Control Study. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 1019–1027. [Google Scholar] [CrossRef]
- Zhang, A.-M.; Bi, R.; Hu, Q.-X.; Fan, Y.; Zhang, Q.; Yao, Y.-G. The OPA1 Gene Mutations Are Frequent in Han Chinese Patients with Suspected Optic Neuropathy. Mol. Neurobiol. 2017, 54, 1622–1630. [Google Scholar] [CrossRef]
- Lehman, N.; Johnson, L. Toxic Optic Neuropathy after Concomitant Use of Melatonin, Zoloft, and a High-Protein Diet. J. Neuroophthalmol. 1999, 19, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Leibold, J.E. The Ocular Toxicity of Ethambutol and Its Relation to Dose. Ann. N. Y. Acad. Sci. 1966, 135, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.K.; Fariduddin, K.; Mahapatra, N.; Bhunia, J.; Mondal, P. Hooch Blindness: A Community Study Report on a Few Indoor Patients of Toxic Optic Neuropathy Following Consumption of Adulterated Alcohol in West Bengal. Nepal J. Ophthalmol. 2012, 4, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Domaç, F.M.; Koçer, A.; Tanidir, R. Optic Neuropathy Related to Hydrogen Peroxide Inhalation. Clin. Neuropharmacol. 2007, 30, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, S.; Pandit, L. Approach to Diagnosis and Management of Optic Neuropathy. Neurol. India 2014, 62, 599–605. [Google Scholar] [PubMed]
- Van Stavern, G.P. Metabolic, Hereditary, Traumatic, and Neoplastic Optic Neuropathies. Contin. Lifelong Learn. Neurol. 2014, 20, 877–906. [Google Scholar] [CrossRef]
- Boyce, S.; Platia, E.; Green, W. Drusen of the Optic Nerve Head. Ann. Ophthalmol. 1978, 10, 695. [Google Scholar]
- Friedman, A.H.; Beckerman, B.; Gold, D.H.; Walsh, J.B.; Gartner, S. Drusen of the Optic Disc. Surv. Ophthamol. 1997, 21, 375–390. [Google Scholar] [CrossRef]
- Friedman, A.H.; Gartner, S.; Modi, S.S. Drusen of the Optic Disc: A Retrospective Study in Cadaver Eyes. Br. J. Ophthalmol. 1975, 59, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Hamann, S.; Malmqvist, L.; Costello, F. Optic Disc Drusen: Understanding an Old Problem from a New Perspective. Acta Ophthalmol. 2018, 96, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Katz, B.J.; Pomeranz, H.D. Visual Field Defects and Retinal Nerve Fiber Layer Defects in Eyes with Buried Optic Nerve Drusen. Am. J. Ophthalmol. 2006, 141, 248–253.e1. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, J.M.; Pomeranz, H.D. Visual Manifestations of Visible and Buried Optic Disc Drusen. J. Neuroophthalmol. 2004, 24, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backhaus, B.; Lorentzen, S.E. Prevalence of Pseudoexfoliation in Non-Glaucomatous Eyes in Denmark. Acta Ophthalmol. 1966, 44, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Auw-Haedrich, C.; Staubach, F.; Witschel, H. Optic Disk Drusen. Surv. Ophthalmol. 2002, 47, 515–532. [Google Scholar] [CrossRef]
- Spencer, W.H. Drusen of the Optic Disc and Aberrant Axoplasmic Transport. Ophthalmology 1978, 85, 21–38. [Google Scholar] [CrossRef]
- Tso, M.O.M. Pathology and Pathogenesis of Drusen of the Optic Nervehead. Ophthalmology 1981, 88, 1066–1080. [Google Scholar] [CrossRef]
- Malmqvist, L.; Li, X.Q.; Eckmann, C.L.; Skovgaard, A.M.; Olsen, E.M.; Larsen, M.; Munch, I.C.; Hamann, S. Optic Disc Drusen in Children: The Copenhagen Child Cohort 2000 Eye Study. J. Neuroophthalmol. 2018, 38, 140–146. [Google Scholar] [CrossRef]
- Malmqvist, L.; Li, X.Q.; Hansen, M.H.; Thomsen, A.K.; Skovgaard, A.M.; Olsen, E.M.; Larsen, M.; Munch, I.C.; Hamann, S. Progression Over 5 Years of Prelaminar Hyperreflective Lines to Optic Disc Drusen in the Copenhagen Child Cohort 2000 Eye Study. J. Neuroophthalmol. 2020, 40, 315–321. [Google Scholar] [CrossRef]
- Duvvuri, B.; Lood, C. Mitochondrial Calcification. Immunometabolism 2021, 3, e210008. [Google Scholar] [CrossRef]
- Peng, T.-I.; Jou, M.-J. Oxidative Stress Caused by Mitochondrial Calcium Overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and Mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Wågström, J.; Malmqvist, L.; Hamann, S. Optic Nerve Head Blood Flow Analysis in Patients with Optic Disc Drusen Using Laser Speckle Flowgraphy. Neuroophthalmology 2020, 45, 92. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhou, X.; Chu, Z.; Stell, L.; Shariati, M.A.; Wang, R.K.; Liao, Y.J. Topographic Quadrant Analysis of Peripapillary Superficial Microvasculature in Optic Disc Drusen. Front. Neurol. 2021, 12, 666359. [Google Scholar] [CrossRef] [PubMed]
- Türker, I.; Doğan, C.; Uzun, S.; Güven, D. Peripapillary Vessel Density in Pediatric Cases with Buried Optic Disk Drusen. Int. Ophthalmol. 2021, 41, 1337. [Google Scholar] [CrossRef]
- Engelke, H.; Shajari, M.; Riedel, J.; Mohr, N.; Priglinger, S.G.; Mackert, M.J. OCT Angiography in Optic Disc Drusen: Comparison with Structural and Functional Parameters. Br. J. Ophthalmol. 2020, 104, 1109–1113. [Google Scholar] [CrossRef]
- Obuchowska, I.; Ustymowicz, A. Blood Flow Disturbances in the Central Retinal Artery in Patients with Bilateral Optic Disc Drusen. Sci. Rep. 2020, 10, 11111. [Google Scholar] [CrossRef]
- Pinto, L.A.; Vandewalle, E.; Marques-Neves, C.; Stalmans, I. Visual Field Loss in Optic Disc Drusen Patients Correlates with Central Retinal Artery Blood Velocity Patterns. Acta Ophthalmol. 2014, 92, e286–e291. [Google Scholar] [CrossRef] [PubMed]
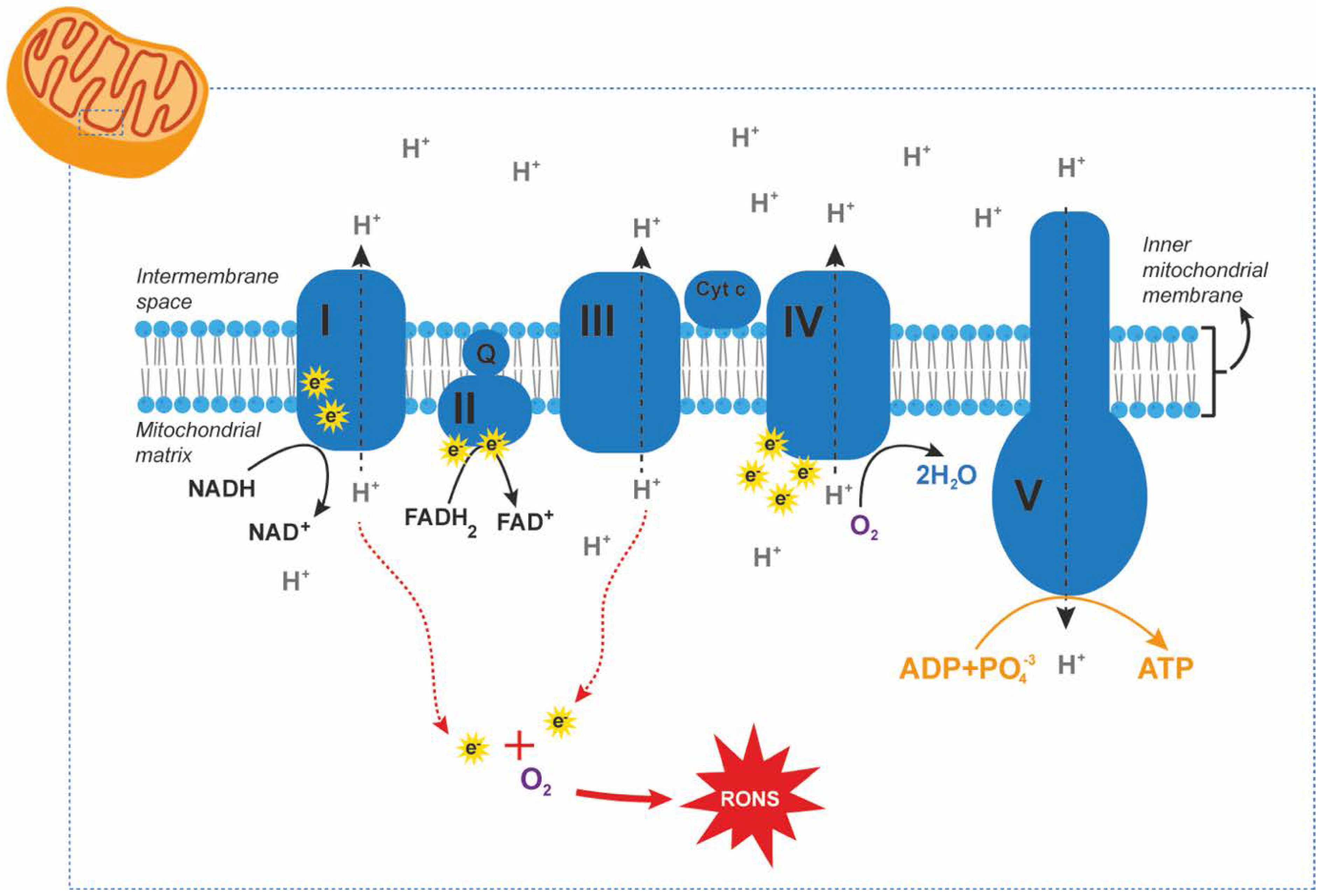
| Type of Glaucoma | Type of Sample | Outcome (Related to Control Group) * | Country | Authors |
|---|---|---|---|---|
| General antioxidant/oxidant status | ||||
| POAG | Blood | Lower TAC | Italy | [45] |
| POAG | Plasma | Lower TAS. Negative correlation with glaucoma severity | Saudi Arabia | [46] |
| POAG | Blood | Lower BAP. Negative correlation with visual acuity | Japan | [47] |
| POAG | Serum | Lower BAP. Positive correlation with RGCs density in young males (<65 years old) | Japan | [48] |
| PEXG | Plasma | Lower TAS | Saudi Arabia | [49,50] |
| PEXG | Serum | Lower TAC | Turkey | [51,52] |
| POAG | Aqueous humor | Lower TAC | Italy | [45] |
| POAG | Aqueous humor | Lower TAC | Spain | [53] |
| POAG | Aqueous humor | Lower TAC | Argentine | [54] |
| PEXG | Aqueous humor | Lower TAC | Turkey | [51] |
| PEXG | Aqueous humor | Lower TRAP | Argentine | [55] |
| PEXG | Aqueous humor | Higher TAS | Turkey | [56] |
| PEXG | Serum | Higher TOS | Turkey | [51,52] |
| PEXG | Aqueous humor | Higher TOS | Turkey | [51] |
| PEXG | Aqueous humor | No changes | Turkey | [56] |
| Antioxidant defense mechanisms | ||||
| POAG | Peripheral blood mononuclear cells | Higher levels of GSSC and lower GSH-to-GSSC ratio. Positive correlation between GSH-to-GSSC ratio and visual field damage | Japan | [57] |
| POAG (IOP ≥ 24 mm Hg) | Blood | Lower levels of GSH and total GSH | England | [58] |
| POAG (IOP ≥ 24 mm Hg) and NTG | Blood | Lower levels of GSH and total GSH in POAG and NTG. Lower redox index in POAG (IOP ≥ 24 mm Hg) | England | [59] |
| PEXG | Plasma | Higher GSH levels | Turkey | [60] |
| POAG | Serum | Higher disulfide, disulfide-to-native thiol ratio, disulfide-to-total thiol ratio | Turkey | [61] |
| POAG | Blood | Downregulation of SOD1 mRNA and upregulation of SOD2 mRNA Upregulation of GPX1 mRNA | Colombia | [62] |
| POAG | Serum | Lower activity of SOD2 but no changes in SOD1 activity | Poland | [63] |
| PEXG | Plasma | Higher total SOD activity | Turkey | [52,64] |
| PEXG | Blood | Lower total SOD activity Lower catalase activity | Turkey | [60] |
| PEXG | Serum | Lower catalase activity | Greece | [65] |
| POAG | Aqueous humor | Increased total SOD activity Increased total GPX activity No changes in catalase activity | Argentine | [54] |
| POAG | Aqueous humor | Increased total SOD activity Increased total GPX activity No changes in catalase activity | Egypt | [66] |
| POAG and PACG | Aqueous humor | Increased total SOD and GPX activity in both POAG and PACG No changes in catalase activity | India | [67] |
| POAG | Aqueous humor | Increased total SOD activity | Spain | [53] |
| PEXG | Aqueous humor | Higher total SOD activity | Argentine | [55] |
| PEXG | Aqueous humor | Lower catalase activity | Greece | [65] |
| PEXG | Aqueous humor | Higher total GPX activity | Argentine | [55] |
| Reactive oxygen and nitrogen species (RONS) | ||||
| POAG | Serum | Higher ferritin levels, especially in men | South Korea | [68,69] |
| PEXG | Plasma | Higher NO levels | Turkey | [52] |
| PEXG | Blood | Lower NO levels | Turkey | [60] |
| POAG and PACG | Aqueous humor | Increased NO levels | Egypt | [66] |
| POAG and PACG | Aqueous humor | Increased NO levels | Taiwan | [70] |
| POAG | Trabecular meshwork | Upregulation of iNOS expression and activity and downregulation of calcium-dependent NOS expression and activity. Positive correlation with visual field defects | Spain | [71] |
| Oxidative stress markers | ||||
| POAG | Plasma | Higher 8-OHdG levels | Saudi Arabia | [72] |
| POAG | Plasma | Higher 8-OHdG levels and lower PARP1 and OGG1 levels. Negative correlation between PARP1 and OGG1 expression and 8-OHdG levels | India | [73] |
| POAG | Blood | Higher 8-OHdG levels | Turkey | [74] |
| PEXG | Plasma | Higher 8-OHdG | Saudi Arabia | [75] |
| POAG | Aqueous humor | Higher 8-OHdG levels | India | [73] |
| POAG | Trabecular meshwork | Higher 8-OHdG levels. Positive correlation with visual field defects | Italy | [76,77] |
| POAG | Blood | Higher MDA levels | Turkey | [74] |
| POAG | Blood | Higher MDA levels | Spain | [45] |
| POAG | Serum | Higher MDA levels | Poland | [63] |
| NTG and HTG | Serum | Oxidation products of linoleic and arachidonic acid increased in both NTG and HTG | Japan | [78] |
| PEXG | Plasma | Higher MDA levels | Turkey | [52] |
| PEXG | Blood | Higher MDA levels | Turkey | [60] |
| POAG | Aqueous humor | Higher MDA levels | Italy | [45] |
| POAG | Aqueous humor | Higher MDA levels | Spain | [79] |
| POAG (excluded NTG) | Aqueous humor | No changes in MDA levels | Spain | [71] |
| POAG (excluded NTG) | Trabecular meshwork | Higher nitrotyrosine immunoreactivity. Positive correlation with higher IOP | Spain | [71] |
| POAG | Blood vessels and glia in the ONH | Higher nitrotyrosine immunoreactivity | Canada | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants 2021, 10, 1538. https://doi.org/10.3390/antiox10101538
Sanz-Morello B, Ahmadi H, Vohra R, Saruhanian S, Freude KK, Hamann S, Kolko M. Oxidative Stress in Optic Neuropathies. Antioxidants. 2021; 10(10):1538. https://doi.org/10.3390/antiox10101538
Chicago/Turabian StyleSanz-Morello, Berta, Hamid Ahmadi, Rupali Vohra, Sarkis Saruhanian, Kristine Karla Freude, Steffen Hamann, and Miriam Kolko. 2021. "Oxidative Stress in Optic Neuropathies" Antioxidants 10, no. 10: 1538. https://doi.org/10.3390/antiox10101538
APA StyleSanz-Morello, B., Ahmadi, H., Vohra, R., Saruhanian, S., Freude, K. K., Hamann, S., & Kolko, M. (2021). Oxidative Stress in Optic Neuropathies. Antioxidants, 10(10), 1538. https://doi.org/10.3390/antiox10101538