1. Introduction
Cardiovascular disease (CVD) is the leading cause of mortality globally, causing an estimated 17.9 million deaths each year, and is expected to account for more than 23.6 million deaths per year by 2030 [
1]. As the main underlying pathology of CVD, atherosclerosis is caused by the chronic accumulation of vascular occlusion plaque in the endothelial layers of arteries, and it eventually results in severe stenosis that limits blood flow and causes significant tissue hypoxia [
2]. Atherogenesis, which is the development process of atherosclerosis, can be defined by four steps in the lesion development process: initiation, promotion, progression, and acute events [
3].
Low-density lipoprotein (LDL) on the inner artery promotes monocyte infiltration into the intima, also known as an “early atherosclerosis event” [
4]. Once differentiated and activated, macrophages absorb excessive oxidized LDL (ox-LDL) and subsequently transform into lipid-laden foam cells [
5]. Once this occurs, it is difficult to control the progression of atherosclerosis. Therefore, prevention of the early atherosclerosis event through consumption of nutraceuticals is a promising strategy.
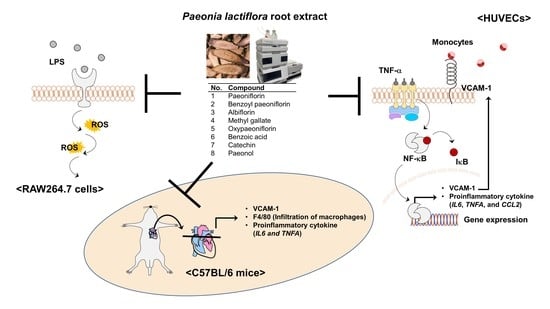
The excessive production of proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin 6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1)/C-C motif chemokine ligand 2 (CCL2), is involved in the disruption of vascular homeostasis. Among them, TNF-α is an important proinflammatory cytokine that triggers vascular inflammation via activation of the NF-κB signaling pathways. Once NF-κB is dissociated from IκB, the inhibitor of NF-κB, it translocates from the cytosol to the nucleus and binds to the specific promoter region and subsequently to transcript vascular cell adhesion molecules and inflammatory cytokines [
6]. Vascular cell adhesion molecule 1 (VCAM-1)/cluster of differentiation 106 (CD106) in the inflamed aortic endothelium contributes to the leukocyte extravasation and subsequent vascular inflammation in vitro and in vivo [
7].
Reactive oxygen species (ROS) play a key role in endothelial dysfunction and atherogenesis [
8]. Multiple studies have shown that antioxidant materials have a protective effect on vascular endothelial cells [
9,
10]. In the initiation stage of atherogenesis, the modification of LDL to ox-LDL in the presence of ROS is a key step, because macrophages recognize ox-LDL as nonself and start phagocytosis, a prerequisite for foam cell formation. Thus, the neutralization of excessive ROS is vital to control the pathogenesis of atherosclerosis.
Paeonia lactiflora Pall. (PLP) is well known in traditional medicine for its anti-inflammatory effects, including the inhibition of macrophage activation and regulation of immune cells or autoimmune diseases [
11]. PLP is divided into shoot and root systems, and its root has long been used as traditional medicine in Korea, China, and other South Asian countries. The root contains several bioactive components, such as albiflorin [
12], paeoniflorin [
13], paeonol [
14], and phenolic compounds, which can be modified by several processing steps for application as nutraceuticals and functional foods [
15]. Parker et al. and Jiang et al. also have reported the pharmacological potential of bioactive constituents of
P. lactiflora and
P. veitchii Lynch [
16] and total glucosides of paeony [
17], respectively. Previous studies have demonstrated the positive effects of
P. lactiflora components, including glycoside and paeoniflorin [
18]. However, the evaluation of the
P. lactiflora extract (PLE) as a functional food material for the treatment of early atherosclerosis remains unclear.
In this study, we studied the effect of PLE on the TNF-α-mediated infiltration of monocytes to the vascular endothelium by inhibition of VCAM-1 expression via regulation of NF-κB signaling pathways in human umbilical vein endothelial cells (HUVECs) and the effect of the oral administration of PLE on TNF-α-induced macrophage infiltration and production of proinflammatory cytokines in vivo. We also quantified eight compounds obtained from freeze-dried PLE using HPLC analysis and evaluated not only the effect of these compounds on TNF-α-mediated infiltration of monocytes to the vascular endothelium in HUVECs but also their effect on LPS-induced excessive ROS production in RAW264.7 cells to compare them. Our findings may help elucidate the preventive mechanism of PLE for early atherosclerosis.
2. Materials and Methods
2.1. Materials
Hyclone (Logan, UT, USA) provided Dulbecco’s modified Eagle’s medium (DMEM), RPMI 1640 medium, and fetal bovine serum (FBS). We obtained EBM-2 medium and EGM-2 Endothelial SingleQuots Kit from Lonza (Basel, Switzerland) and 2-mercaptoethanol from Gibco (Grand Island, NY, USA). We purchased recombinant human TNF-α from BD Pharmingen (San Diego, CA, USA) and recombinant mouse TNF-α from NKMAX (Seongnam-si, Gyeonggi-do, Korea). Sigma-Aldrich (St. Louis, MO, USA) provided 2′,7′-dichlorofluorescein diacetate (DCFH-DA) and analytical-grade reference standards (methyl gallate, catechin hydrate, paeoniflorin, benzoic acid, and paeonol). We used horseradish peroxidase (HRP)-conjugated pierce goat antirabbit IgG (H+L), pierce goat antimouse IgG (H+L), and Alexa Fluor 488 and 594 goat antirabbit IgG (H+L) for the secondary antibodies, purchasing these from Thermo Fisher Scientific Inc. (Eugene, OR, USA). The PLE reference standards, oxypaeoflorin, benzoyl paeoniflorin, and albiflorin, were purchased from Ensol Biosciences Inc. (Daejeon, Korea), Biosynth Carbosynth (San Diego, CA, USA), and FUJIFILM Wako Pure Chemical Corporation (Richmond, VA, USA).
2.2. Sample Preparation and Extraction Procedures
P. lactiflora root was obtained from the Bonghwa Herbal Crop Research Institue, GBARES, Bonghwa, Korea, in February 2019. The specimen was identified by a botanist, Young Jin Seo. The dried material was pulverized and then passed through an 8-mesh sieve. The powdered material (10 g) was extracted at 80% ethanol in a shaking incubator at room temperature for 24 h and a solvent-to-sample ratio of 50 to 1. The extract was filtered through quantitative filter papers (GVS, Zola Predosa, Bologna, Italy) before evaporation. The extract was then concentrated by rotary evaporation to eliminate ethanol. Then, the extract was freeze-dried in the range of 0.4–0.8 torr below –35 °C for 46 h and then stored at −20 °C until use. We prepared bamboo (
Phyllostachys pubescens) leaf extracts as a positive control as described previously [
19] and diluted them with distilled water immediately before use.
2.3. Cell Culture
We purchased human umbilical vein endothelial cells (HUVECs) and THP-1 cells, a human monocytic cell line, from Lonza (Basel, Switzerland), along with RAW264.7 cells, a mouse macrophage, from the Korean Cell Line Bank (KCLB; Seoul, Korea). We maintained HUVECs in an EBM-2 medium containing EGM-2 Endothelial SingleQuots Kit, passaged at 70−80 confluence, and cultured until passage 10. We maintained THP-1 in an RPMI 1640 medium supplemented with 10% FBS and 0.1% 2-mercaptoethanol; THP-1 cells were passaged between 2–6 × 105 cells/mL. We maintained the RAW264.7 cells in DMEM, including 10% FBS; these cells were passaged at 70−80 confluence. We incubated all cells at 37 °C and 5% CO2 in a CO2 incubator (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Monocyte–Endothelial Cell Adhesion Assay
We performed the monocyte–endothelial cell adhesion assay as previously described [
20]. We seeded the HUVECs at 1.5 × 10
5 cells/mL into 96-well plates and incubated overnight to reach confluent monolayers. PLE and each compound were prepared with dimethyl sulfoxide (DMSO) as a stock solution. DMSO was not used at a final concentration of >0.1% (
v/
v) and was stored at −20 °C until use. We treated the HUVECs with 25, 50, and 100 μg/mL of PLE or compounds (50 μM) for 1 h and then stimulated them with 10 ng/mL TNF-α for 5 h. We prepared the fluorescently labeled THP-1 cells to be added to the activated HUVECs using 2 μM calcein AM (Sigma-Aldrich, St. Louis, MO, USA) for 15 min at 37 °C in phosphate-buffered saline (PBS). After adding the THP-1 cells to the HUVECs, we incubated these for 1 h, washing out the unbound THP-1 cells with PBS three times and measuring the monocyte adhesion at Ex = 485 and Em = 538 nm using a fluorescent plate reader (SpectraMax; Molecular Devices Corporation, Sunnyvale, CA, USA). Parthenolide was used as a positive control because it is a well-known agent to assess the anti-inflammatory effect via inhibition of NF-ĸB activation.
2.5. Western Blot Assay
We seeded the HUVECs at 5 × 104 cells/mL and RAW264.7 cells at 3 × 105 cells/mL in 100 mm dishes and cultured these for 24 h. We pretreated the cells with PLE with various concentrations (25, 50, and 100 μg/mL) or compounds (50 μM) for 1 h and then treated them with TNF-α (10 ng/mL) or LPS (1 μg/mL). We washed the cells twice using ice-cold PBS, scraped them with the cell lysis buffer (Cell Signaling Technologies, Beverly, MA, USA) containing protease and phosphatase inhibitor (Thermo Fisher Scientific), and then collected them. After 30 min, we centrifuged the collected cells at 13,000× g for 15 min. We then transferred the cell lysates into prechilled tubes and used these for the Western blot assay.
For protein quantification, we determined the cell lysate by reference to a standard curve of bovine serum albumin. We measured 30 μg of lysate protein, using the DC protein assay kit (Bio-rad Inc., Hercules, CA, USA). We separated the proteins with 10% sodium dodecyl sulfate–polyacrylamide gel (SDS−PAGE) electrophoretically and then transferred these to a polyvinylidene difluoride (PVDF) membrane (Millipore, Temecula, CA, USA). We blocked the transferred proteins using TBST containing 5% skim milk (BD biosciences) for 1 h at room temperature. After blocking, we incubated each membrane with specific primary antibodies overnight at 4 °C. After washing three times with TBST, we incubated horseradish peroxidase (HRP)-conjugated secondary antibodies against the species of primary antibodies for 1 h at room temperature. We detected the protein bands using a chemiluminescence detection kit (ATTO, Tokyo, Japan) and visualized the bands using GeneGnome XRQ NPC (Syngene, Cambridge, UK). We immunoblotted the samples for p-p65 (Ser536), p65, IκBα, p-IκB kinase (IKKα/β) (Ser176/180), IKKα, IKKβ, α/β-tubulin, HO-1, Keap-1, Nrf2 (1:1000, Cell Signaling Technologies, Beverly, MA, USA), anti-lamin B1 (1:10,000, Abcam, Cambridge, MA, UK), and VCAM-1 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
2.6. Separation of Cytoplasmic and Nuclear Fractions
We seeded the HUVECs at 5 × 104 cells/mL in 100 mm dishes and cultured them for 24 h. We pretreated the cells with PLE for 1 h and then treated them with TNF-α (10 ng/mL) for 15 min. We washed the cells twice with PBS and then fractionated them using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) according to the manufacturer’s manual. We determined the separated cytoplasmic and nuclear proteins using Western blot assay.
2.7. Animals and Experiment Design
Central Lab Animal Inc. (Seoul, Korea) supplied 10-week-old male C57BL/6 mice (23−25 g). All mice were housed in a controlled environment with a 12 h light/dark cycle and maintained in cages in an air-conditioned room (23 ± 2 °C); we provided the mice free access to a standard chow diet and water. We randomly divided 25 mice into five groups (n = 5/group). In the control group (normal mice), we injected the mice intraperitoneally (i.p.) with PBS. In the TNF-α-treated group, we injected the mice i.p. with TNF-α at 25 μg/kg in PBS. We orally administered the extracts to the PLE low-dose (100 mg/kg body weight/day) treated group, PLE high-dose (500 mg/kg/day) treated group, and bamboo leaf extract (BLE; 100 mg/kg/day) treated group for 3 days. Then, we injected the mice i.p. with TNF-α daily for 7 consecutive days for early atherosclerosis 1 h after the last oral administration. At the end of 10 days, mice were anesthetized, and the aorta and heart were exposed and isolated.
2.8. Immunofluorescence
For the in vitro immunofluorescence (IF), we seeded the HUVECs at 2 × 104 cells/mL in an 8-well-chamber slide glass and then cultured them overnight. We pretreated the cells with PLE for 1 h and then with TNF-α (10 ng/mL) for 15 min. We fixed the cells with 4% formaldehyde in PBS for 15 min after washing them with PBS. Then, we permeated the methanol into the cells at −20 °C for 15 min. After washing, we blocked the cells with a blocking buffer consisting of 5% FBS and 0.3% Tween 20 for 1 h at room temperature. We incubated the cells with specific primary antibodies in an antibody buffer overnight at 4 °C and then with goat polyclonal secondary antibody to rabbit IgG H&L (Alexa Fluor 488). We performed nuclear staining with a Vectashield antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA), observed the cells under a fluorescence microscope, and recorded the images using LAS X (Leica Microsystems, Wetzlar, Hessen, Germany).
For the in vivo IF, we embedded the mouse heart and aorta in an optimal cutting temperature compound (FSC 22; Leica Microsystems, Lake, IL, USA), freezing and cryosectioning them. We obtained the histological data on 10 μm cryosections with a CM1850 cryostat (Leica Microsystems, Nussloch, Germany). We fixed the frozen tissue sections in 4% formaldehyde in PBS for 15 min, washed them three times with PBS, and blocked them with nonspecific binding using 5% FBS and 0.3% Tween 20 in PBS for 1 h at room temperature. We specifically stained the mouse aortic intimal surfaces using primary antibodies, including F4/80 and VCAM-1, overnight and washed the sections three times with PBS; then, we performed the same procedure as done with the in vitro IF protocol.
2.9. Quantitative Real-Time Polymerase Chain Reaction
We seeded the HUVECs at 5 × 10
4 cells/mL in 100 mm dishes and cultured these for 24 h. We pretreated the cells with PLE or compounds (50 μM) for 1 h and then cultured them with TNF-α (10 ng/mL) for 24 h. We isolated the cells for RNA, using the RNAiso Plus (Takara Bio Inc., Kyoto, Japan) according to the manufacturer’s instructions. Then, we prepared the synthesis of the cDNA using the ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan). We performed the real-time polymerase chain reaction (qRT-PCR) reactions with a CFX Connect real-time PCR detection system (Bio-rad) using SYBR Green real-time PCR master mix (Toyobo Co., Ltd., Osaka, Japan) according to the manufacturer’s manual.
Table 1 describes the specific primers. We used the comparative Ct method for the data analyses using CFX Maestro Software (Bio-rad Inc.) and IBM SPSS 25.0 software package for Windows (SPSS Inc., Chicago, IL, USA), normalizing the values for each gene to
GAPDH expression levels.
2.10. Dichlorofluorescein Diacetate Assay
We evaluated the ROS production by RAW264.7 cells exposed to LPS with a 2′,7′-dichlorofluorescein diacetate (DCF-DA) assay. We seeded the RAW264.7 cells at 2 × 105 cells/mL in a 96-well plate overnight. We pretreated the cells with 50 μM of each compound in a 200 μL medium for 1 h. An effective ROS inhibitor, N-acetyl cysteine (NAC, 1 mM (32.64 μg/200 μL)), was used as a positive control. Subsequently, we stimulated the RAW264.7 cells by 1 μg/mL LPS for 24 h, washed them with PBS, and treated them with 20 μM of DCF-DA for 30 min in the dark. We then washed the cells twice with PBS. We measured ROS production at Ex = 485 and Em = 538 nm, using a fluorescent plate reader (SpectraMax; Molecular Devices Corporation, Sunnyvale, CA, USA).
2.11. Determination of PLE Component Content
We injected the samples using a high-performance liquid chromatography (HPLC) system (Agilent 1260 Infinity II Series, Santa Clara, CA, USA) equipped with a quaternary pump, an autosampler, and a diode array detector (DAD). A Unison Imtakt US-C18 column (250 × 4.6 mm, 5 μm) at 25 °C was used for the separation of the individual components of PLE. The reference standards, including methyl gallate, oxypaeoflorin, catechin, albiflorin, paeoniflorin, benzoic acid, benzoyl paeoniflorin, and paeonol, as well as PLE, were diluted with 70% ethanol. The range of the standard solutions was 0.4–50 µg/mL (0.4, 2, 10, 25 and 50 µg/mL). Then, 5 μL of the diluted standards and PLE were injected, and they were analyzed at a flow rate of 0.8 mL/min. We monitored the ultraviolet absorbance of the effluent at 240 nm. The HPLC mobile phase consisted of mobile phases A (0.01% phosphoric acid/water) and B (acetonitrile (ACN)). The gradient program was set as follows: 0 min, 5% B; 0–15 min, 5–45% B; 15–30 min, 45% B; 30–32 min, 45–5% B; 32–40 min, 5% B. The column was equilibrated with 70% ACN for 20 min at the end of the analysis, and it was confirmed that there were no residual analytes in the column.
The integrated peaks of PLE were quantified and identified by comparing their retention times against those of the corresponding standard peaks. All samples were analyzed by comparing the peak area with the external calibration curve from a neat standard solution. The content of each PLE component was estimated using Equation (1):
where
Pspl is the concentration of each component,
Aj is the
jth measurement of the area of the
ith calibration standard,
B1 is the slope of the calibration curve,
Bo is the intercept of the calibration curve, and
F is the dilution factor.
2.12. Statistical Analysis
We present the results as mean ± standard deviation (SD). We used the IBM SPSS 20.0 software package for the data analysis. We determined the significance of differences using a one-way analysis of variance considering the results significant at the 95% confidence level.
4. Discussion
CVD, the leading cause of death worldwide, is showing a slightly decreasing trend in developed countries but is still emerging as a social problem, with an increasing incidence rate in developing countries. Various statin treatments are prescribed for CVD, but patients with CVD do not develop symptoms until the final stage of clogged blood vessels, so treatment is limited.
In our previously published report, we classified atherogenesis, the development of atherosclerosis, as a major cause of CVD into initiation, promotion, progression, and acute events [
3]. Therefore, we tried to develop nutraceuticals for the prevention of CVD. Among these stages, because the foam cell-mediated necrotic core is created in the progression stage and is difficult to control, we focused on the promotion stage and recruitment of monocytes and infiltration to the inner artery.
To develop nutraceuticals against atherogenesis, we have screened 20 species of botanical extracts; we selected PLE because it exhibited the greatest inhibitory effect on TNF-α-induced monocyte adhesion to vascular endothelial cells (data not shown). We reextracted in consideration of the in vivo experiment, performed the same experiment as before, confirmed that it showed similar efficacy, and then selected it as the final material.
Li et al. prepared total glycosides of paeony (TGP) capsules and confirmed their antiatherosclerotic effect in rats [
25]. A recent study showed that a combination of total glucosides of
P. lactiflora Pall. and total ligustici phenolic acids from
Ligusticum chuanxiong Hort. has synergistic effects on focal cerebral ischemia in vitro and in vivo [
18]. These data suggest that the intake of enhanced
P. lactiflora components has a positive effect on vascular diseases such as atherosclerosis. However, these results did not confirm the activity of PLE or whether the promotion stage in atherogenesis was regulated.
We confirmed the preventive effect of PLE on TNF-α-induced atherogenesis by demonstrating the inhibition of the interaction between monocytes and vascular endothelium with similar contents of BLE and levels of
VCAM1 mRNA and protein expression in HUVECs. In our previous studies,
Lespedeza cuneata extract and erythorbyl laurate were shown to exert antiatherosclerotic activities via inhibition of the interaction between HUVECs and THP-1 and VCAM-1 expression in vitro and in vivo [
20,
26]. These results indicate that PLE has protective properties for atherogenesis via the inhibition of monocyte adhesion to vascular endothelium. As the main signaling pathway to activate vascular inflammation, NF-κB plays a critical role in the gene expression of VCAM-1 and inflammatory cytokines including TNF-α and IL-6. As we expected, PLE suppressed the activity of TNF-α-induced p65, a subunit of NF-κB, translocation from the cytosol to the nucleus by suppressing phosphorylation of IKK and p65 in HUVECs.
G. You et al. confirmed that the content of Radix Paeoniae Alba, the dried root of
P. lactiflora Pall., shows differences in chemical composition and antioxidant effects based on processing or excipient addition [
27]. To evaluate the compound compositions of PLE and its effect on atherosclerotic markers, we identified and quantified PLE compounds using HPLC. A previous study demonstrated that TGP contains 15 monoterpene glycosides, including paeoniflorin, albiflorin, oxypaeoniflorin, benzoylpaeoniflorin, benzoyloxypeoniflorin, lactiflorin, albiflorin R1, benzoylalbiflorin, galloylpaeoniflorin, and paeoniflorin sulfonate [
17]. Interestingly, using HPLC-DAD analysis, we found that the composition of PLE compounds is different from that of TGP components and newly identified gallate, catechin, and benzoic acid. As shown by the representative chromatograms of
P. lactiflora root extract in
Figure 3, paeoniflorin has a 15-fold higher concentration in PLE than the other compounds. In addition, when compared with the reported paeoniflorin content of
P. lactiflora roots, the paeoniflorin content in
P.
lactiflora was >8%, which was higher than that of paeoniflorin, which is 2.0–4.0% in plants in the Paeoniaceae family [
28]. Additionally, Yang Y. et al. identified that the paeoniflorin contents among the 20
Paeonia species ranged from 20.14 to 39.29 mg/g, which were lower than the paeoniflorin content of 83.47 mg/g in this study [
29]. These variations in the phytochemical concentration of
P.
lactiflora are affected by regional changes, such as in temperature, soil, and rainfall, or seasonal changes, such as harvest time [
30].
Although studies reported that paeonol and paeoniflorin are the main compounds in PLE that ameliorate vascular inflammation [
31,
32], few studies have evaluated the inhibitory effects of other PLE compounds on arteriosclerotic factors. We confirmed that not only paeonol and paeoniflorin but also other compounds showed inhibitory effects against the capacity of monocytes to adhere to the vascular endothelium. Among them, benzoic acid showed the highest inhibition capacity for THP-1 adhesion to HUVECs, followed by catechin and gallate. qRT-PCR results have also shown that gallate and benzoic acid had the highest inhibitory effects on TNF-α-induced
VCAM1 gene expression in HUVECs. Proinflammatory cytokines accelerate the influx and activation of inflammatory cells, such as differentiation from monocytes to macrophages [
7,
33]. Each PLE component had a distinct inhibition effect on TNF-α-induced
CCL2,
TNFA, and
IL6 mRNA expression in HUVECs. Benzoic acid showed the strongest inhibitory effect on
CCL2; paeoniflorin, benzoic acid, and gallate showed the strongest inhibitory effects on
TNFA; and gallate showed the strongest inhibitory effect on
IL6 mRNA expression in HUVECs. Benzoic acid, a plant phenolic acid derivative, is an antimicrobial preservative in food [
34], but the effect against atherosclerosis-related vascular diseases is still unclear. Our study showed that benzoic acid could contribute to early atherosclerosis by inhibiting monocyte adhesion to the endothelium and abnormal
CCL2 mRNA expression. ROS also play a critical role in vascular inflammation and subsequent progression of atherogenesis [
8]. Multiple studies have suggested that antioxidant botanical materials can be promising preventive antiatherosclerotic agents [
35]. Paeoniflorin has the strongest antioxidant effect on LPS-induced ROS production in RAW264.7 cells. Additionally, except for paeonol, all PLE compounds exert antioxidant effects. These results indicate that even though paeoniflorin has the highest content among PLE compounds and exhibits ameliorating atherosclerosis factors, other ingredients also contribute to the control of vascular inflammation of PLE.
We conducted animal experiments to directly verify whether the in vitro efficacy is also shown in vivo. TNF-α as a stimulus in vivo can mediate endothelial activation such as aortic endothelial injury [
36]. We already confirmed that intraperitoneal injection of TNF-α induced vascular inflammation, such as macrophage infiltration and abnormal mRNA expression of VCAM-1 and inflammatory cytokines such as IL-6 and TNF-α in the mouse aorta [
37]. We found that oral administration of PLE significantly suppressed TNF-α-induced mRNA expression of proinflammatory cytokines, including
IL6 and
TNFA, in the mouse aorta. Additionally, PLE blocked F4/80-positive monocyte infiltration into the intima and suppressed VCAM-1 expression in the mouse aortic root. These in vivo results proved that oral administration of PLE, not only single compounds, suppresses against the early stage of atherosclerosis via the regulation of monocyte infiltration into the vascular endothelium.
Collectively, we demonstrated that the protective effect of PLE is due to a combination of PLE compounds and does not only depend on specific compounds such as paeoniflorin and paeonol. Additionally, PLE has a protective effect on vascular inflammation, including monocyte infiltration to the inner artery and abnormal expression of cytokines, and has an antioxidant effect via inhibition of the NF-κB signaling pathways.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}