Localization of Free and Bound Metal Species through X-Ray Synchrotron Fluorescence Microscopy in the Rodent Brain and Their Relation to Behavior


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
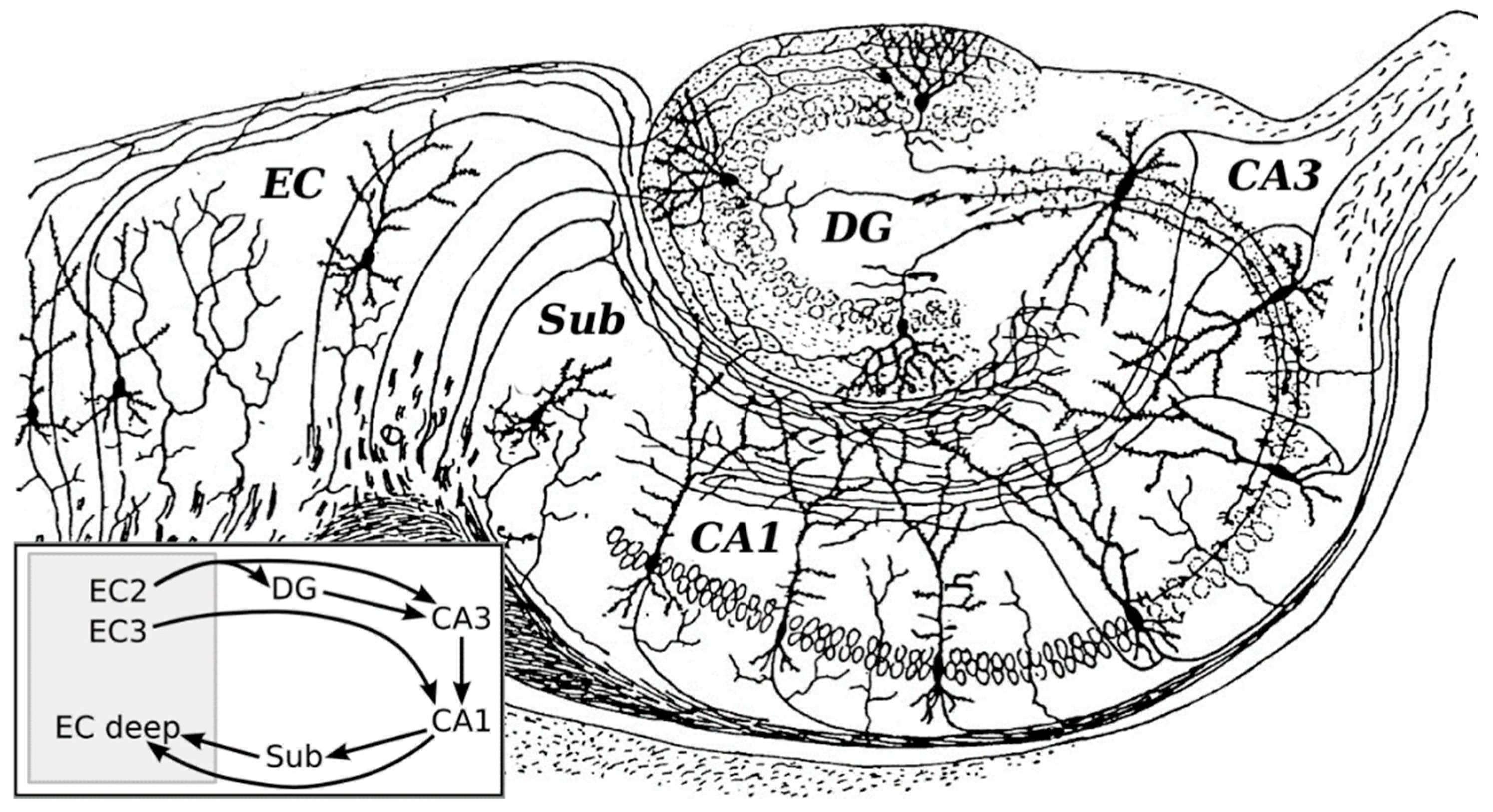
2. The Hippocampal Trisynaptic Circuit and Long-Term Potentiation (LTP)
3. Metal Localization and Their Roles in Learning and Memory
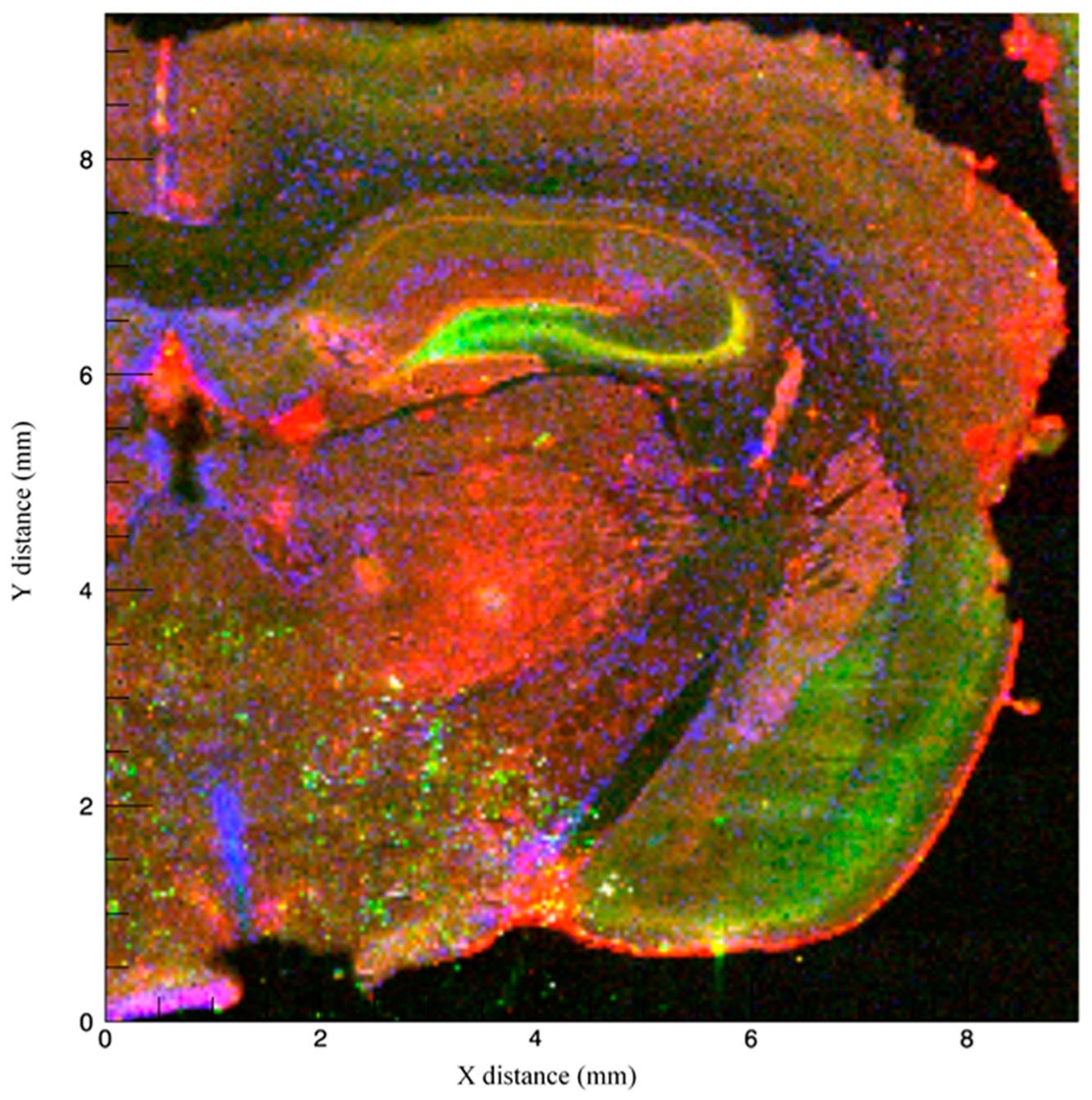
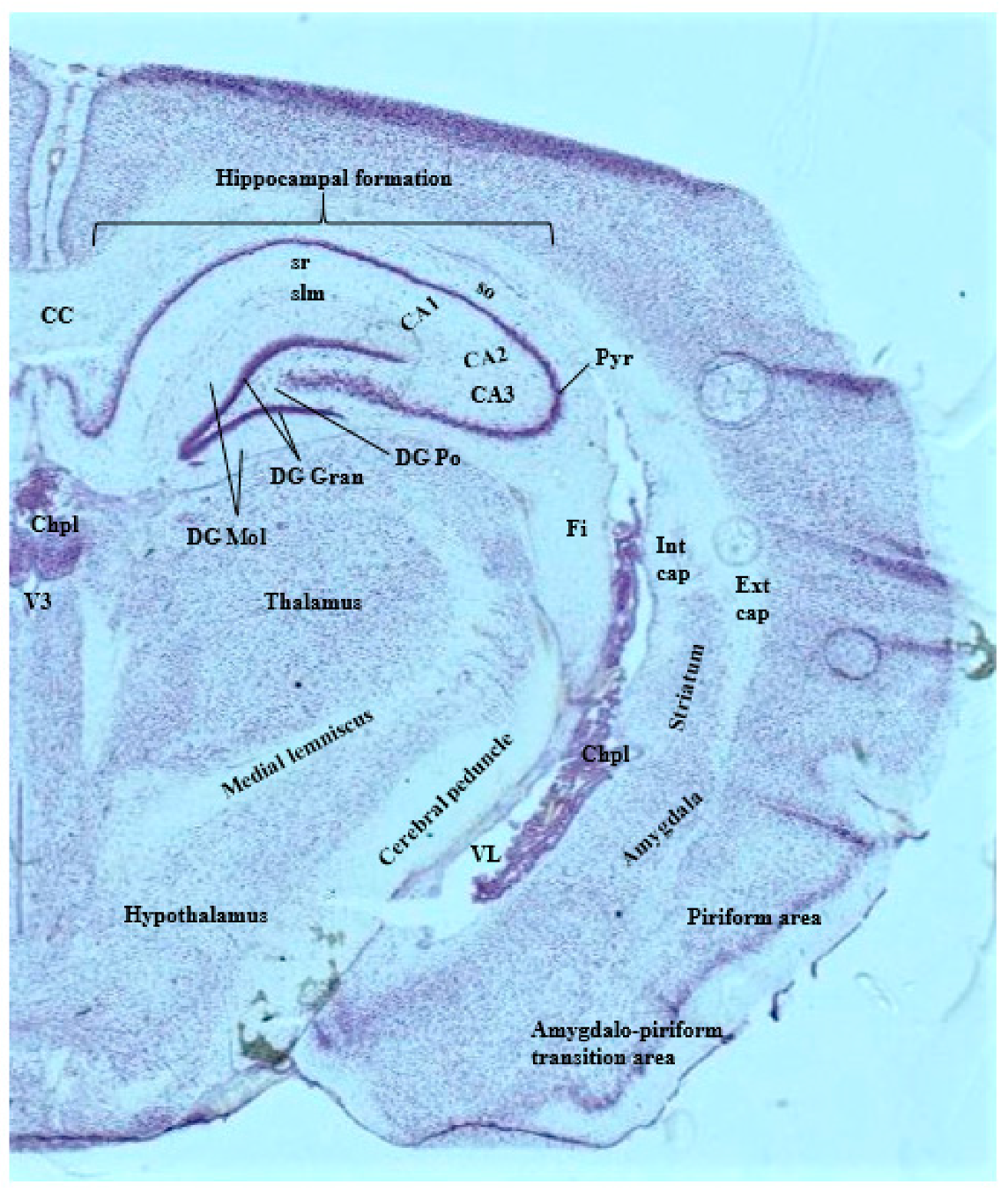
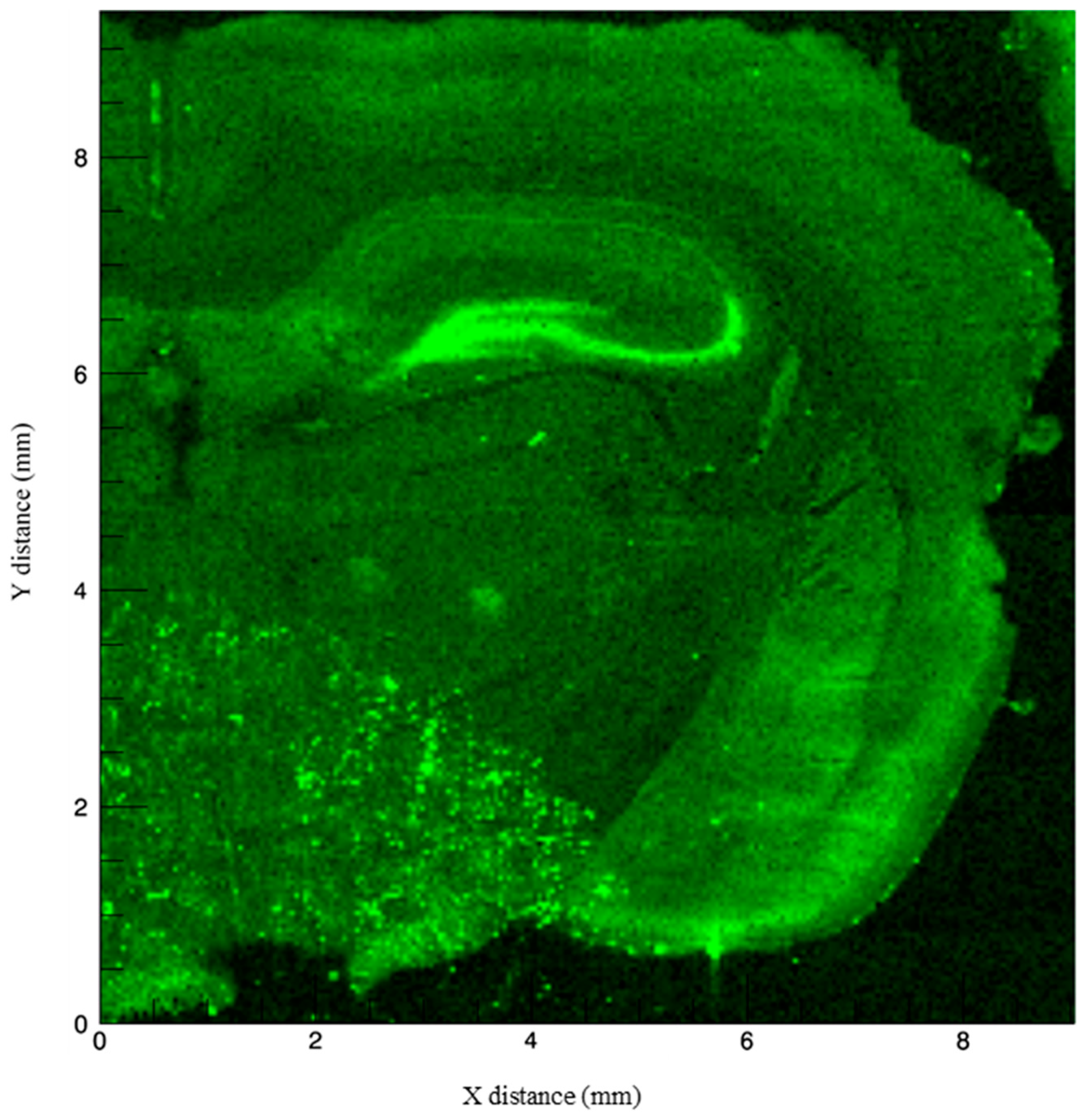
3.1. Imaging Methods to Infer Localization
3.2. Properities and Localization of Zinc
Zinc, LTP Signaling, and Learning and Memory
3.3. Properities and Localization of Copper
3.3.1. Copper, LTP Signaling, and Learning and Memory
3.3.2. Interactions between Zinc and Copper
3.4. Properities and Localization of Iron
3.4.1. Iron, LTP Signaling, and Learning and Memory
3.4.2. Interactions among Zinc, Copper, and Iron
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef]
- Bertinato, J.; L’Abbé, M.R. Maintaining copper homeostasis: Regulation of copper-trafficking proteins in response to copper deficiency or overload. J. Nutr. Biochem. 2004, 15, 316–322. [Google Scholar] [CrossRef]
- Levenson, C.W. Zinc: The new antidepressant? Nutr. Rev. 2006, 64, 39–42. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron homeostasis in health and disease. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Keen, C.L.; Hanna, L.A.; Lanoue, L.; Uriu-Adams, J.Y.; Rucker, R.B.; Clegg, M.S. Developmental consequences of trace mineral deficiencies in rodents: Acute and long-term effects. J. Nutr. 2003, 133, 1477S–1480S. [Google Scholar] [CrossRef]
- Osredkar, J.; Sustar, N. Copper and zinc, biological role and significance of copper/zinc imbalance. J. Clin. S 2011, 3. [Google Scholar] [CrossRef]
- Qian, J.; Noebels, J.L. Exocytosis of vesicular zinc reveals persistent depression of neurotransmitter release during metabotropic glutamate receptor long-term depression at the hippocampal CA3-CA1 synapse. J. Neurosci. 2006, 26, 6089–6095. [Google Scholar] [CrossRef]
- Ramon, Y.C. Histologie Du Système Nerveux De L’homme Et Des Vertébrés; Maloine: Paris, France, 1911. [Google Scholar]
- Stepan, J.; Dine, J.; Eder, M. Functional optical probing of the hippocampal trisynaptic circuit in vitro: Network dynamics, filter properties, and polysynaptic induction of CA1 LTP. Front. Neurosci. 2015, 9, 160. [Google Scholar] [CrossRef]
- Nakazawa, K.; McHugh, T.J.; Wilson, M.A.; Tonegawa, S. NMDA receptors, place cells and hippocampal spatial memory. Nat. Rev. Neurosci. 2004, 5, 361–372. [Google Scholar] [CrossRef]
- File:CajalHippocampus (modified).png. Available online: https://commons.wikimedia.org/wiki/File:CajalHippocampus_(modified).png (accessed on 27 March 2019).
- Andersen, P. Organization of Hippocampal Neurons and Their Interconnections. In The Hippocampus; Isaacson, R.L., Pribram, K.H., Eds.; Springer: Boston, MA, USA, 1975. [Google Scholar]
- Bliss, T.V.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 357–374. [Google Scholar] [CrossRef]
- Lømo, T. The discovery of long-term potentiation. Phil. Trans. R. Soc. Lond. 2003, 358, 617–620. [Google Scholar] [CrossRef]
- Lømo, T. Frequency potentiation of excitatory synaptic activity in the dentate area of the hippocampal formation. Acta Physiol. Scand. 1966, 68 (Suppl. 277), 128. [Google Scholar]
- Hebb, D.O. The Organization of Behavior: A Neuropsychological Theory; John Wiley & Sons: New York, NY, USA, 1949. [Google Scholar]
- Lowel, S.; Singer, W. Selection of intrinsic horizontal connections in the visual cortex by correlated neuronal activity. Science 1992, 255, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Moriyoshi, K.; Masu, M.; Ishii, T.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Molecular cloning and characterization of the rat NMDA receptor. Nature 1991, 354, 31–37. [Google Scholar] [CrossRef]
- Nicoll, R.A.; Roche, K.W. Long-term potentiation: Peeling the onion. Neuropharmacology 2013, 74, 18–22. [Google Scholar] [CrossRef]
- Giese, K.P.; Fedorov, N.B.; Filipkowski, R.K.; Silva, A.J. Autophosphorylation at Thr286 of the calcium-calmodulin kinase II in LTP and learning. Science 1998, 279, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Takamiya, K.; He, K.; Song, L.; Huganir, R.L. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol. 2009, 103, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Mayford, M.; Wang, J.; Kandel, E.R.; O’Dell, T.J. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 1995, 81, 891–904. [Google Scholar] [CrossRef]
- Mayford, M.; Bach, M.E.; Huang, Y.-Y.; Wang, L.; Hawkins, R.D.; Kandel, E.R. Control of memory formation through regulated expression of a CaMKII transgene. Science 1996, 274, 1678–1683. [Google Scholar] [CrossRef]
- Morris, M.R.G.; Anderson, E.; Lynch, S.G.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 1986, 319, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef]
- Whitlock, R.J.; Heynen, J.A.; Shuler, G.M.; Bear, F.M. Learning induces long-term potentiation in the hippocampus. Science 2006, 313, 1093–1097. [Google Scholar] [CrossRef]
- Okada, T.; Yamada, N.; Tsuzuki, K.; Horikawa, H.P.M.; Tanaka, K.; Ozawa, S. Long-term potentiation in the hippocampal CA1 area and dentate gyrus plays different roles in spatial learning. Eur. J. Neurosci. 2003, 17, 341–349. [Google Scholar] [CrossRef]
- Zamanillo, D.; Sprengel, R.; Hvalby, Ø.; Jensen, V.; Burnashev, N.; Rozov, A.; Kaiser, K.M.; Koster, H.J.; Brochardt, T.; Worley, P.; et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 1999, 284, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory amino acids in synaptic transmission in the schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.W.; Cotman, C.W. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl-D-aspartate antagonists. Neurosci. Lett. 1986, 70, 132–137. [Google Scholar] [CrossRef]
- Mellor, J.; Nicoll, R.A. Hippocampal mossy fiber LTP is independent of postsynaptic calcium. Nat. Neurosci. Brief Commun. 2001, 4, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A.; Malenka, R.C. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 1995, 377, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A.; Schmitz, D. Synaptic plasticity at hippocampal mossy fiber synapses. Nat. Rev. Neurosci. 2005, 6, 863–876. [Google Scholar] [CrossRef]
- Yeckel, M.F.; Kapur, A.; Johnston, D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat. Neurosci. 1999, 2, 625–633. [Google Scholar] [CrossRef]
- Derrick, B.E.; Martinez, J.L. Opioid receptor activation is one factor underlying the frequency dependence of mossy fiber LTP induction. J. Neurosci. 1994, 14, 4359–4367. [Google Scholar] [CrossRef] [PubMed]
- Wallis, J.L.; Irvine, M.W.; Jane, D.E.; Lodge, D.; Collingridge, G.L.; Bortolotto, Z.A. An interchangeable role for kainate and metabotropic glutamate receptors in the induction of rat hippocampal mossy fiber long-term potentiation in vivo. Hippocampus 2015, 25, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Jonas, P.; Major, G.; Sakmann, B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J. Physiol. 1993, 472, 615–663. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Nicoll, R.A. Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature 1995, 376, 256. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.B.; Castillo, P.E. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron 2008, 57, 108–120. [Google Scholar] [CrossRef]
- Nakazawa, K.; Sun, L.D.; Quirk, M.C.; Rondi-Reig, L.; Wilson, M.A.; Tonegawa, S. Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron 2003, 38, 305–315. [Google Scholar] [CrossRef]
- Rajji, T.; Chapman, D.; Eichenbaum, H.; Greene, R. The role of CA3 hippocampal NMDA receptors in paired associate learning. J. Neurosci. 2006, 26, 908–915. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Bauer, E.P.; LeDoux, J.E. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J. Neurosci. 1999, 19, 10512–10519. [Google Scholar] [CrossRef]
- Eder, M.; Zieglgänsberger, W.; Dodt, H.U. Neocortical long-term potentiation and long-term depression: Site of expression investigated by infrared-guided laser stimulation. J. Neurosci. 2002, 22, 7558–7568. [Google Scholar] [CrossRef]
- Rioult-Pedotti, M.S.; Friedman, D.; Donoghue, J.P. Learning-induced LTP in neocortex. Science 2000, 290, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Toyoizumi, T.; Miller, K.D. Equalization of ocular dominance columns induced by an activity-dependent learning rule and the maturation of inhibition. J. Neurosci. 2009, 29, 6514–6525. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: Burlington, MA, USA, 2007. [Google Scholar]
- Allen Brain Institute for Brain Science (2011). Allen Mouse Brain Atlas. Available online: http://mouse.brain-map.org/experiment/thumbnails/100048576?image_type=atlas (accessed on 12 December 2018).
- Huang, E.P. Metal ions and synaptic transmission: Think zinc. Proc. Natl. Acad. Sci. USA 1997, 94, 13386–13387. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Yamaguchi-Iwai, Y.; Sasaki, R.; Nagao, M. Overview of mammalian zinc transporters. Cell. Mol. Life Sci. CMLS 2004, 61, 49–68. [Google Scholar] [CrossRef]
- Kasarskis, E.J. Zinc metabolism in normal and zinc-deficient rat brain. Exp. Neurol. 1984, 85, 114–127. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Clausell, J.; Danscher, G. Intravesicular localization of zinc in rat telencephalic boutons. A histochemical study. Brain Res. 1985, 337, 91–98. [Google Scholar] [CrossRef]
- Morris, D.R.; Levenson, C.W. Ion channels and zinc: Mechanisms of neurotoxicity and neurodegeneration. J. Toxicol. 2012, 2012, 6. [Google Scholar] [CrossRef]
- Assaf, S.Y.; Chung, S.H. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984, 308, 734. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Giblin, L.J.; Krężel, A.; McAdoo, D.J.; Muelle, R.N.; Zeng, Y.; Balayi, R.V.; Masalha, R.; Thompson, R.B.; Fierke, C.A.; et al. Concentrations of extracellular free zinc (pZn) e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 2006, 198, 285–293. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Koh, J.Y.; Aizenman, E.; Bush, A.I.; Hershfinkel, M. The neurophysiology and pathology of brain zinc. J. Neurosci. 2011, 31, 16076–16085. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.R. Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J. Neurosci. 2003, 23, 6847–6855. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.; Cunningham, M.G. Zinc: The brain’s dark horse. Synapse 2009, 63, 1029–1049. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Pensalfini, A.; Frazzini, V.; Sensi, S.L. New therapeutic targets in Alzheimer’s disease: Brain deregulation of calcium and zinc. Cell Death Dis. 2011, 2, e176. [Google Scholar] [CrossRef] [PubMed]
- Kalappa, B.I.; Anderson, C.T.; Goldberg, J.M.; Lippard, S.J.; Tzounopoulos, T. AMPA receptor inhibition by synaptically released zinc. Proc. Natl. Acad. Sci. USA 2015, 112, 15749–15754. [Google Scholar] [CrossRef] [PubMed]
- Amico-Ruvio, S.A.; Murthy, S.E.; Smith, T.P.; Popescu, G.K. Zinc effects on NMDA receptor gating kinetics. Biophys. J. 2011, 100, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.T.; Radford, R.J.; Zastrow, M.L.; Zhang, D.Y.; Apfel, U.P.; Lippard, S.J.; Tzounopoulos, T. Modulation of extrasynaptic NMDA receptors by synaptic and tonic zinc. Proc. Natl. Acad. Sci. USA 2015, 112, E2705–E2714. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, G.L.; Mayer, M.L. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature 1987, 328, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Barberis, A.; Cherubini, E.; Mozrzymas, J.W. Zinc inhibits miniature GABAergic currents by allosteric modulation of GABAA receptor gating. J. Neurosci. 2000, 20, 8618–8627. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Koh, J.; Choi, D.W. Zinc selectively blocks the action of N-methyl-D-aspartate on cortical neurons. Science 1987, 236, 589–593. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration: Role of zinc ion availability. Prog. Neurobiol. 2005, 75, 367–390. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Bush, A.I. Synaptically released zinc: Physiological functions and pathological effects. Biometals 2001, 14, 353–366. [Google Scholar] [CrossRef]
- Ketterman, J.K.; Li, Y.V. Presynaptic evidence for zinc release at the mossy fiber synapse of rat hippocampus. J. Neurosci. Res. 2008, 86, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Molnár, P.; Nadler, J.V. Lack of effect of mossy fiber-released zinc on granule cell GABAA receptors in the pilocarpine model of epilepsy. J. Neurophysiol. 2001, 85, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Sindreu, C.B.; Varoqui, H.; Erickson, J.D.; Pérez-Clausell, J. Boutons containing vesicular zinc define a subpopulation of synapses with low AMPAR content in rat hippocampus. Cereb. Cortex 2003, 13, 823–829. [Google Scholar] [CrossRef]
- Christensen, M.K.; Geneser, F.A. Distribution of neurons of origin of zinc-containing projections in the amygdala of the rat. Anat. Embryol. 1995, 191, 227–237. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H.; Imano, S.; Oku, N. Increases in extracellular zinc in the amygdala in acquisition and recall of fear experience and their roles in response to fear. Neuroscience 2010, 168, 715–722. [Google Scholar] [CrossRef]
- Takeda, A.; Tamano, H.; Ogawa, T.; Takada, S.; Nakamura, M.; Fujii, H.; Ando, M. Intracellular Zn2+ signaling in the dentate gyrus is required for object recognition memory. Hippocampus 2014, 24, 1404–1412. [Google Scholar] [CrossRef]
- Tamano, H.; Minamino, T.; Fujii, H.; Takada, S.; Nakamura, M.; Ando, M.; Takeda, A. Blockade of intracellular Zn2+ signaling in the dentate gyrus erases recognition memory via impairment of maintained LTP. Hippocampus 2015, 25, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Murakami, T.; Nakada, H.; Minamino, T.; Koike, Y. Weakened intracellular Zn2+-buffering in the aged dentate gyrus and its involvement in erasure of maintained LTP. Mol. Neurobiol. 2018, 55, 3856–3865. [Google Scholar] [CrossRef]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef]
- Li, Y.; Hough, C.J.; Frederickson, C.J.; Sarvey, J.M. Induction of mossy fiber→ Ca3 long-term potentiation requires translocation of synaptically released Zn2+. J. Neurosci. 2001, 21, 8015–8025. [Google Scholar] [CrossRef]
- Besser, L.; Chorin, E.; Sekler, I.; Silverman, W.F.; Atkin, S.; Russell, J.T.; Hershfinkel, M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009, 29, 2890–2901. [Google Scholar] [CrossRef]
- Ceccom, J.; Halley, H.; Daumas, S.; Lassalle, J.M. A specific role for hippocampal mossy fiber’s zinc in rapid storage of emotional memories. Learn Mem. 2014, 21, 287–297. [Google Scholar] [CrossRef]
- Takeda, A.; Sakurada, N.; Kanno, S.; Minami, A.; Oku, N. Response of extracellular zinc in the ventral hippocampus against novelty stress. J. Neurochem. 2006, 99, 670–676. [Google Scholar] [CrossRef]
- North, R.A. P2X receptors. Phil. Trans. R. Soc. B 2016, 371, 20150427. [Google Scholar] [CrossRef]
- Schwiebert, E.M.; Liang, L.; Cheng, N.-L.; Williams, C.R.; Olteanu, D.; Welty, E.A.; Zsembery, A. Extracellular zinc and ATP-gated P2X receptor calcium entry channels: New zinc receptors as physiological sensory and therapeutic targets. Purinergic Signal. 2005, 1, 299–310. [Google Scholar] [CrossRef]
- Zsembery, Á.; Fortenberry, J.A.; Liang, L.; Bebok, Z.; Tucker, T.A.; Boyce, A.T.; Braunstein, G.M.; Welty, E.; Bell, P.D.; Sorcher, E.J.; et al. Extracellular zinc and ATP restore chloride secretion across cystic fibrosis airway epithelia by triggering calcium entry. J. Biol. Chem. 2004, 279, 10720–10729. [Google Scholar] [CrossRef]
- Lorca, R.A.; Rozas, C.; Loyola, S.; Moreira-Ramos, S.; Zeise, M.L.; Kirkwood, A.; Huidobro-Toro, J.P.; Morales, B. Zinc enhances long-term potentiation through P2X receptor modulation in the hippocampal CA1 region. Eur. J. Neurosci. 2011, 33, 1175–1185. [Google Scholar] [CrossRef]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef]
- Linkous, D.H.; Flinn, J.M.; Koh, J.Y.; Lanzirotti, A.; Bertsch, P.M.; Jones, B.F.; Giblin, L.J.; Frederickson, C.J. Evidence that the ZnT3 protein controls the total amount of elemental zinc in synaptic vesicles. J. Histochem. Cytochem. 2008, 56, 3–6. [Google Scholar] [CrossRef]
- Levenson, C.W.; Morris, D. Zinc and neurogenesis: Making new neurons from development to adulthood. Adv. Nutr. 2011, 2, 96–100. [Google Scholar] [CrossRef]
- Gao, H.L.; Zheng, W.; Xin, N.; Chi, Z.H.; Wang, Z.Y.; Chen, J.; Wang, Z.Y. Zinc deficiency reduces neurogenesis accompanied by neuronal apoptosis through caspase-dependent and –independent signaling pathways. Neurotox Res. 2009, 16, 416–425. [Google Scholar] [CrossRef]
- Suh, S.W.; Won, S.J.; Hamby, A.M.; Yoo, B.H.; Fan, Y.; Sheline, C.T.; Tamano, H.; Takeda, A.; Liu, J. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J. Cereb. Blood Flow Metab. 2009, 29, 1579–1588. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014, 28, 474–481. [Google Scholar] [CrossRef]
- Flinn, J.M.; Hunter, D.; Linkous, D.H.; Lanzirotti, A.; Smith, L.N.; Brightwell, J.; Jones, B.F. Enhanced zinc consumption causes memory deficits and increased brain levels of zinc. Physiol. Behav. 2005, 83, 793–803. [Google Scholar] [CrossRef]
- Craven, K.M.; Kochen, W.R.; Hernandez, C.M.; Flinn, J.M. Zinc exacerbates tau pathology in a tau mouse model. J. Alzheimer’s Dis. 2018, 64, 1–14, (Preprint). [Google Scholar] [CrossRef]
- Linkous, D.H.; Adlard, P.A.; Wanschura, P.B.; Conko, K.M.; Flinn, J.M. The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimer’s Dis. 2009, 18, 565–579. [Google Scholar] [CrossRef]
- Railey, A.M.; Micheli, T.L.; Wanschura, P.B.; Flinn, J.M. Alterations in fear response and spatial memory in pre- and post-natal zinc supplemented rats: Remediation by copper. Physiol. Behav. 2010, 100, 95–100. [Google Scholar] [CrossRef]
- Flinn, J.M.; Bozzelli, P.L.; Adlard, P.A.; Railey, A.M. Spatial memory deficits in a mouse model of late-onset Alzheimer’s disease are caused by zinc supplementation and correlate with amyloid-beta levels. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Lippi, S.L.P.; Smith, M.L.; Flinn, J.M. A novel hAPP/htau mouse model of Alzheimer’s disease: Inclusion of APP with tau exacerbates behavioral deficits and zinc administration heightens tangle pathology. Front. Aging Neurosci. 2018, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Boroujeni, S.T.; Naghdi, N.; Shahbazi, M.; Farrokhi, A.; Bagherzadeh, F.; Kazemnejad, A.; Javadian, M. The effect of severe zinc deficiency and zinc supplement on spatial learning and memory. Biol. Trace Elem. Res. 2009, 130, 48–61. [Google Scholar] [CrossRef]
- Corona, C.; Masciopinto, F.; Silvestri, E.; Del Viscovo, A.; Lattanzio, R.; La Sorda, R.; Ciavardelli, D.; Goglia, F.; Piantelli, M.; Canzoniero, L.M.T.; et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 2010, 1, e91. [Google Scholar] [CrossRef]
- Takeda, A. Zinc signaling in the hippocampus and its relation to pathogenesis of depression. J. Trace Elem. Med. Biol. 2012, 26, 80–84. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 9, 36–45. [Google Scholar] [CrossRef]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-M.; Gybina, A.A.; Pyatskowit, J.W.; Gitschier, J.; Prohaska, J.R. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J. Nutr. 2006, 136, 21–26. [Google Scholar] [CrossRef]
- Zatta, P.; Frank, A. Copper deficiency and neurological disorders in man and animals. Brain Res. Rev. 2007, 54, 19–33. [Google Scholar] [CrossRef]
- Bolognin, S.; Pasqualetto, F.; Mucignat-Caretta, C.; Scancar, J.; Milacic, R.; Zambenedetti, P.; Cozzi, B.; Zatta, P. Effects of a copper-deficient diet on the biochemistry, neural morphology, and behavior of aged mice. PLoS ONE 2012, 7, e47063. [Google Scholar] [CrossRef]
- Peters, C.; Muñoz, B.; Sepúlveda, F.J.; Urrutia, J.; Quiroz, M.; Luza, S.; De Ferrari, G.V.; Aguayo, L.G.; Opazo, C. Biphasic effects of copper on neurotransmission in rat hippocampal neurons. J. Neurochem. 2011, 119, 78–88. [Google Scholar] [CrossRef]
- Davis, M.; Myers, K.M. The role of glutamate and gamma-aminobutyric acid in fear extinction: Clinical implications for exposure therapy. Biol. Psychiatry 2002, 52, 998–1007. [Google Scholar] [CrossRef]
- Davis, M. Role of NMDA receptors and MAP kinase in the amygdala in extinction of fear: Clinical implications for exposure therapy. Eur. J. Neurosci. 2002, 16, 395–398. [Google Scholar] [CrossRef]
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608. [Google Scholar] [CrossRef]
- Yates, M.A.; Juraska, J.M. Increases in size and myelination of the rat corpus callosum during adulthood are maintained into old age. Brain Res. 2007, 1142, 13–18. [Google Scholar] [CrossRef]
- Pushkar, Y.; Robison, G.; Sullivan, B.; Zheng, W.; Fu, S.X.; Kohne, M.; Jiang, W.; Rohr, S.; Lai, B.; Marcus, M.A.; et al. Aging results in copper accumulations in GFAP-positive cells in the subventricular zone. Aging Cell 2013, 12, 823–832. [Google Scholar] [CrossRef]
- Fu, S.; Jiang, W.; Zheng, W. Age-dependent increase of brain copper levels and expressions of copper regulatory proteins in the subventricular zone and choroid plexus. Front. Mol. Neurosci. 2015, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; O’Neal, S.; Hong, L.; Jiang, W.; Zheng, W. Elevated adult neurogenesis in brain subventricular zone following in vivo manganese exposure: Roles of copper and DMT1. Toxicol. Sci. 2015, 143, 482–498. [Google Scholar] [CrossRef]
- Doreulee, N.; Yanovsky, Y.; Haas, H.L. Suppression of long-term potentiation in hippocampal slices by copper. Hippocampus 1997, 7, 666–669. [Google Scholar] [CrossRef]
- Goldschmith, A.; Infante, C.; Leiva, J.; Motles, E.; Palestini, M. Interference of chronically ingested copper in long-term potentiation (LTP) of rat hippocampus. Brain Res. 2005, 1056, 176–182. [Google Scholar] [CrossRef]
- Leiva, J.; Palestini, M.; Infante, C.; Goldschmidt, A.; Motles, E. Copper suppresses hippocampus LTP in the rat, but does not alter learning or memory in the morris water maze. Brain Res. 2009, 1256, 69–75. [Google Scholar] [CrossRef]
- Jand, A.; Taheri-nejad, M.R.; Mosleh, M.; Palizvan, M.R. Low, but not high, doses of copper sulfate impair synaptic plasticity in the hippocampal CA1 region in vivo. Biol. Trace Elem. Res. 2017, 185, 143–147. [Google Scholar] [CrossRef]
- Sakimura, K.; Kutsuwada, T.; Ito, I.; Manabe, T.; Takayama, C.; Kushiya, E.; Yagi, T.; Aizawa, S.; Inoue, Y.; Sugiyama, H.; et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor ε1 subunit. Nature 1995, 373, 151. [Google Scholar] [CrossRef]
- Ortiz, O.; Delgado-García, J.M.; Espadas, I.; Bahí, A.; Trullas, R.; Dreyer, J.L.; Gruart, A.; Moratalla, R. Associative learning and CA3–CA1 synaptic plasticity are impaired in D1R null, Drd1a−/− mice and in hippocampal siRNA silenced Drd1a mice. J. Neurosci. 2010, 30, 12288–12300. [Google Scholar] [CrossRef]
- Chhetri, S.K.; Mills, R.J.; Shaunak, S.; Emsley, H.C.A. Copper deficiency. Br. Med. J. 2014, 348, g3691. [Google Scholar] [CrossRef]
- Knight, J. Metal heads. N. Sci. 2000, 167, 36–39. [Google Scholar]
- Maret, W.; Sanstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 201, 3–18. [Google Scholar] [CrossRef]
- Nations, S.P.; Boyer, P.J.; Love, L.A.; Burritt, M.F.; Butz, J.A.; Wolfe, G.I.; Hynan, L.S.; Reisch, J.; Trivdei, J.R. Denture cream: An unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology 2008, 71, 639–643. [Google Scholar] [CrossRef]
- Railey, A.M.; Groeber, C.M.; Flinn, J.M. The effect of metals on spatial memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 24, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162. [Google Scholar] [CrossRef]
- Salvador, G.A. Iron in neuronal function and dysfunction. BioFactors 2010, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Connor, J.R.; Jones, B.C. Iron in the brain. Nutr. Rev. 1993, 51, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.-H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef]
- Hallgren, B.; Sourander, P. The effect of age on the non-haemin iron in the human brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef]
- Rajan, K.S.; Manian, A.A.; Davis, J.M.; Dekirmenjian, H. Metal chelates of 1-dopa for improved replenishment of dopaminergic pools. Brain Res. 1976, 107, 317–331. [Google Scholar] [CrossRef]
- Aoki, S.; Okada, Y.; Nishimura, K.; Barkovich, A.J.; Kjos, B.O.; Brasch, R.C.; Norman, D. Normal deposition of brain iron in childhood and adolescence: MR imaging at 1.5 T. Radiology 1989, 172, 381–385. [Google Scholar] [CrossRef]
- Pino, J.M.V.; da Luz, M.H.M.; Antunes, H.K.M.; de Campos Giampá, S.Q.; Martins, V.R.; Lee, K.S. Iron-restricted diet affects brain ferritin levels, dopamine metabolism and cellular prion protein in a region-specific manner. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and myelination: The role of Iron. GLIA 2009, 57, 467–478. [Google Scholar] [CrossRef]
- Lozoff, B.; Georgieff, M.K. Iron deficiency and brain development. Semin. Pediatr. Neurol. 2006, 13, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, E.K.; Claussen, A.H.; Scott, K.G. Early childhood anemia and mild or moderate mental retardation. Am. J. Clin. Nutr. 1999, 69, 115–119. [Google Scholar] [CrossRef]
- Pollitt, E. Early iron deficiency anemia and later mental retardation. Am. J. Clin. Nutr. 1999, 69, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Su, T.P.; Chen, Y.S.; Hsu, J.W.; Huang, K.L.; Chang, W.H.; Chen, T.J.; Bai, Y.M. Association between psychiatric disorders and iron deficiency anemia among children and adolescents: A nationwide population-based study. BMC Psychiatry 2013, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Nichol, H.; Liu, S.; Cheng, Y.C.N.; Haacke, E.M. Measuring iron in the brain using quantitative susceptibility mapping and X-ray fluorescence imaging. Neuroimage 2013, 78, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.S.; Tkac, I.; Magid, R.; O’Connor, M.B.; Andrews, N.C.; Schallert, T.; Gunshin, H.; Georgieff, M.K.; Petryk, A. Iron is essential for neuron development and memory function in mouse hippocampus. J. Nutr. 2009, 139, 672–679. [Google Scholar] [CrossRef]
- Fretham, S.J.; Carlson, E.S.; Georgieff, M.K. The role of iron in learning and memory. Adv. Nutr. 2011, 2, 112–121. [Google Scholar] [CrossRef]
- Jorgenson, L.A.; Wobken, J.D.; Georgieff, M.K. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev. Neurosci. 2003, 25, 412–420. [Google Scholar] [CrossRef]
- Robison, G.; Zakharova, T.; Fu, S.; Jiang, W.; Fulper, R.; Barrea, R.; Zheng, W.; Pushkar, Y. X-ray fluorescence imaging of the hippocampal formation after manganese exposure. Metallomics 2013, 5, 1554–1565. [Google Scholar] [CrossRef]
- Cheah, J.H.; Kim, S.F.; Hester, L.D.; Clancy, K.W.; Patterson, S.E., III; Papadopoulos, V.; Snyder, S.H. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 2006, 51, 431–440. [Google Scholar] [CrossRef]
- Pelizzoni, I.; Macco, R.; Morini, M.F.; Zacchetti, D.; Grohovaz, F.; Codazzi, F. Iron handling in hippocampal neurons: Activity-dependent iron entry and mitochondria-mediated neurotoxicity. Aging Cell 2011, 10, 172–183. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, H.; Xie, J. New insights into the crosstalk between NMDARs and iron: Implications for understanding pathology of neurological diseases. Front. Mol. Neurosci. 2017, 10, 71. [Google Scholar] [CrossRef]
- White, R.S.; Bhattacharya, A.K.; Chen, Y.; Byrd, M.; McMullen, M.F.; Siegel, S.J.; Carlson, G.C.; Kim, S.F. Lysosomal iron modulates NMDA receptor-mediate excitation via small GTPase, Dexras1. Mol. Brain 2016, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Humeres Al Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Núñez, M.T. Iron mediates N-Methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef] [PubMed]
- Georgieff, M.K. The role of iron in neurodevelopment: Fetal iron deficiency and the developing hippocampus. Biochem. Soc. Trans. 2008, 36 Pt 6, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; Sousa, N.; Bessa, J.M.; Sousa, J.C.; Marques, F. Metabolism and adult neurogenesis: Towards an understanding of the role of lipocalin-2 and iron-related oxidative stress. Neurosci. Biobehav. Rev. 2018, 95, 73–84. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Eccleston, P.A.; Silberberg, D.H. The differentiation of oligodendrocytes in a serum-free hormone-supplemented medium. Dev. Brain Res. 1984, 16, 1–9. [Google Scholar] [CrossRef]
- Tran, P.V.; Carlson, E.S.; Fretham, S.J.; Georgieff, M.K. Early-life iron deficiency anemia alters neurotrophic factor expression and hippocampal neuron differentiation in male rats. J. Nutr. 2008, 138, 2495–2501. [Google Scholar] [CrossRef]
- Lozoff, B. Early iron deficiency has brain and behavior effects consistent with dopaminergic dysfunction–3. J. Nutr. 2011, 141, 740S–746S. [Google Scholar] [CrossRef] [PubMed]
- Brigman, J.L.; Wright, T.; Talani, G.; Prasad-Mulcare, S.; Jinde, S.; Seabold, G.K.; Mathur, P.; Davis, M.I.; Bock, R.; Gustin, R.M.; et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density and disrupts learning. J. Neurosci. 2010, 30, 4590–4600. [Google Scholar] [CrossRef] [PubMed]
- Felt, B.T.; Beard, J.L.; Schallert, T.; Shao, J.; Aldridge, J.W.; Connor, J.R.; Georgieff, M.K.; Lozoff, B. Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav. Brain Res. 2006, 171, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Kwik-Uribe, C.L.; Golub, M.S.; Keen, C.L. Chronic marginal iron intakes during early development in mice alter brain iron concentrations and behavior despite postnatal iron supplementation. J. Nutr. 2000, 130, 2040–2048. [Google Scholar] [CrossRef]
- Beard, J.; Erikson, K.M.; Jones, B.C. Neonatal iron deficiency results in irreversible changes in dopamine function in rats. J. Nutr. 2003, 133, 1174–1179. [Google Scholar] [CrossRef]
- Dunnett, S.B.; Torres, E.M.; Annett, L.E. A lateralised grip strength test to evaluate unilateral nigrostriatal lesions in rats. Neurosci. Lett. 1998, 246, 1–4. [Google Scholar] [CrossRef]
- Maaroufi, K.; Ammari, M.; Jeljeli, M.; Roy, V.; Sakly, M.; Abdelmelek, H. Impairment of emotional behavior and spatial learning in adult Wistar rats by ferrous sulfate. Physiol. Behav. 2009, 96, 343–349. [Google Scholar] [CrossRef]
- Rodrigue, K.M.; Daugherty, A.M.; Haacke, E.M.; Raz, N. The role of hippocampal iron concentration and hippocampal volume in age-related differences in memory. Cereb. Cortex 2013, 23, 1533–1541. [Google Scholar] [CrossRef]
- Ha, J.H.; Doguer, C.; Wang, X.; Flores, S.R.; Collins, J.F. High-iron consumption impairs growth and causes copper-deficiency anemia in weanling Sprague-Dawley rats. PLoS ONE 2016, 11, e0161033. [Google Scholar] [CrossRef]
- Espinoza, A.; Le Blanc, S.; Olivares, M.; Pizarro, F.; Ruz, M.; Arredondo, M. Iron, copper, and zinc transport: Inhibition of divalent metal transporter 1 (DMT1) and human copper transporter 1 (hCTR1) by shRNA. Biol. Trace. Elem. Res. 2012, 146, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C. Interaction of metal ions with neurotransmitter receptors and potential role in neurodiseases. Biometals 2014, 27, 1097–1113. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neely, C.L.C.; Lippi, S.L.P.; Lanzirotti, A.; Flinn, J.M. Localization of Free and Bound Metal Species through X-Ray Synchrotron Fluorescence Microscopy in the Rodent Brain and Their Relation to Behavior. Brain Sci. 2019, 9, 74. https://doi.org/10.3390/brainsci9040074
Neely CLC, Lippi SLP, Lanzirotti A, Flinn JM. Localization of Free and Bound Metal Species through X-Ray Synchrotron Fluorescence Microscopy in the Rodent Brain and Their Relation to Behavior. Brain Sciences. 2019; 9(4):74. https://doi.org/10.3390/brainsci9040074
Chicago/Turabian StyleNeely, Caroline L. C., Stephen L. P. Lippi, Antonio Lanzirotti, and Jane M. Flinn. 2019. "Localization of Free and Bound Metal Species through X-Ray Synchrotron Fluorescence Microscopy in the Rodent Brain and Their Relation to Behavior" Brain Sciences 9, no. 4: 74. https://doi.org/10.3390/brainsci9040074
APA StyleNeely, C. L. C., Lippi, S. L. P., Lanzirotti, A., & Flinn, J. M. (2019). Localization of Free and Bound Metal Species through X-Ray Synchrotron Fluorescence Microscopy in the Rodent Brain and Their Relation to Behavior. Brain Sciences, 9(4), 74. https://doi.org/10.3390/brainsci9040074